
Targeting Myocardial Fibrosis—A Magic Pill in Cardiovascular Medicine?


Abstract
:1. Introduction
2. The Extracellular Matrix—From Physiology to Pathophysiology
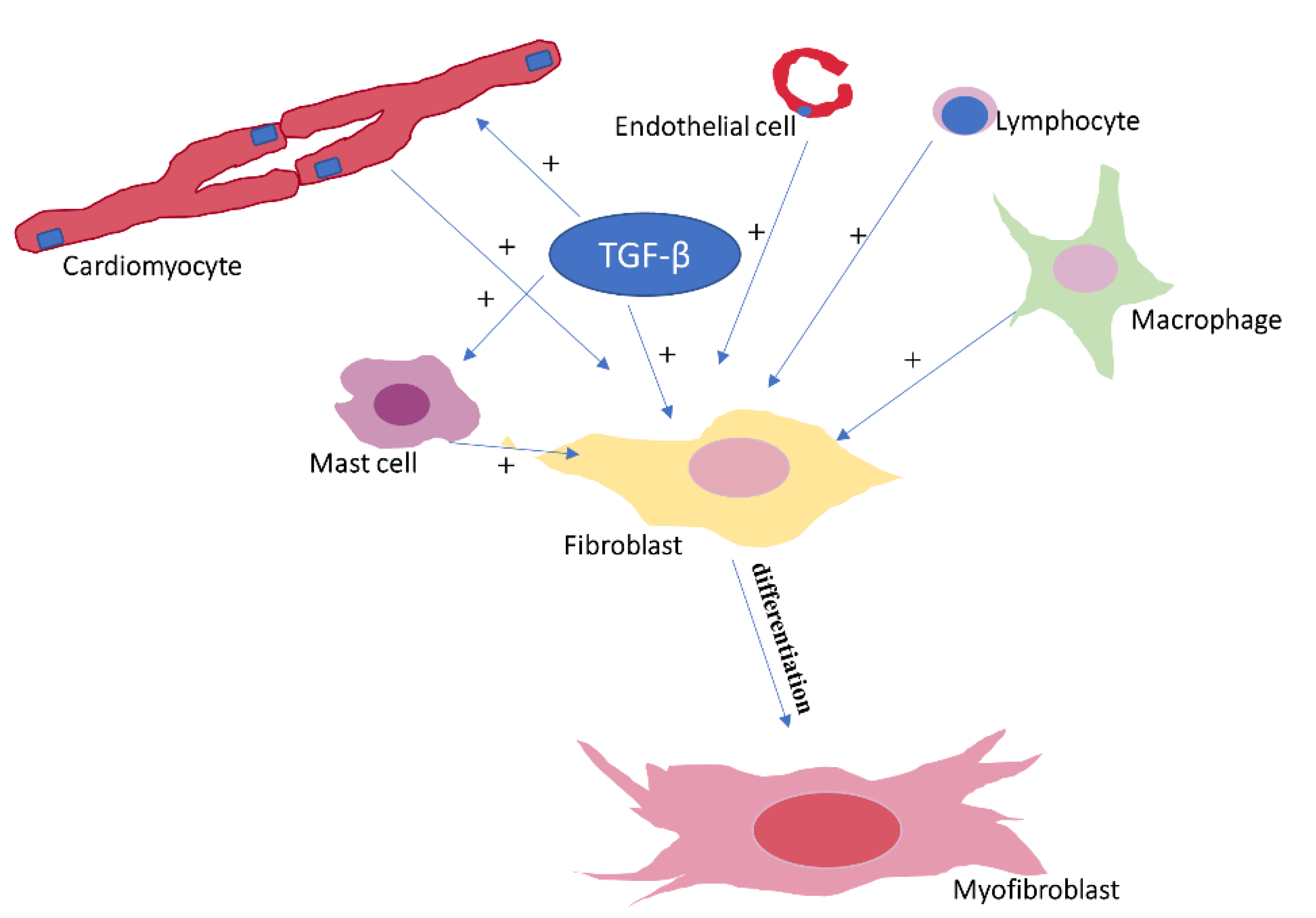
2.1. Cellular Components Involved in Cardiac Fibrosis
2.2. Extracellular Components Involved in Cardiac Fibrosis
2.3. Types of Cardiac Fibrosis
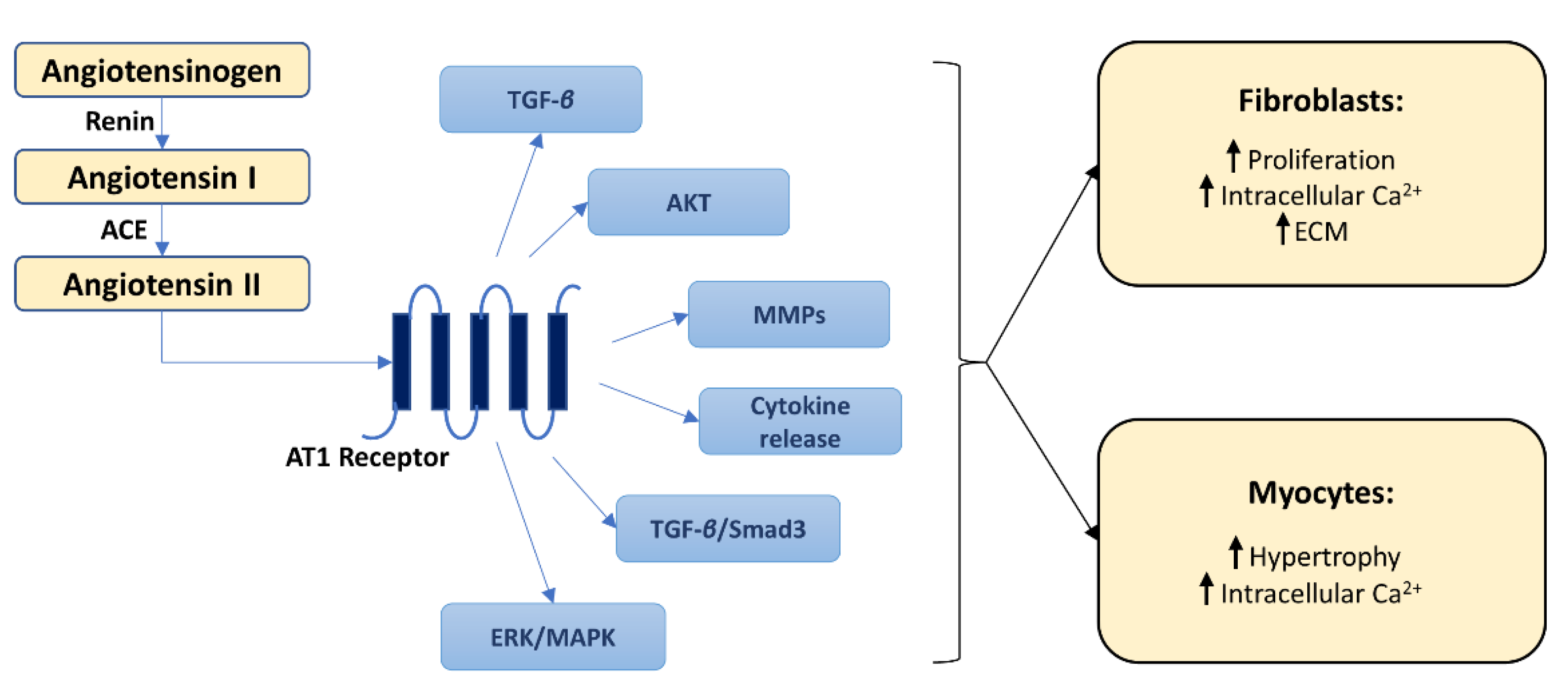
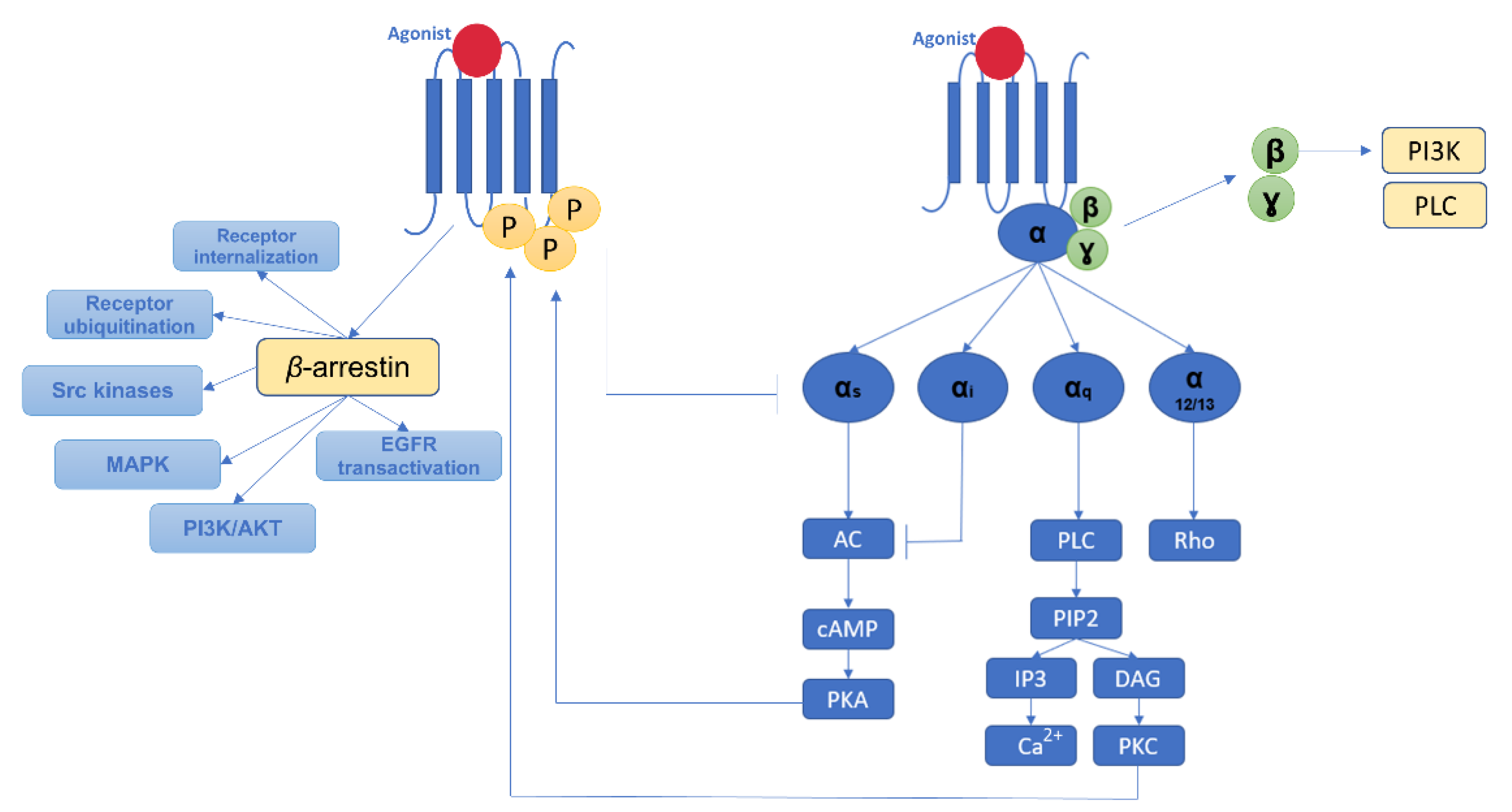
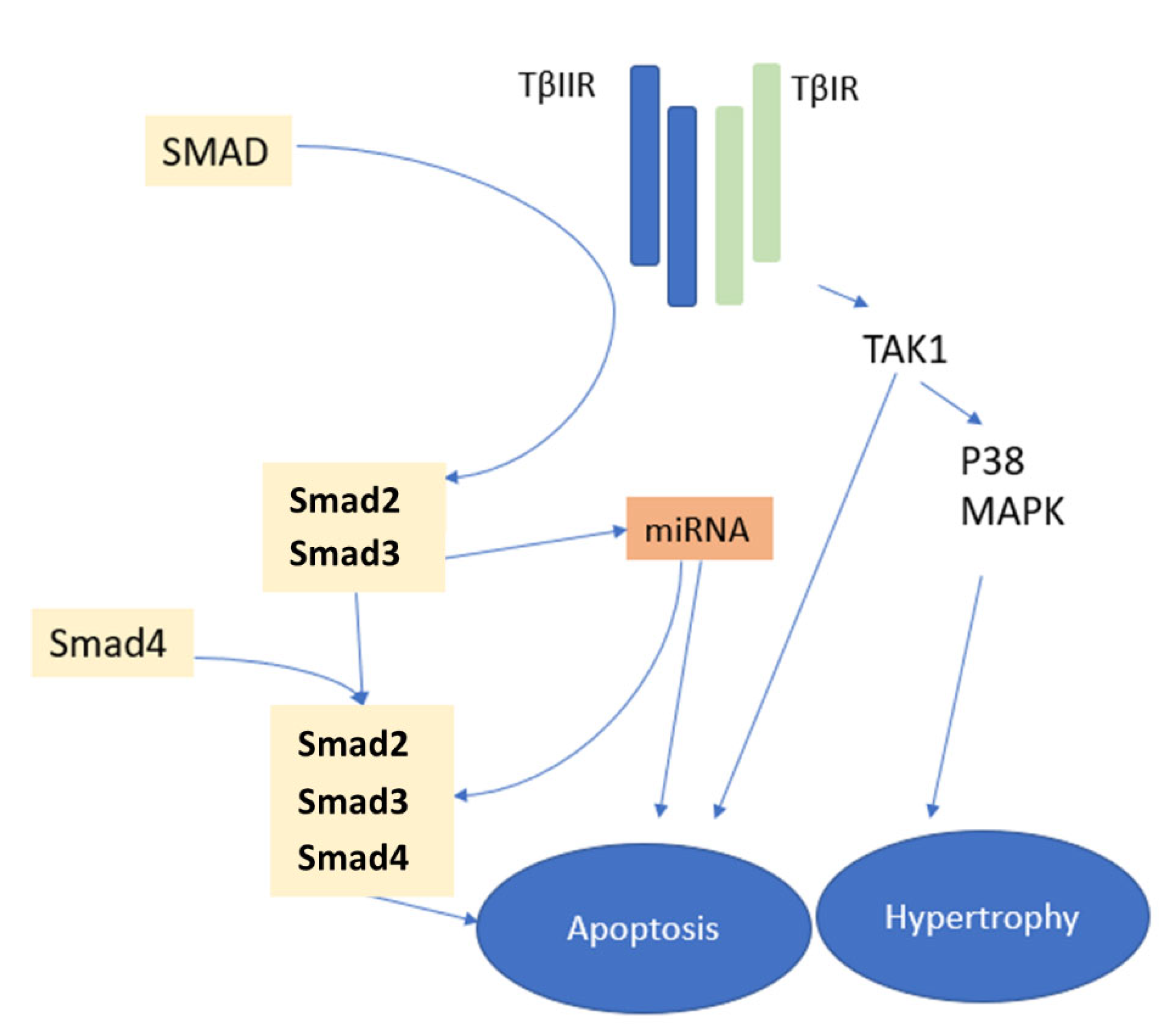
2.4. Molecular Pathways of Myocardial Fibrosis
3. Identification and Quantification of Myocardial Fibrosis
3.1. Invasive Methods to Investigate Cardiac Fibrosis
3.2. Non-Invasive Methods to Evaluate Cardiac Fibrosis
4. Targeting Myocardial Fibrosis—A Magic Pill in Cardiovascular Medicine?
4.1. Cardiac Antifibrotic Effects of Non-Antifibrotic Drugs
4.2. Novel Targets for Cardiac Fibrosis Prevention and Therapy
4.3. Targeted Blockade—Aiming to Obtain a ‘Better Scar’
4.4. Indirect Blockade of Fibrosis via Stimulation of Myocardial Regeneration/Repair
4.5. Modulation of Collagen Turnover
5. Gaps in Knowledge, Ongoing and Future Research
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef]
- Torrent-Guasp, F.; Kocica, M.J.; Corno, A.F.; Komeda, M.; Carreras-Costa, F.; Flotats, A.; Cosin-Aguillar, J.; Wen, H. Towards new understanding of the heart structure and function. Eur. J. Cardio-Thorac. Surg. 2005, 27, 191–201. [Google Scholar] [CrossRef]
- Burlew, B.S.; Weber, K.T. Connective Tissue and the Heart. Functional significance and regulatory mechanisms. Cardiol. Clin. 2000, 18, 435–442. [Google Scholar] [CrossRef]
- Spinale, F.G. Myocardial Matrix Remodeling and the Matrix Metalloproteinases: Influence on Cardiac Form and Function. Physiol. Rev. 2007, 87, 1285–1342. [Google Scholar] [CrossRef] [PubMed]
- Gaudesius, G.; Miragoli, M.; Thomas, S.P.; Rohr, S. Coupling of Cardiac Electrical Activity Over Extended Distances by Fibroblasts of Cardiac Origin. Circ. Res. 2003, 93, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Willems, I.E.; Havenith, M.G.; De Mey, J.G.; Daemen, M.J. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am. J. Pathol. 1994, 145, 868–875. [Google Scholar] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.-L.; Gabbiani, G. The Myofibroblast: One Function, Multiple Origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.-J.; Dangerfield, A.L.; Rattan, S.G.; Bathe, K.L.; Cunnington, R.H.; Raizman, J.E.; Bedosky, K.M.; Freed, D.H.; Kardami, E.; Dixon, I.M.C. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: Expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev. Dyn. 2010, 239, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2012, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, P.; Ren, G.; Nagar, H.; Kraemer, D.; Mendoza, L.; Michael, L.H.; Caughey, G.H.; Entman, M.L.; Frangogiannis, N.G. Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. J. Pathol. 2004, 205, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Baldeviano, G.C.; Barin, J.G.; Talor, M.V.; Srinivasan, S.; Bedja, D.; Zheng, D.; Gabrielson, K.; Iwakura, Y.; Rose, N.R.; Cihakova, D. Interleukin-17A Is Dispensable for Myocarditis but Essential for the Progression to Dilated Cardiomyopathy. Circ. Res. 2010, 106, 1646–1655. [Google Scholar] [CrossRef]
- Tang, T.-T.; Yuan, J.; Zhu, Z.-F.; Zhang, W.-C.; Xiao, H.; Xia, N.; Yan, X.-X.; Nie, S.-F.; Liu, J.; Zhou, S.-F.; et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 2011, 107, 232. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From inflammation to fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Grabmaier, U.; Uhl, A.; Gess, S.; Boehm, F.; Zehrer, A.; Pick, R.; Salvermoser, M.; Czermak, T.; Pircher, J.; et al. Midkine drives cardiac inflammation by promoting neutrophil trafficking and NETosis in myocarditis. J. Exp. Med. 2019, 216, 350–368. [Google Scholar] [CrossRef]
- Adiarto, S.; Heiden, S.; Vignon-Zellweger, N.; Nakayama, K.; Yagi, K.; Yanagisawa, M.; Emoto, N. ET-1 from endothelial cells is required for complete angiotensin II-induced cardiac fibrosis and hypertrophy. Life Sci. 2012, 91, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Kofler, S.; Nickel, T.; Weis, M. Role of cytokines in cardiovascular diseases: A focus on endothelial responses to inflammation. Clin. Sci. 2005, 108, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Bedja, D.; Koitabashi, N.; Xing, D.; Chen, J.; Fox-Talbot, K.; Rouf, R.; Chen, S.; Steenbergen, C.; Harmon, J.W.; et al. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of TGF-β signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E841–E850. [Google Scholar] [CrossRef]
- Piek, A.; de Boer, R.A.; Silljé, H.H.W. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Dolmatova, E.; Spagnol, G.; Boassa, D.; Baum, J.R.; Keith, K.; Ambrosi, C.; Kontaridis, M.I.; Sorgen, P.L.; Sosinsky, G.E.; Duffy, H.S. Cardiomyocyte ATP release through pannexin 1 aids in early fibroblast activation. Am. J. Physiol. Circ. Physiol. 2012, 303, H1208–H1218. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.J.; Morgan, J.; Bienvenu, L.A.; Fletcher, E.K.; Cranston, G.A.; Shen, J.Z.; Reichelt, M.E.; Delbridge, L.M.; Young, M.J. Cardiomyocyte Mineralocorticoid Receptors Are Essential for Deoxycorticosterone/Salt-Mediated Inflammation and Cardiac Fibrosis. Hypertension 2012, 60, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Koitabashi, N.; Danner, T.; Zaiman, A.L.; Pinto, Y.M.; Rowell, J.; Mankowski, J.; Zhang, D.; Nakamura, T.; Takimoto, E.; Kass, D.A. Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload. J. Clin. Investig. 2011, 121, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Kurisu, S.; Ozono, R.; Oshima, T.; Kambe, M.; Ishida, T.; Sugino, H.; Matsuura, H.; Chayama, K.; Teranishi, Y.; Iba, O.; et al. Cardiac Angiotensin II Type 2 Receptor Activates the Kinin/NO System and Inhibits Fibrosis. Hypertension 2003, 41, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Sen, S. Alteration of Cardiac Collagen Phenotypes in Hypertensive Hypertrophy: Role of Blood Pressure. J. Mol. Cell. Cardiol. 1993, 25, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Sen, S. Alteration of collagen phenotypes in ischemic cardiomyopathy. J. Clin. Investig. 1991, 88, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Naugle, J.E.; Olson, E.R.; Zhang, X.; Mase, S.E.; Pilati, C.F.; Maron, M.B.; Folkesson, H.G.; Horne, W.I.; Doane, K.J.; Meszaros, J.G. Type VI collagen induces cardiac myofibroblast differentiation: Implications for postinfarction remodeling. Am. J. Physiol. Circ. Physiol. 2006, 290, H323–H330. [Google Scholar] [CrossRef] [PubMed]
- Graham-Brown, M.P.M.; Patel, A.S.; Stensel, D.J.; March, D.S.; Marsh, A.-M.; McAdam, J.; McCann, G.P.; Burton, J.O. Imaging of Myocardial Fibrosis in Patients with End-Stage Renal Disease: Current Limitations and Future Possibilities. BioMed Res. Int. 2017, 2017, 5453606. [Google Scholar] [CrossRef]
- Iwanaga, Y.; Aoyama, T.; Kihara, Y.; Onozawa, Y.; Yoneda, T.; Sasayama, S. Excessive activation of matrix metalloproteinases coincides with left ventricular remodeling during transition from hypertrophy to heart failure in hypertensive rats. J. Am. Coll. Cardiol. 2002, 39, 1384–1391. [Google Scholar] [CrossRef]
- Arisha, M.M.; Girerd, N.; Chauveau, S.; Bresson, D.; Scridon, A.; Bonnefoy, E.; Chevalier, P. In-Hospital Heart Rate Turbulence and Microvolt T-Wave Alternans Abnormalities for Prediction of Early Life-Threatening Ventricular Arrhythmia after Acute Myocardial Infarction. Ann. Noninvasive Electrocardiol. 2013, 18, 530–537. [Google Scholar] [CrossRef]
- Brilla, C.G.; Zhou, G.; Matsubara, L.; Weber, K.T. Collagen Metabolism in Cultured Adult Rat Cardiac Fibroblasts: Response to Angiotensin II and Aldosterone. J. Mol. Cell. Cardiol. 1994, 26, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2012, 10, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fan, D.; Wang, C.; Wang, J.-Y.; Cui, X.; Wu, D.; Zhou, Y.; Wu, L.-L. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-β1 pathways in cardiac fibroblasts. Cardiovasc. Res. 2011, 91, 80–89. [Google Scholar] [CrossRef]
- Ock, S.; Ham, W.; Kang, C.W.; Kang, H.; Lee, W.S.; Kim, J. IGF-1 protects against angiotensin II-induced cardiac fibrosis by targeting αSMA. Cell Death Dis. 2021, 12, 688. [Google Scholar] [CrossRef] [PubMed]
- Rompe, F.; Artuc, M.; Hallberg, A.; Alterman, M.; Ströder, K.; Thöne-Reineke, C.; Reichenbach, A.; Schacherl, J.; Dahlöf, B.; Bader, M.; et al. Direct Angiotensin II Type 2 Receptor Stimulation Acts Anti-Inflammatory Through Epoxyeicosatrienoic Acid and Inhibition of Nuclear Factor κB. Hypertension 2010, 55, 924–931. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-Induced Inflammation in the Rat Heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Nakaya, M.; Chikura, S.; Watari, K.; Mizuno, N.; Mochinaga, K.; Mangmool, S.; Koyanagi, S.; Ohdo, S.; Sato, Y.; Ide, T.; et al. Induction of Cardiac Fibrosis by β-Blocker in G Protein-independent and G Protein-coupled Receptor Kinase 5/β-Arrestin2-dependent Signaling Pathways. J. Biol. Chem. 2012, 287, 35669–35677. [Google Scholar] [CrossRef]
- Communal, C.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Opposing Effects of β1- and β2-Adrenergic Receptors on Cardiac Myocyte Apoptosis: Role of a pertussis toxin-sensitive G protein. Circulation. Circulation 1999, 100, 2210–2212. [Google Scholar] [CrossRef]
- Benjamin, I.J.; Jalil, J.E.; Tan, L.B.; Cho, K.; Weber, K.T.; Clark, W.A. Isoproterenol-induced myocardial fibrosis in relation to myocyte necrosis. Circ. Res. 1989, 65, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.-N.; Kiriazis, H.; Ruggiero, D.; Gao, X.-M.; Su, Y.; Jian, A.; Han, L.-P.; McMullen, J.R.; Du, X.-J. Spontaneous ventricular tachyarrhythmias in β2-adrenoceptor transgenic mice in relation to cardiac interstitial fibrosis. Am. J. Physiol. Circ. Physiol. 2015, 309, H946–H957. [Google Scholar] [CrossRef] [PubMed]
- Hermida, N.; Michel, L.Y.; Esfahani, H.; Dubois-Deruy, E.; Hammond, J.; Bouzin, C.; Markl, A.; Colin, H.; Van Steenbergen, A.; De Meester, C.; et al. Cardiac myocyte β3-adrenergic receptors prevent myocardial fibrosis by modulating oxidant stress-dependent paracrine signaling. Eur. Heart J. 2017, 39, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Masuyama, T.; Sakata, Y.; Mano, T.; Nishikawa, N.; Kondo, H.; Akehi, N.; Kuzuya, T.; Miwa, T.; Hori, M. Roles of renin–angiotensin and endothelin systems in development of diastolic heart failure in hypertensive hearts. Cardiovasc. Res. 2000, 47, 274–283. [Google Scholar] [CrossRef]
- Kulasekaran, P.; Scavone, C.A.; Rogers, D.S.; Arenberg, D.A.; Thannickal, V.J.; Horowitz, J.C. Endothelin-1 and Transforming Growth Factor-β1 Independently Induce Fibroblast Resistance to Apoptosis via AKT Activation. Am. J. Respir. Cell Mol. Biol. 2009, 41, 484–493. [Google Scholar] [CrossRef]
- Wray, D.W.; Nishiyama, S.K.; Richardson, R.S. Role of α1-adrenergic vasoconstriction in the regulation of skeletal muscle blood flow with advancing age. Am. J. Physiol. Circ. Physiol. 2009, 296, H497–H504. [Google Scholar] [CrossRef]
- Sun, M.; Chen, M.; Dawood, F.; Zurawska, U.; Li, J.Y.; Parker, T.S.; Kassiri, Z.; Kirshenbaum, L.A.; Arnold, M.; Khokha, R.; et al. Tumor Necrosis Factor-α Mediates Cardiac Remodeling and Ventricular Dysfunction After Pressure Overload State. Circulation 2007, 115, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Torre-Amione, G.; Warren, M.S.; Whitmore, J.; Soran, O.Z.; Feldman, A.M.; Mann, U.L. Results of Targeted Anti–Tumor Necrosis Factor Therapy with Etanercept (ENBREL) in Patients with Advanced Heart Failure. Circulation 2001, 103, 1044–1047. [Google Scholar] [CrossRef]
- Okuno, T.; Andoh, A.; Bamba, S.; Araki, Y.; Fujiyama, Y.; Fujimiya, M.; Bamba, T. Interleukin-1β and Tumor Necrosis Factor-α Induce Chemokine and Matrix Metalloproteinase Gene Expression in Human Colonic Subepithelial Myofibroblasts. Scand. J. Gastroenterol. 2002, 37, 317–324. [Google Scholar] [CrossRef]
- Zhang, W.; Chancey, A.L.; Tzeng, H.-P.; Zhou, Z.; Lavine, K.J.; Gao, F.; Sivasubramanian, N.; Barger, P.M.; Mann, D.L. The Development of Myocardial Fibrosis in Transgenic Mice with Targeted Overexpression of Tumor Necrosis Factor Requires Mast Cell–Fibroblast Interactions. Circulation 2011, 124, 2106–2116. [Google Scholar] [CrossRef]
- Brønnum, H.; Eskildsen, T.; Andersen, D.C.; Schneider, M.; Sheikh, S.P. IL-1β suppresses TGF-β-mediated myofibroblast differentiation in cardiac fibroblasts. Growth Factors 2013, 31, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Lekakis, J.P.; Nikolaou, M.; Paraskevaidis, I.; Andreadou, I.; Kaplanoglou, T.; Katsimbri, P.; Skarantavos, G.; Soucacos, P.N.; Kremastinos, D.T. Inhibition of Interleukin-1 by Anakinra Improves Vascular and Left Ventricular Function in Patients with Rheumatoid Arthritis. Circulation 2008, 117, 2662–2669. [Google Scholar] [CrossRef] [PubMed]
- Lai, N.C.; Gao, M.H.; Tang, E.; Tang, R.; Guo, T.; Dalton, N.D.; Deng, A.; Tang, T. Pressure overload-induced cardiac remodeling and dysfunction in the absence of interleukin 6 in mice. Lab. Investig. 2012, 92, 1518–1526. [Google Scholar] [CrossRef]
- Banerjee, I.; Fuseler, J.W.; Intwala, A.R.; Baudino, T.A. IL-6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. Am. J. Physiol. Circ. Physiol. 2009, 296, H1694–H1704. [Google Scholar] [CrossRef]
- Brooks, W.W.; Conrad, C.H. Myocardial Fibrosis in Transforming Growth Factor β1Heterozygous Mice. J. Mol. Cell. Cardiol. 2000, 32, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Joyce, J.; Margulies, K.B.; Tsuda, T. Enhanced Bioactive Myocardial Transforming Growth Factor-β in Advanced Human Heart Failure. Circ. J. 2014, 78, 2711–2718. [Google Scholar] [CrossRef]
- Rosenkranz, S.; Flesch, M.; Amann, K.; Haeuseler, C.; Kilter, H.; Seeland, U.; Schlüter, K.-D.; Böhm, M. Alterations of β-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-β1. Am. J. Physiol. Circ. Physiol. 2002, 283, H1253–H1262. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-β Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.-J.; et al. Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Huang, S.; Chen, B.; Su, Y.; Alex, L.; Humeres, C.; Shinde, A.V.; Conway, S.J.; Frangogiannis, N.G. Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart. J. Mol. Cell. Cardiol. 2019, 132, 84–97. [Google Scholar] [CrossRef]
- Kong, P.; Shinde, A.V.; Su, Y.; Russo, I.; Chen, B.; Saxena, A.; Conway, S.J.; Graff, J.M.; Frangogiannis, N.G. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation 2018, 137, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Bageghni, S.A.; Hemmings, K.E.; Zava, N.; Denton, C.P.; Porter, K.E.; Ainscough, J.F.X.; Drinkhill, M.J.; Turner, N.A. Cardiac fibroblast-specific p38α MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism. FASEB J. 2018, 32, 4941–4954. [Google Scholar] [CrossRef] [PubMed]
- Cosme, J.; Guo, H.; Hadipour-Lakmehsari, S.; Emili, A.; Gramolini, A.O. Hypoxia-Induced Changes in the Fibroblast Secretome, Exosome, and Whole-Cell Proteome Using Cultured, Cardiac-Derived Cells Isolated from Neonatal Mice. J. Proteome Res. 2017, 16, 2836–2847. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Milano, G.; Vassalli, G. Beneficial effects of exosomes secreted by cardiac-derived progenitor cells and other cell types in myocardial ischemia. Stem Cell Investig. 2017, 4, 93. [Google Scholar] [CrossRef]
- Yue, Y.; Garikipati, V.N.S.; Verma, S.K.; Goukassian, D.A.; Kishore, R. Interleukin-10 Deficiency Impairs Reparative Properties of Bone Marrow-Derived Endothelial Progenitor Cell Exosomes. Tissue Eng. Part A 2017, 23, 1241–1250. [Google Scholar] [CrossRef]
- Liu, T.; Song, D.; Dong, J.; Zhu, P.; Liu, J.; Liu, W.; Ma, X.; Zhao, L.; Ling, S. Current Understanding of the Pathophysiology of Myocardial Fibrosis and Its Quantitative Assessment in Heart Failure. Front. Physiol. 2017, 8, 238. [Google Scholar] [CrossRef]
- López, B.; González, A.; Ravassa, S.; Beaumont, J.; Moreno, M.U.; José, G.S.; Querejeta, R.; Díez, J. Circulating Biomarkers of Myocardial Fibrosis: The Need for a Reappraisal. J. Am. Coll. Cardiol. 2015, 65, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Gudowska-Sawczuk, M.; Gruszewska, E.; Panasiuk, A.; Cylwik, B.; Swiderska, M.; Flisiak, R.; Szmitkowski, M.; Chrostek, L. High serum N-terminal propeptide of procollagen type III concentration is associated with liver diseases. Gastroenterol. Rev. 2017, 12, 203–207. [Google Scholar] [CrossRef]
- Liu, S.; Wu, Q.; Zhang, S.; Wang, Z.; Liu, H.; Teng, L.; Xiao, P.; Lu, Y.; Wang, X.; Dong, C.; et al. Serum Galectin-3 levels and all-cause and cardiovascular mortality in maintenance hemodialysis patients: A prospective cohort study. BMC Nephrol. 2022, 23, 5. [Google Scholar] [CrossRef]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Lok, S.I.; Nous, F.M.; Van Kuik, J.; Van Der Weide, P.; Winkens, B.; Kemperman, H.; Huisman, A.; Lahpor, J.R.; De Weger, R.A.; De Jonge, N. Myocardial fibrosis and pro-fibrotic markers in end-stage heart failure patients during continuous-flow left ventricular assist device support. Eur. J. Cardio-Thorac. Surg. 2015, 48, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.; Stirrat, C.; Semple, S.; Newby, D.; Dweck, M.; Mirsadraee, S. Assessment of myocardial fibrosis with T1 mapping MRI. Clin. Radiol. 2016, 71, 768–778. [Google Scholar] [CrossRef]
- Flett, A.S.; Hayward, M.P.; Ashworth, M.T.; Hansen, M.; Taylor, A.M.; Elliott, P.; McGregor, C.; Moon, J. Equilibrium Contrast Cardiovascular Magnetic Resonance for the Measurement of Diffuse Myocardial Fibrosis: Preliminary Validation in Humans. Circulation. Circulation 2010, 122, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Mondillo, S.; Galderisi, M.; Mele, D.; Cameli, M.; Lomoriello, V.S.; Zacà, V.; Ballo, P.; D’Andrea, A.; Muraru, D.; Losi, M.; et al. Speckle-Tracking Echocardiography. J. Ultrasound Med. 2011, 30, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Popovic, Z.; Kwon, D.H.; Mishra, M.; Buakhamsri, A.; Greenberg, N.L.; Thamilarasan, M.; Flamm, S.D.; Thomas, J.D.; Lever, H.M.; Desai, M.Y. Association Between Regional Ventricular Function and Myocardial Fibrosis in Hypertrophic Cardiomyopathy Assessed by Speckle Tracking Echocardiography and Delayed Hyperenhancement Magnetic Resonance Imaging. J. Am. Soc. Echocardiogr. 2008, 21, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, R.; Fujimoto, S.; Saito, Y.; Nakamura, S. Non-Invasive Quantitation of Myocardial Fibrosis Using Combined Tissue Harmonic Imaging and Integrated Backscatter Analysis in Dilated Cardiomyopathy. Cardiology 2007, 108, 11–17. [Google Scholar] [CrossRef]
- Zannad, F.; Alla, F.; Dousset, B.; Perez, A.; Pitt, B. Limitation of Excessive Extracellular Matrix Turnover May Contribute to Survival Benefit of Spironolactone Therapy in Patients with Congestive Heart Failure: Insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation 2000, 102, 2700–2706. [Google Scholar] [CrossRef] [PubMed]
- Brilla, C.G.; Funck, R.C.; Rupp, H. Lisinopril-Mediated Regression of Myocardial Fibrosis in Patients with Hypertensive Heart Disease. Circulation 2000, 102, 1388–1393. [Google Scholar] [CrossRef]
- Shibasaki, Y.; Nishiue, T.; Masaki, H.; Tamura, K.; Matsumoto, N.; Mori, Y.; Nishikawa, M.; Matsubara, H.; Iwasaka, T. Impact of the Angiotensin II Receptor Antagonist, Losartan, on Myocardial Fibrosis in Patients with End-Stage Renal Disease: Assessment by Ultrasonic Integrated Backscatter and Biochemical Markers. Hypertens. Res. 2005, 28, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.R.; Jhund, P.S.; Baicu, C.F.; Claggett, B.L.; Pieske, B.; Voors, A.A.; Prescott, M.F.; Shi, V.; Lefkowitz, M.; McMurray, J.J.; et al. Plasma Biomarkers Reflecting Profibrotic Processes in Heart Failure with a Preserved Ejection Fraction: Data from the Prospective Comparison of ARNI with ARB on Management of Heart Failure With Preserved Ejection Fraction Study. Circ. Heart Fail. 2016, 9, e002551. [Google Scholar] [CrossRef]
- Abulhul, E.; McDonald, K.; Martos, R.; Phelan, D.; Spiers, J.P.; Hennessy, M.; Baugh, J.; Watson, C.; O’Loughlin, C.; Ledwidge, M. Long-Term Statin Therapy in Patients with Systolic Heart Failure and Normal Cholesterol: Effects on Elevated Serum Markers of Collagen Turnover, Inflammation, and B-Type Natriuretic Peptide. Clin. Ther. 2012, 34, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Ashton, E.; Windebank, E.; Skiba, M.; Reid, C.; Schneider, H.; Rosenfeldt, F.; Tonkin, A.; Krum, H. Why did high-dose rosuvastatin not improve cardiac remodeling in chronic heart failure? Mechanistic insights from the UNIVERSE study. Int. J. Cardiol. 2011, 146, 404–407. [Google Scholar] [CrossRef]
- Lewis, G.A.; Dodd, S.; Clayton, D.; Bedson, E.; Eccleson, H.; Schelbert, E.B.; Naish, J.H.; Jimenez, B.D.; Williams, S.G.; Cunnington, C.; et al. Pirfenidone in heart failure with preserved ejection fraction: A randomized phase 2 trial. Nat. Med. 2021, 27, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Hocher, B.; Godes, M.; Olivier, J.; Weil, J.; Eschenhagen, T.; Slowinski, T.; Neumayer, H.-H.; Bauer, C.; Paul, M.; Pinto, Y.M. Inhibition of left ventricular fibrosis by tranilast in rats with renovascular hypertension. J. Hypertens. 2002, 20, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Visnagri, A.; Kandhare, A.D.; Ghosh, P.; Bodhankar, S.L. Endothelin receptor blocker bosentan inhibits hypertensive cardiac fibrosis in pressure overload-induced cardiac hypertrophy in rats. Cardiovasc. Endocrinol. 2013, 2, 85–97. [Google Scholar] [CrossRef]
- Soylu, K.; Cerik, I.B.; Aksan, G.; Nar, G.; Meric, M. Evaluation of ivabradine in left ventricular dyssynchrony and reverse remodeling in patients with chronic heart failure. J. Arrhythmia 2020, 36, 762–767. [Google Scholar] [CrossRef]
- Giannetta, E.; Isidori, A.M.; Galea, N.; Carbone, I.; Mandosi, E.; Vizza, C.D.; Naro, F.; Morano, S.; Fedele, F.; Lenzi, A. Chronic Inhibition of cGMP Phosphodiesterase 5A Improves Diabetic Cardiomyopathy: A Randomized, Controlled Clinical Trial Using Magnetic Resonance Imaging with Myocardial Tagging. Circulation 2012, 125, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Östman-Smith, I. Reduction by oral propranolol treatment of left ventricular hypertrophy secondary to pressure-overload in the rat. J. Cereb. Blood Flow Metab. 1995, 116, 2703–2709. [Google Scholar] [CrossRef]
- Sandmann, S.; Bohle, R.M.; Dreyer, T.; Unger, T. The T-type calcium channel blocker mibefradil reduced interstitial and perivascular fibrosis and improved hemodynamic parameters in myocardial infarction-induced cardiac failure in rats. Virchows Arch. 2000, 436, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Zhi, H.; Luptak, I.; Alreja, G.; Shi, J.; Guan, J.; Metes-Kosik, N.; Joseph, J. Effects of Direct Renin Inhibition on Myocardial Fibrosis and Cardiac Fibroblast Function. PLoS ONE 2013, 8, e81612. [Google Scholar] [CrossRef]
- Yamamoto, K.; Mano, T.; Yoshida, J.; Sakata, Y.; Nishikawa, N.; Nishio, M.; Ohtani, T.; Hori, M.; Miwa, T.; Masuyama, T. ACE inhibitor and angiotensin II type 1 receptor blocker differently regulate ventricular fibrosis in hypertensive diastolic heart failure. J. Hypertens. 2005, 23, 393–400. [Google Scholar] [CrossRef]
- Cleland, J.G.; Tendera, M.; Adamus, J.; Freemantle, N.; Polonski, L.; Taylor, J. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur. Heart J. 2006, 27, 2338–2345. [Google Scholar] [CrossRef]
- Lim, D.-S.; Lutucuta, S.; Bachireddy, P.; Youker, K.; Evans, A.; Entman, M.; Roberts, R.; Marian, A.J. Angiotensin II Blockade Reverses Myocardial Fibrosis in a Transgenic Mouse Model of Human Hypertrophic Cardiomyopathy. Circulation 2001, 103, 789–791. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef]
- McDiarmid, A.K.; Swoboda, P.P.; Erhayiem, B.; Bounford, K.A.; Bijsterveld, P.; Tyndall, K.; Fent, G.J.; Garg, P.; Dobson, L.E.; Musa, T.A.; et al. Myocardial Effects of Aldosterone Antagonism in Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2020, 9, e011521. [Google Scholar] [CrossRef]
- Girerd, N.; Ferreira, J.P.; Rossignol, P.; Zannad, F. A tentative interpretation of the TOPCAT trial based on randomized evidence from the brain natriuretic peptide stratum analysis. Eur. J. Heart Fail. 2016, 18, 1411–1414. [Google Scholar] [CrossRef]
- Liu, F.; Chen, Y.; Feng, X.; Teng, Z.; Yuan, Y.; Bin, J. Effects of Beta-Blockers on Heart Failure with Preserved Ejection Fraction: A Meta-Analysis. PLoS ONE 2014, 9, e90555. [Google Scholar] [CrossRef]
- Kobayashi, M.; Machida, N.; Mitsuishi, M.; Yamane, Y. β-blocker improves survival, left ventricular function, and myocardial remodeling in hypertensive rats with diastolic heart failure. Am. J. Hypertens. 2004, 17, 1112–1119. [Google Scholar] [CrossRef]
- Ciulla, M.M.; Paliotti, R.; Esposito, A.; Diìez, J.; Loópez, B.; Dahlöf, B.; Nicholls, M.G.; Smith, R.D.; Gilles, L.; Magrini, F.; et al. Different Effects of Antihypertensive Therapies Based on Losartan or Atenolol on Ultrasound and Biochemical Markers of Myocardial Fibrosis: Results of a Randomized Trial. Circulation 2004, 110, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Sandmann, S.; Claas, R.; Cleutjens, J.P.M.; Daemen, M.J.A.P.; Unger, T. Calcium Channel Blockade Limits Cardiac Remodeling and Improves Cardiac Function in Myocardial Infarction-Induced Heart Failure in Rats. J. Cardiovasc. Pharmacol. 2001, 37, 64–77. [Google Scholar] [CrossRef]
- Teng, G.; Svystonyuk, D.; Mewhort, H.E.M.; Turnbull, J.D.; Belke, D.D.; Duff, H.J.; Fedak, P.W.M. Tetrandrine reverses human cardiac myofibroblast activation and myocardial fibrosis. Am. J. Physiol. Circ. Physiol. 2015, 308, H1564–H1574. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-A.; Kim, Y.-J.; Lee, H.-W.; Kim, D.-H.; Kim, H.-K.; Chang, H.-J.; Sohn, D.-W.; Oh, B.-H.; Park, Y.-B. Effect of Rosuvastatin on Cardiac Remodeling, Function, and Progression to Heart Failure in Hypertensive Heart with Established Left Ventricular Hypertrophy. Hypertension 2009, 54, 591–597. [Google Scholar] [CrossRef]
- Chang, Y.-Y.; Wu, Y.-W.; Lee, J.-K.; Lin, Y.-M.; Lin, Y.-H.; Kao, H.-L.; Hung, C.-S.; Lin, H.-J. Effects of 12 weeks of atorvastatin therapy on myocardial fibrosis and circulating fibrosis biomarkers in statin-naïve patients with hypertension with atherosclerosis. J. Investig. Med. 2016, 64, 1194–1199. [Google Scholar] [CrossRef]
- Krum, H.; Ashton, E.; Reid, C.; Kalff, V.; Rogers, J.; Amarena, J.; Singh, B.; Tonkin, A. Double-Blind, Randomized, Placebo-Controlled Study of High-Dose HMG CoA Reductase Inhibitor Therapy on Ventricular Remodeling, Pro-Inflammatory Cytokines and Neurohormonal Parameters in Patients with Chronic Systolic Heart Failure. J. Card. Fail. 2007, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.; Richard, V.; Derumeaux, G.; Hogie, M.; Henry, J.P.; Lallemand, F.; Compagnon, P.; Macé, B.; Comoy, E.; Letac, B.; et al. Role of Endogenous Endothelin in Chronic Heart Failure: Effect of Long-Term Treatment with an Endothelin Antagonist on Survival, Hemodynamics, and Cardiac Remodeling. Circulation 1997, 96, 1976–1982. [Google Scholar] [CrossRef]
- Prasad, S.K.; Dargie, H.J.; Smith, G.C.; Barlow, M.M.; Grothues, F.; Groenning, B.A.; Cleland, J.G.; Pennell, D.J. Comparison of the dual receptor endothelin antagonist enrasentan with enalapril in asymptomatic left ventricular systolic dysfunction: A cardiovascular magnetic resonance study. Heart 2006, 92, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Anand, I.; McMurray, J.; Cohn, J.N.; Konstam, M.A.; Notter, T.; Quitzau, K.; Ruschitzka, F.; Lüscher, T.F. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the Endothelin A Receptor Antagonist Trial in Heart Failure (EARTH): Randomised, double-blind, placebo-controlled trial. Lancet 2004, 364, 347–354. [Google Scholar] [CrossRef]
- Park, S.; Nguyen, N.B.; Pezhouman, A.; Ardehali, R. Cardiac fibrosis: Potential therapeutic targets. Transl. Res. 2019, 209, 121–137. [Google Scholar] [CrossRef]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming Growth Factor-β Function Blocking Prevents Myocardial Fibrosis and Diastolic Dysfunction in Pressure-Overloaded Rats. Circulation 2002, 106, 130–135. [Google Scholar] [CrossRef]
- Frantz, S.; Hu, K.; Adamek, A.; Wolf, J.; Sallam, A.; Maier, S.K.; Lonning, S.; Ling, H.; Ertl, G.; Bauersachs, J. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res. Cardiol. 2008, 103, 485–492. [Google Scholar] [CrossRef]
- Engebretsen, K.V.; Skårdal, K.; Bjørnstad, S.; Marstein, H.S.; Skrbic, B.; Sjaastad, I.; Christensen, G.; Bjørnstad, J.L.; Tønnessen, T. Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J. Mol. Cell. Cardiol. 2014, 76, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Takemura, G.; Kosai, K.-I.; Li, Y.; Takahashi, T.; Esaki, M.; Yuge, K.; Miyata, S.; Maruyama, R.; Mikami, A.; et al. Postinfarction Gene Therapy Against Transforming Growth Factor-β Signal Modulates Infarct Tissue Dynamics and Attenuates Left Ventricular Remodeling and Heart Failure. Circulation 2005, 111, 2430–2437. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Thorikay, M.; Deckers, M.; van Dinther, M.; Grygielko, E.; Gellibert, F.; de Gouville, A.; Huet, S.; Dijke, P.T.; Laping, N. Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008, 73, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Ohtomo, T.; Ninomiya-Tsuji, J.; Tsuchiya, M. A dominant negative TAK1 inhibits cellular fibrotic responses induced by TGF-β. Biochem. Biophys. Res. Commun. 2003, 307, 332–337. [Google Scholar] [CrossRef]
- See, F.; Thomas, W.; Way, K.; Tzanidis, A.; Kompa, A.; Lewis, D.; Itescu, S.; Krum, H. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J. Am. Coll. Cardiol. 2004, 44, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Ding, C.; Wilson, E.; Marcus, G.M.; Olgin, J.E. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm 2010, 7, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Kelly, D.J.; Mifsud, S.A.; Zhang, Y.; Cox, A.J.; See, F.; Krum, H.; Wilkinson-Berka, J.; Gilbert, R.E. Tranilast attenuates cardiac matrix deposition in experimental diabetes: Role of transforming growth factor-β. Cardiovasc. Res. 2005, 65, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Huang, H.; Liu, J.; Wang, Y.; Lu, Z.; Xu, Z. Adverse Events of Pirfenidone for the Treatment of Pulmonary Fibrosis: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2012, 7, e47024. [Google Scholar] [CrossRef]
- Meng, J.; Qin, Y.; Chen, J.; Wei, L.; Huang, X.-R.; Yu, X.; Lan, H.-Y. Treatment of Hypertensive Heart Disease by Targeting Smad3 Signaling in Mice. Mol. Ther. Methods Clin. Dev. 2020, 18, 791–802. [Google Scholar] [CrossRef]
- Chen, K.; Mehta, J.L.; Li, D.; Joseph, L.; Joseph, J. Transforming Growth Factor β Receptor Endoglin Is Expressed in Cardiac Fibroblasts and Modulates Profibrogenic Actions of Angiotensin II. Circ. Res. 2004, 95, 1167–1173. [Google Scholar] [CrossRef]
- Kapur, N.K.; Wilson, S.; Yunis, A.A.; Qiao, X.; Mackey, E.; Paruchuri, V.; Baker, C.; Aronovitz, M.J.; Karumanchi, S.A.; Letarte, M.; et al. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation 2012, 125, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Hang, P.; Zhao, J.; Cai, B.; Tian, S.; Huang, W.; Guo, J.; Sun, C.; Li, Y.; Du, Z. Brain-Derived Neurotrophic Factor Regulates TRPC3/6 Channels and Protects Against Myocardial Infarction in Rodents. Int. J. Biol. Sci. 2015, 11, 536–545. [Google Scholar] [CrossRef]
- Gallini, R.; Lindblom, P.; Bondjers, C.; Betsholtz, C.; Andrae, J. PDGF-A and PDGF-B induces cardiac fibrosis in transgenic mice. Exp. Cell Res. 2016, 349, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Pontén, A.; Li, X.; Thorén, P.; Aase, K.; Sjöblom, T.; Östman, A.; Eriksson, U. Transgenic Overexpression of Platelet-Derived Growth Factor-C in the Mouse Heart Induces Cardiac Fibrosis, Hypertrophy, and Dilated Cardiomyopathy. Am. J. Pathol. 2003, 163, 673–682. [Google Scholar] [CrossRef]
- Chen, Y.; Surinkaew, S.; Naud, P.; Qi, X.-Y.; Gillis, M.-A.; Shi, Y.-F.; Tardif, J.-C.; Dobrev, D.; Nattel, S. JAK-STAT signalling and the atrial fibrillation promoting fibrotic substrate. Cardiovasc. Res. 2017, 113, 310–320. [Google Scholar] [CrossRef]
- Liao, C.-H.; Akazawa, H.; Tamagawa, M.; Ito, K.; Yasuda, N.; Kudo, Y.; Yamamoto, R.; Ozasa, Y.; Fujimoto, M.; Wang, P.; et al. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J. Clin. Investig. 2010, 120, 242–253. [Google Scholar] [CrossRef]
- Koitabashi, N.; Arai, M.; Kogure, S.; Niwano, K.; Watanabe, A.; Aoki, Y.; Maeno, T.; Nishida, T.; Kubota, S.; Takigawa, M.; et al. Increased Connective Tissue Growth Factor Relative to Brain Natriuretic Peptide as a Determinant of Myocardial Fibrosis. Hypertension 2007, 49, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Grotendorst, G.R.; Rahmanie, H.; Duncan, M.R. Combinatorial signaling pathways determine fibroblast proliferation and myofibroblast differentiation. FASEB J. 2004, 18, 469–479. [Google Scholar] [CrossRef]
- Panek, A.N.; Posch, M.G.; Alenina, N.; Ghadge, S.K.; Erdmann, B.; Popova, E.; Perrot, A.; Geier, C.; Morano, R.D.I.; Bader, M.; et al. Connective Tissue Growth Factor Overexpression in Cardiomyocytes Promotes Cardiac Hypertrophy and Protection against Pressure Overload. PLoS ONE 2009, 4, e6743. [Google Scholar] [CrossRef]
- Yoon, P.O.; Lee, M.-A.; Cha, H.; Jeong, M.H.; Kim, J.; Jang, S.P.; Choi, B.Y.; Jeong, D.; Yang, D.K.; Hajjar, R.J.; et al. The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. J. Mol. Cell. Cardiol. 2010, 49, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Vainio, L.E.; Szabó, Z.; Lin, R.; Ulvila, J.; Yrjölä, R.; Alakoski, T.; Piuhola, J.; Koch, W.J.; Ruskoaho, H.; Fouse, S.D.; et al. Connective Tissue Growth Factor Inhibition Enhances Cardiac Repair and Limits Fibrosis After Myocardial Infarction. JACC Basic Transl. Sci. 2019, 4, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Noe, A.; Chandra, P.; Al-Fayoumi, S.; Ligueros-Saylan, M.; Sarangapani, R.; Maahs, S.; Ksander, G.M.; Rigel, D.F.; Jeng, A.Y.; et al. Pharmacokinetics and Pharmacodynamics of LCZ696, a Novel Dual-Acting Angiotensin Receptor-Neprilysin Inhibitor (ARNi). J. Clin. Pharmacol. 2010, 50, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Martens, P.; Beliën, H.; Dupont, M.; Vandervoort, P.; Mullens, W. The reverse remodeling response to sacubitril/valsartan therapy in heart failure with reduced ejection fraction. Cardiovasc. Ther. 2018, 36, e12435. [Google Scholar] [CrossRef] [PubMed]
- Pfau, D.; Thorn, S.L.; Zhang, J.; Mikush, N.; Renaud, J.; Klein, R.; Dekemp, R.A.; Wu, X.; Hu, X.; Sinusas, A.J.; et al. Angiotensin Receptor Neprilysin Inhibitor Attenuates Myocardial Remodeling and Improves Infarct Perfusion in Experimental Heart Failure. Sci. Rep. 2019, 9, 5791. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. The Path to an Angiotensin Receptor Antagonist-Neprilysin Inhibitor in the Treatment of Heart Failure. J. Am. Coll. Cardiol. 2015, 65, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, X.; Zhang, C.; Zhang, C.; Bu, P. Lcz696 Alleviates Myocardial Fibrosis After Myocardial Infarction Through the sFRP-1/Wnt/β-Catenin Signaling Pathway. Front. Pharmacol. 2021, 12, 724147. [Google Scholar] [CrossRef] [PubMed]
- Januzzi, J.L.; Prescott, M.F.; Butler, J.; Felker, G.M.; Maisel, A.S.; McCague, K.; Camacho, A.; Piña, I.L.; Rocha, R.A.; Shah, A.M.; et al. Association of Change in N-Terminal Pro–B-Type Natriuretic Peptide Following Initiation of Sacubitril-Valsartan Treatment with Cardiac Structure and Function in Patients with Heart Failure with Reduced Ejection Fraction. JAMA 2019, 322, 1085–1095. [Google Scholar] [CrossRef]
- Zile, M.R.; O’Meara, E.; Claggett, B.; Prescott, M.F.; Solomon, S.D.; Swedberg, K.; Packer, M.; McMurray, J.J.; Shi, V.; Lefkowitz, M.; et al. Effects of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients with HFrEF. J. Am. Coll. Cardiol. 2019, 73, 795–806. [Google Scholar] [CrossRef]
- Solomon, S.D.; Zile, M.; Pieske, B.; Voors, A.; Shah, A.; Kraigher-Krainer, E.; Shi, V.; Bransford, T.; Takeuchi, M.; Gong, J.; et al. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: A phase 2 double-blind randomised controlled trial. Lancet 2012, 380, 1387–1395. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Junbo Ge, D.P.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin–Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Takahashi, M.; Izawa, A.; Ise, H.; Hongo, M.; Kolattukudy, P.E.; Ikeda, U. Cardiac Overexpression of Monocyte Chemoattractant Protein-1 in Transgenic Mice Prevents Cardiac Dysfunction and Remodeling After Myocardial Infarction. Circ. Res. 2006, 99, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Haudek, S.B.; Cheng, J.; Du, J.; Wang, Y.; Hermosillo-Rodriguez, J.; Trial, J.; Taffet, G.E.; Entman, M.L. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2010, 49, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, J.G.; Villalba, M.; Busnadiego, O.; López-Olañeta, M.M.; Sandoval, P.; Snabel, J.; López-Cabrera, M.; Erler, J.; Hanemaaijer, R.; Lara-Pezzi, E.; et al. Matrix cross-linking lysyl oxidases are induced in response to myocardial infarction and promote cardiac dysfunction. Cardiovasc. Res. 2015, 109, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Roell, W.; Lewalter, T.; Sasse, P.; Tallini, Y.N.; Choi, B.-R.; Breitbach, M.; Doran, R.; Becher, U.M.; Hwang, S.-M.; Bostani, T.; et al. Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature 2007, 450, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Eloff, B.C.; Gilat, E.; Wan, X.; Rosenbaum, D.S. Pharmacological Modulation of Cardiac Gap Junctions to Enhance Cardiac Conduction: Evidence Supporting a Novel Target for Antiarrhythmic Therapy. Circulation 2003, 108, 3157–3163. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, X.-R.; Wei, L.-H.; Chung, A.C.; Yu, C.-M.; Lan, H.-Y. miR-29b as a Therapeutic Agent for Angiotensin II-induced Cardiac Fibrosis by Targeting TGF-β/Smad3 signaling. Mol. Ther. 2014, 22, 974–985. [Google Scholar] [CrossRef]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Li, S.-H.; Guo, J.; Mihic, A.; Wu, J.; Sun, L.; Davis, K.; Weisel, R.D.; Li, R.-K. Role of miR-145 in cardiac myofibroblast differentiation. J. Mol. Cell. Cardiol. 2013, 66, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.A.; Damon, B.; Mironov, V.; Kasyanov, V.; Ramamurthi, A.; Moreno-Rodriguez, R.; Trusk, T.; Potts, J.D.; Goodwin, R.L.; Davis, J.; et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J. Cell. Biochem. 2007, 101, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wu, H.; Xia, W.; Chen, X.; Zhu, S.; Zhang, S.; Shao, Y.; Ma, W.; Yang, D.; Zhang, J. Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J. Cardiol. 2014, 63, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Zhu, Y.; Jiang, H.; D’Amore, A.; Sakaguchi, H.; Tchao, J.; Tobita, K.; Wagner, W.R. Timing effect of intramyocardial hydrogel injection for positively impacting left ventricular remodeling after myocardial infarction. Biomaterials 2015, 83, 182–193. [Google Scholar] [CrossRef]
- French, K.M.; Maxwell, J.T.; Bhutani, S.; Ghosh-Choudhary, S.; Fierro, M.J.; Johnson, T.D.; Christman, K.L.; Taylor, W.R.; Davis, M.E. Fibronectin and Cyclic Strain Improve Cardiac Progenitor Cell Regenerative PotentialIn Vitro. Stem Cells Int. 2016, 2016, 8364382. [Google Scholar] [CrossRef]
- Traverse, J.H.; Henry, T.D.; Dib, N.; Patel, A.N.; Pepine, C.; Schaer, G.L.; DeQuach, J.A.; Kinsey, A.M.; Chamberlin, P.; Christman, K.L. First-in-Man Study of a Cardiac Extracellular Matrix Hydrogel in Early and Late Myocardial Infarction Patients. JACC Basic Transl. Sci. 2019, 4, 659–669. [Google Scholar] [CrossRef]
- Wang, F.; Li, Z.; Tamama, K.; Sen, C.K.; Guan, J. Fabrication and Characterization of Prosurvival Growth Factor Releasing, Anisotropic Scaffolds for Enhanced Mesenchymal Stem Cell Survival/Growth and Orientation. Biomacromolecules 2009, 10, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Pozzobon, M.; Bollini, S.; Iop, L.; De Gaspari, P.; Chiavegato, A.; Rossi, C.A.; Giuliani, S.; Leon, F.F.; Elvassore, N.; Sartore, S.; et al. Human Bone Marrow-Derived CD133+ Cells Delivered to a Collagen Patch on Cryoinjured Rat Heart Promote Angiogenesis and Arteriogenesis. Cell Transplant. 2010, 19, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Hill, K.L.; Li, Q.; Suntharalingam, P.; Mansoor, A.; Wang, X.; Jameel, M.N.; Zhang, P.; Swingen, C.; Kaufman, D.S.; et al. A Fibrin Patch-Based Enhanced Delivery of Human Embryonic Stem Cell-Derived Vascular Cell Transplantation in a Porcine Model of Postinfarction Left Ventricular Remodeling. Stem Cells 2010, 29, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-K.; Zhen, Y.-Y.; Leu, S.; Tsai, T.-H.; Chang, L.-T.; Sheu, J.-J.; Chen, Y.-L.; Chua, S.; Chai, H.-T.; Lu, H.-I.; et al. Direct implantation versus platelet-rich fibrin-embedded adipose-derived mesenchymal stem cells in treating rat acute myocardial infarction. Int. J. Cardiol. 2014, 173, 410–423. [Google Scholar] [CrossRef]
- Engler, A.; Krieger, C.; Johnson, C.P.; Raab, M.; Tang, H.-Y.; Speicher, D.W.; Sanger, J.W.; Sanger, J.M.; Discher, D.E. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: Scar-like rigidity inhibits beating. J. Cell Sci. 2008, 121 Pt 22, 3794–3802. [Google Scholar] [CrossRef]
- Miyagawa, S.; Saito, A.; Sakaguchi, T.; Yoshikawa, Y.; Yamauchi, T.; Imanishi, Y.; Kawaguchi, N.; Teramoto, N.; Matsuura, N.; Iida, H.; et al. Impaired Myocardium Regeneration with Skeletal Cell Sheets—A Preclinical Trial for Tissue-Engineered Regeneration Therapy. Transplantation 2010, 90, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, S.; Domae, K.; Yoshikawa, Y.; Fukushima, S.; Nakamura, T.; Saito, A.; Sakata, Y.; Hamada, S.; Toda, K.; Pak, K.; et al. Phase I Clinical Trial of Autologous Stem Cell–Sheet Transplantation Therapy for Treating Cardiomyopathy. J. Am. Heart Assoc. 2017, 6, e003918. [Google Scholar] [CrossRef] [PubMed]
- Lagoutte, P.; Bettler, E.; Goff, S.V.-L.; Moali, C. Procollagen C-proteinase enhancer-1 (PCPE-1), a potential biomarker and therapeutic target for fibrosis. Matrix Biol. Plus 2021, 11, 100062. [Google Scholar] [CrossRef] [PubMed]
- Morel, P.S.; Duvivier, V.; Bertin, F.; Provost, N.; Hammoutene, A.; Hubert, E.-L.; Gonzalez, A.; Tupinon-Mathieu, I.; Paradis, V.; Delerive, P. Procollagen C-Proteinase Enhancer-1 (PCPE-1) deficiency in mice reduces liver fibrosis but not NASH progression. PLoS ONE 2022, 17, e0263828. [Google Scholar] [CrossRef]
- Baicu, C.F.; Zhang, Y.; Van Laer, A.O.; Renaud, L.; Zile, M.R.; Bradshaw, A.D. Effects of the absence of procollagen C-endopeptidase enhancer-2 on myocardial collagen accumulation in chronic pressure overload. Am. J. Physiol. Circ. Physiol. 2012, 303, H234–H240. [Google Scholar] [CrossRef]
- Zhou, R.; Wang, C.; Wen, C.; Wang, D. miR-21 promotes collagen production in keloid via Smad7. Burns 2017, 43, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, W.; Xu, M.; Huang, H.; Wang, J.; Chen, X. Micro-RNA 21Targets Dual Specific Phosphatase 8 to Promote Collagen Synthesis in High Glucose–Treated Primary Cardiac Fibroblasts. Can. J. Cardiol. 2014, 30, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Lekgabe, E.D.; Kiriazis, H.; Zhao, C.; Xu, Q.; Moore, X.L.; Su, Y.; Bathgate, R.; Du, X.-J.; Samuel, C.S. Relaxin Reverses Cardiac and Renal Fibrosis in Spontaneously Hypertensive Rats. Hypertension 2005, 46, 412–418. [Google Scholar] [CrossRef]
- Sassoli, C.; Chellini, F.; Pini, A.; Tani, A.; Nistri, S.; Nosi, D.; Zecchi-Orlandini, S.; Bani, D.; Formigli, L. Relaxin Prevents Cardiac Fibroblast-Myofibroblast Transition via Notch-1-Mediated Inhibition of TGF-β/Smad3 Signaling. PLoS ONE 2013, 8, e63896. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Metra, M.; Teerlink, J.R.; Unemori, E.; Felker, G.M.; Voors, A.A.; Filippatos, G.; Greenberg, B.; Teichman, S.L.; Severin, T.; et al. Design of the RELAXin in Acute Heart Failure Study. Am. Heart J. 2012, 163, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G.; Villarreal, F. Targeting matrix metalloproteinases in heart disease: Lessons from endogenous inhibitors. Biochem. Pharmacol. 2014, 90, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Eckhouse, S.R.; Purcell, B.P.; McGarvey, J.R.; Lobb, D.; Logdon, C.B.; Doviak, H.; O’Neill, J.W.; Shuman, J.A.; Novack, C.P.; Zellars, K.N.; et al. Local Hydrogel Release of Recombinant TIMP-3 Attenuates Adverse Left Ventricular Remodeling After Experimental Myocardial Infarction. Sci. Transl. Med. 2014, 6, 223ra21. [Google Scholar] [CrossRef] [PubMed]
- Delinière, A.; Baranchuk, A.; Giai, J.; Bessiere, F.; Maucort-Boulch, D.; Defaye, P.; Marijon, E.; Le Vavasseur, O.; Dobreanu, D.; Scridon, A.; et al. Prediction of ventricular arrhythmias in patients with a spontaneous Brugada type 1 pattern: The key is in the electrocardiogram. Europace 2019, 21, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Scridon, A.; Şerban, R.C. Laboratory Monitoring: A Turning Point in the Use of New Oral Anticoagulants. Ther. Drug Monit. 2016, 38, 12–21. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Vagnozzi, R.J.; Johansen, A.K.; Maliken, B.D.; Prasad, V.; Boyer, J.G.; Brody, M.; Schips, T.; Kilian, K.K.; et al. Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart. JCI Insight 2019, 4, e128722. [Google Scholar] [CrossRef] [PubMed]
- Şerban, R.C.; Scridon, A. Data Linking Diabetes Mellitus and Atrial Fibrillation—How Strong Is the Evidence? From Epidemiology and Pathophysiology to Therapeutic Implications. Can. J. Cardiol. 2018, 34, 1492–1502. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technology | Advantages | Disadvantages |
|---|---|---|
| Echocardiography |
|
|
| Cardiac magnetic resonance |
|
|
| Endomyocardial biopsy |
|
|
| Therapeutic Class | Drug | Study Type | Species | Duration | Underlying CVD | Results | References |
|---|---|---|---|---|---|---|---|
| RAAS inhibitors | Spironolactone (12.5–50.0 mg/day) | Placebo-controlled randomized trial | Human | 6 months | HFrEF | Reduced PINP/PIIINP | [77] |
| Lisinopril (5–20 mg/day) | Double-blind randomized trial | Human | 6 months | Hypertensive heart disease | Reduced CVF and improved diastolic function | [78] | |
| Enalapril (5 mg/day) | Double-blind, randomized controlled clinical trial | Human | 6 months | HFpEF-ESRF | Reduced PICP | [79] | |
| Losartan (50 mg/day) | Double-blind, randomized controlled clinical trial | Human | 6 months | HFpEF-ESRF | Reduced CVF and improved diastolic function in severe fibrosis | [79] | |
| Angiotensin receptor neprilysin inhibitor | Sacubitril-valsartan (200mg bid) | Double-blind, randomized controlled clinical trial | Human | 9 months | HFpEF | No significant change in PIIINP/MMP2 | [80] |
| Statins | Atorvastatin (40 mg/day) | Randomized open label study | Human | 6 months | HFrEF | Reduction in PIIINP levels | [81] |
| Rosuvastatin (40 mg/day) | Double-blind, randomized, placebo-controlled study | Human | 6 months | HFrEF | No significant change in PINP/PIIINP | [82] | |
| Pyridones | Pirfenidone | Double-blind, randomized, placebo-controlled study | Human | 52 weeks | HFpEF | Ongoing | [83] |
| Mast cell degranulation inhibitor | Tranilast (400 mg/kg/day) | Experimental | Rat | 12 weeks | 2K1C renovascular hypertension | Decreased fibrotic area to total left ventricular area ratio | [84] |
| Endothelin receptor blocker | Bosentan (100 mg/kg/day) | Experimental | Rat | 4 weeks | Myocardial hypertrophy | Decreased histological interstitial and perivascular fibrosis | [85] |
| Pacemaker current inhibitor | Ivabradine (5 mg bid) | Double-blind, randomized, placebo-controlled study | Human | 8 months | HFrEF | Reversed LV volumes and increased LVEF | [86] |
| Phosphodiesterase type 5 inhibitors | Sildenafil (100 mg/day) | Double-blind, randomized, placebo-controlled study | Human | 3 months | Type 2 diabetes | Improved LV contraction parameters and reduced TGF-β and MCP-1 | [87] |
| Beta-blocker | Propranolol (40 mg/kg/day) | Preclinical | Rat | 10 weeks | Left ventricular pressure overload, hypertrophy | No significant reduction in interstitial fibrosis | [88] |
| Calcium channel blockers | Mibefradil (10 mg/kg/d ay) | Preclinical | Rat | 6 weeks | Myocardial infarction | Decreased infarct size and perivascular fibrosis | [89] |
| Therapeutic Target | Strategy |
|---|---|
| Cell transplantation | Direct remuscularization Stimulation of endogenous cardiovascular progenitor cells |
| TGF-β signaling | Suppression of TGF-β1 TGFβRII plasmid transfection ALK5 inhibition TGFβRII inhibition |
| Biomaterials | Hydrogel (alginate, polyester-VEGF, decellularized ECM, gelatin-HGF) Patch (alginate-neonatal rat cardiomyocytes, decellularized ECM) Glue (fibrin-fibroblast growth factor) Scaffold (fibrin–endothelial cells–smooth muscle cells) |
| Direct reprogramming | GMT (retrovirus/lentivirus) GMHT (retrovirus) miRNAs (miR-1, miR-133, miR-208, miR-499) Chemical/small molecule cocktails |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scridon, A.; Balan, A.I. Targeting Myocardial Fibrosis—A Magic Pill in Cardiovascular Medicine? Pharmaceutics 2022, 14, 1599. https://doi.org/10.3390/pharmaceutics14081599
Scridon A, Balan AI. Targeting Myocardial Fibrosis—A Magic Pill in Cardiovascular Medicine? Pharmaceutics. 2022; 14(8):1599. https://doi.org/10.3390/pharmaceutics14081599
Chicago/Turabian StyleScridon, Alina, and Alkora Ioana Balan. 2022. "Targeting Myocardial Fibrosis—A Magic Pill in Cardiovascular Medicine?" Pharmaceutics 14, no. 8: 1599. https://doi.org/10.3390/pharmaceutics14081599