
Metabolic Pathways of Leishmania Parasite: Source of Pertinent Drug Targets and Potent Drug Candidates


Abstract
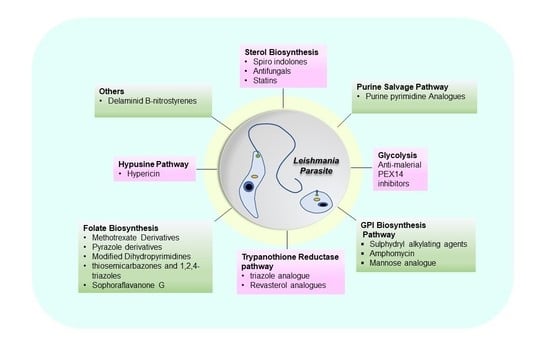
:
1. Introduction
2. Drug Candidates Targeting Metabolic Pathways of Leishmania
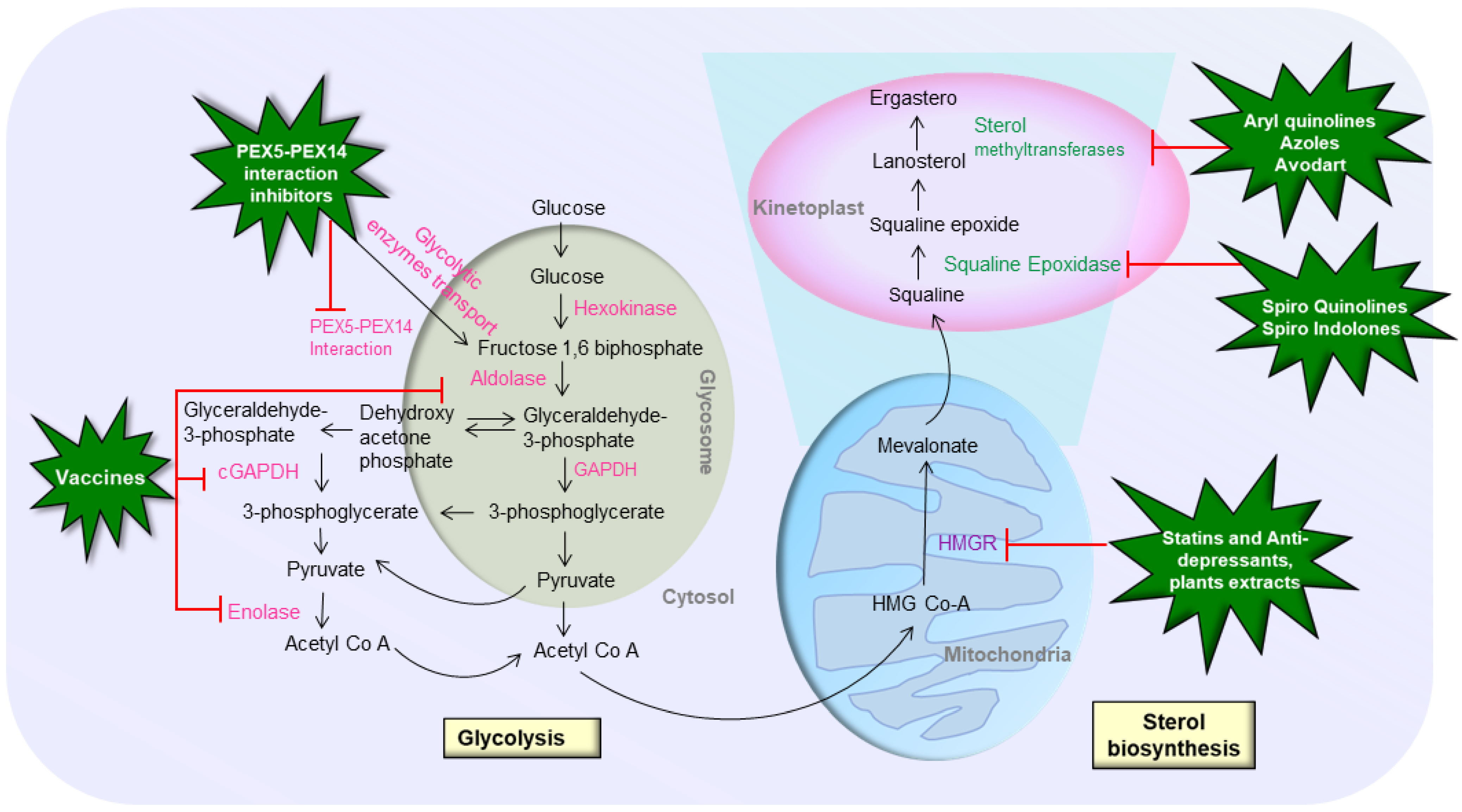
2.1. Sterol Biosynthetic Pathway
2.2. Purine Salvage Pathway
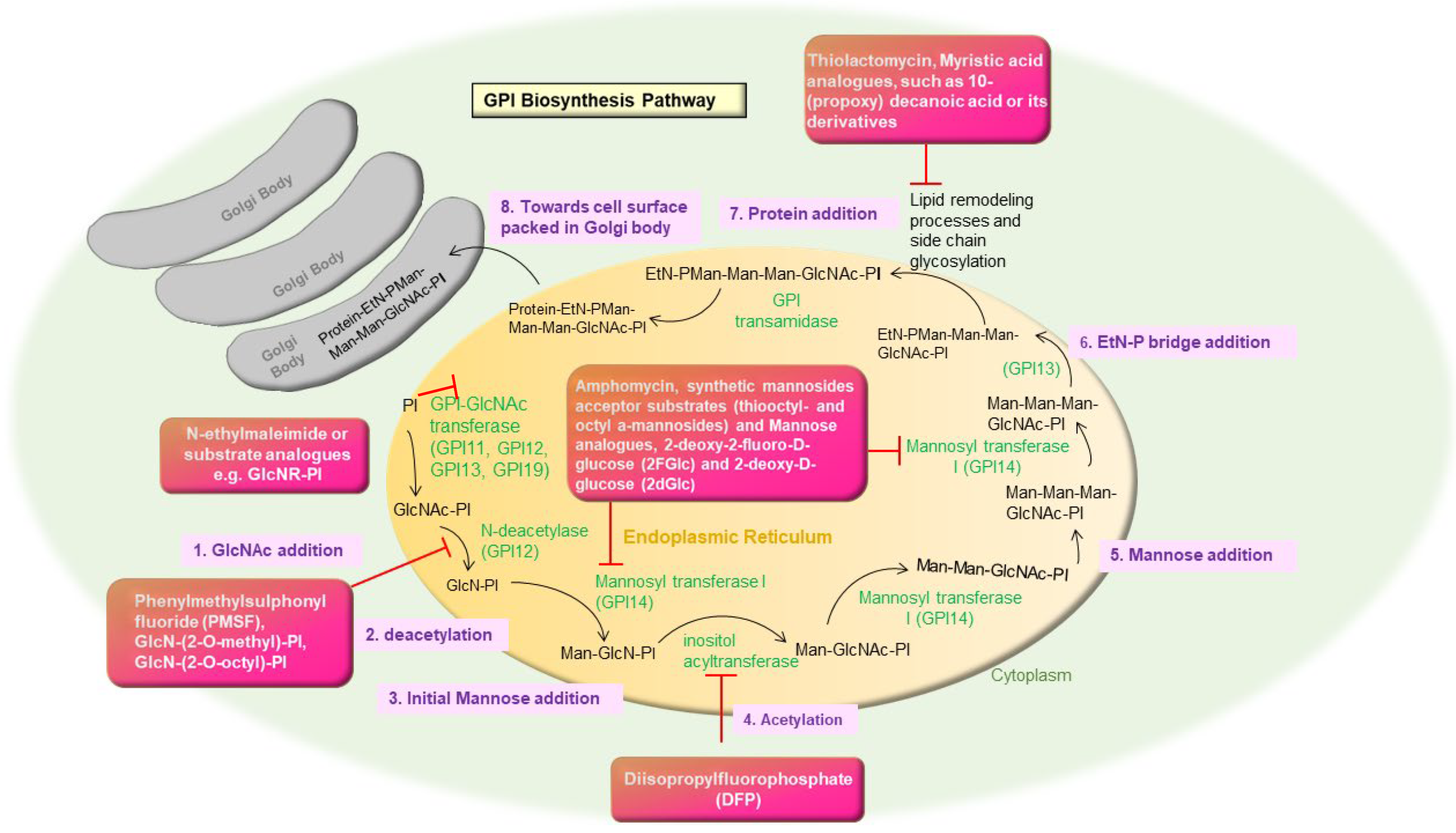
2.3. Glycosyl Phosphatidyl Inositol (GPI) Biosynthesis
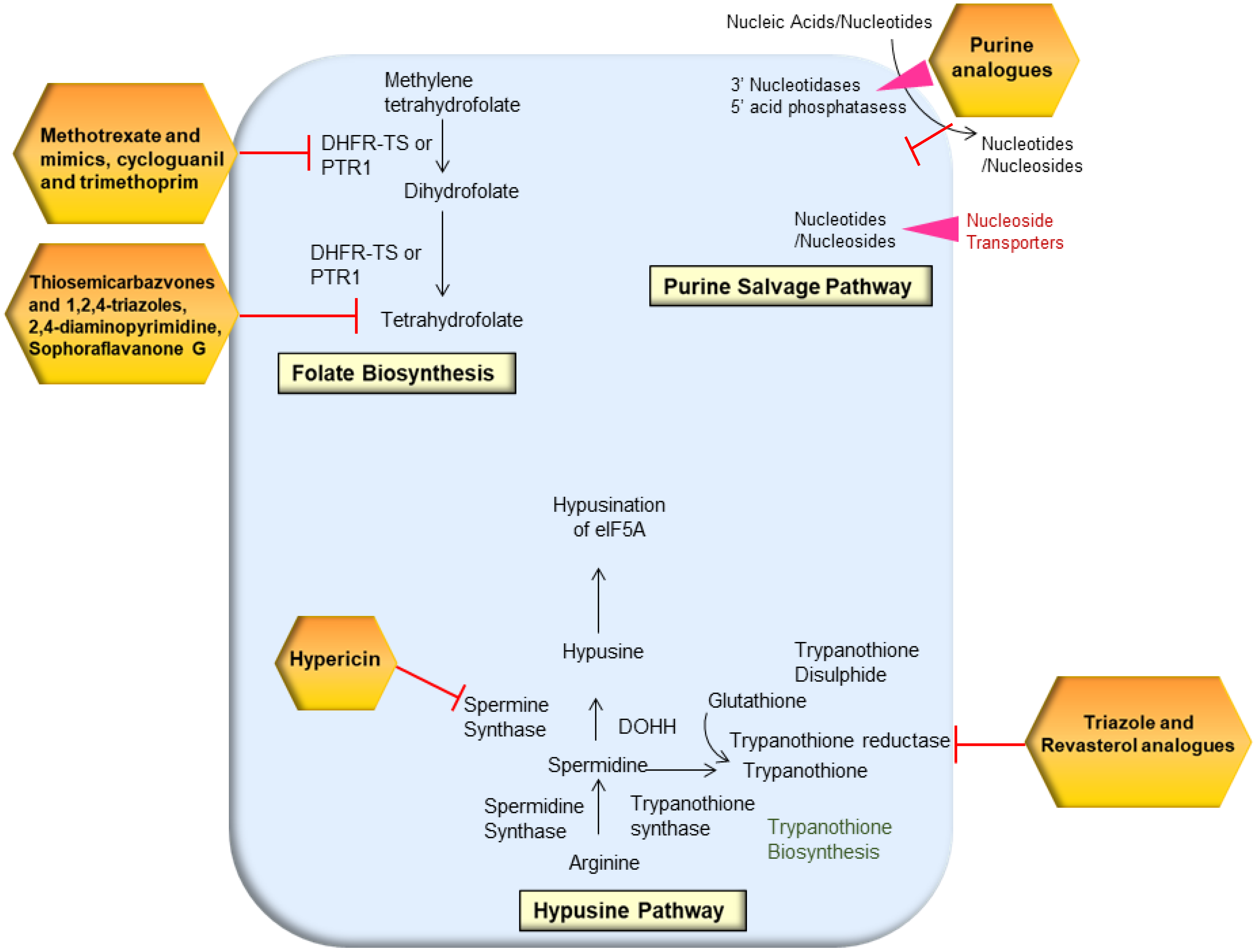
2.4. Folate Biosynthesis Pathway
2.5. Trypanothione Pathway
2.6. Hypusine Pathway
2.7. Other Pathways of Leishmania Parasite Targeted by Various Drug Candidates
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duthie, M.S.; Goto, Y.; Ghosh, P.; Mondal, D. Impact of sequelae of visceral leishmaniasis and their contribution to ongoing transmission of Leishmania donovani. Pathog. Dis. 2019, 77, ftz057. [Google Scholar] [CrossRef] [Green Version]
- Sahu, U.; Khare, P. Interferon-γ: A key cytokine in leishmaniasis. In Pathogenesis, Treatment and Prevention of Leishmaniasis; Academic Press: Cambridge, MA, USA, 2021; pp. 197–208. [Google Scholar] [CrossRef]
- Samant, M.; Sahu, U.; Pandey, S.C.; Khare, P. Role of Cytokines in Experimental and Human Visceral Leishmaniasis. Front. Cell. Infect. Microbiol. 2021, 11, 624009. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Chaudhury, A.; Maji, A.K.; Sir, U.N. Brahmachari and his battle against Kala-Azar. Trop. Parasitol. 2021, 11, 89–91. [Google Scholar] [CrossRef]
- Verhaar, A.P.; Wildenberg, M.E.; Peppelenbosch, M.P.; Hommes, D.W.; Brink, G.R.V.D. Repurposing Miltefosine for the Treatment of Immune-Mediated Disease? J. Pharmacol. Exp. Ther. 2014, 350, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Shah, P.; Baharia, R.K.; Tandon, R.; Khare, P.; Sundar, S.; Sahasrabuddhe, A.A.; Siddiqi, M.I.; Dube, A. Over-Expression of 60s Ribosomal L23a Is Associated with Cellular Proliferation in SAG Resistant Clinical Isolates of Leishmania donovani. PLoS Negl. Trop. Dis. 2013, 7, e2527. [Google Scholar] [CrossRef]
- Chawla, B.; Madhubala, R. Drug targets in Leishmania. J. Parasit. Dis. 2010, 34, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Braga, S.S. Multi-target drugs active against leishmaniasis: A paradigm of drug repurposing. Eur. J. Med. Chem. 2019, 183, 111660. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.; Ochoa, R.; Asela, C.; Muskus, C. Repurposing of known drugs for leishmaniasis treatment using bioinformatic predictions, in vitro validations and pharmacokinetic simulations. J. Comput.-Aided Mol. Des. 2019, 33, 845–854. [Google Scholar] [CrossRef]
- de Souza, M.L.; da Costa, L.A.G.; Silva, E.D.O.; de Sousa, A.L.M.D.; dos Santos, W.M.; Neto, P.J.R. Recent strategies for the development of oral medicines for the treatment of visceral leishmaniasis. Drug Dev. Res. 2020, 81, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Torres-Santos, E.C.; Andrade-Neto, V.V.; Cunha-Junior, E.F.; Faioes, V.D.S.; Martins, T.P.; Silva, R.L.; Leon, L.L. Leishmaniasis treatment update of possibilities for drug repurposing. Front. Biosci. 2018, 23, 967–996. [Google Scholar] [CrossRef] [Green Version]
- Roatt, B.M.; de Oliveira Cardoso, J.M.; De Brito, R.C.F.; Coura-Vital, W.; de Oliveira Aguiar-Soares, R.D.; Reis, A.B. Recent advances and new strategies on leishmaniasis treatment. Appl. Microbiol. Biotechnol. 2020, 104, 8965–8977. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Panic, G.; Duthaler, U.; Speich, B.; Keiser, J. Repurposing drugs for the treatment and control of helminth infections. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 185–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santana, W.; de Oliveira, S.S.C.; Ramos, M.H.; Santos, A.L.S.; Dolabella, S.S.; Souto, E.B.; Severino, P.; Jain, S. Exploring Innovative Leishmaniasis Treatment: Drug Targets from Pre-Clinical to Clinical Findings. Chem. Biodivers. 2021, 18, e2100336. [Google Scholar] [CrossRef] [PubMed]
- Khare, P.; Gupta, A.K.; Gajula, P.K.; Sunkari, K.Y.; Jaiswal, A.K.; Das, S.; Bajpai, P.; Chakraborty, T.K.; Dube, A.; Saxena, A.K. Identification of Novel S-Adenosyl-l-Homocysteine Hydrolase Inhibitors through Homology-Model-Based Virtual Screening, Synthesis, and Biological Evaluation. J. Chem. Inf. Model. 2012, 52, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Dinesh, N.; Soumya, N.; Singh, S. Antileishmanial effect of mevastatin is due to interference with sterol metabolism. Parasitol. Res. 2015, 114, 3873–3883. [Google Scholar] [CrossRef]
- Dinesh, N.; Pallerla, D.S.R.; Kaur, P.K.; Babu, N.K.; Singh, S. Exploring Leishmania donovani 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) as a potential drug target by biochemical, biophysical and inhibition studies. Microb. Pathog. 2014, 66, 14–23. [Google Scholar] [CrossRef]
- Saha, A.K.; Mukherjee, T.; Bhaduri, A. Mechanism of action of amphotericin B on Leishmania donovani promastigotes. Mol. Biochem. Parasitol. 1986, 19, 195–200. [Google Scholar] [CrossRef]
- Pinto-Martinez, A.K.; Rodriguez-Durán, J.; Serrano-Martin, X.; Hernandez-Rodriguez, V.; Benaim, G. Mechanism of Action of Miltefosine on Leishmania donovani Involves the Impairment of Acidocalcisome Function and the Activation of the Sphingosine-Dependent Plasma Membrane Ca2+ Channel. Antimicrob. Agents Chemother. 2018, 62, e01614-17. [Google Scholar] [CrossRef] [Green Version]
- Emami, S.; Tavangar, P.; Keighobadi, M. An overview of azoles targeting sterol 14α-demethylase for antileishmanial therapy. Eur. J. Med. Chem. 2017, 135, 241–259. [Google Scholar] [CrossRef]
- Rottmann, M.; McNamara, C.; Yeung, B.K.S.; Lee, M.C.S.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D.M.; Dharia, N.V.; Tan, J.; et al. Spiroindolones, a new and potent chemotype for the treatment of malaria. Science 2011, 329, 1175–1180. [Google Scholar] [CrossRef] [Green Version]
- Scala, A.; Cordaro, M.; Grassi, G.; Piperno, A.; Barberi, G.; Cascio, A.; Risitano, F. Direct synthesis of C3-mono-functionalized oxindoles from N-unprotected 2-oxindole and their antileishmanial activity. Bioorganic Med. Chem. 2014, 22, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Acharya, C.; Pal, U.; Chowdhury, S.R.; Sarkar, K.; Maiti, N.C.; Jaisankar, P.; Majumder, H.K. A Novel Spirooxindole Derivative Inhibits the Growth of Leishmania donovani Parasites both In Vitro and In Vivo by Targeting Type IB Topoisomerase. Antimicrob. Agents Chemother. 2016, 60, 6281–6293. [Google Scholar] [CrossRef] [Green Version]
- Leañez, J.; Nuñez, J.; García-Marchan, Y.; Sojo, F.; Arvelo, F.; Rodriguez, D.; Buscema, I.; Alvarez-Aular, A.; Forero, J.S.B.; Kouznetsov, V.V.; et al. Experimental Parasitology Anti-leishmanial effect of spiro dihydroquinoline-oxindoles on volume regulation decrease and sterol biosynthesis of Leishmania braziliensis. Exp. Parasitol. 2019, 198, 31–38. [Google Scholar] [CrossRef]
- Moghaddam, F.M.; Saberi, V.; Karimi, A. Highly diastereoselective cascade [5 + 1] double Michael reaction, a route for the synthesis of spiro(thio)oxindoles. Sci. Rep. 2021, 11, 22834. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, K.E.J.; Bravo, S.M.M.; Cricco, J.A. Role of Heme and Heme-Proteins in Trypanosomatid Essential Metabolic Pathways. Enzym. Res. 2011, 2011, 22834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friggeri, L.; Hargrove, T.Y.; Rachakonda, G.; Blobaum, A.L.; Fisher, P.; de Oliveira, G.M.; da Silva, C.F.; Soeiro, M.D.N.C.; Nes, W.D.; Lindsley, C.W.; et al. Sterol 14α-Demethylase Structure-Based Optimization of Drug Candidates for Human Infections with the Protozoan Trypa-nosomatidae Laura. J. Med. Chem. 2018, 61, 10910–10921. [Google Scholar] [CrossRef]
- Tabrez, S.; Rahman, F.; Ali, R.; Akand, S.K.; Alaidarous, M.A.; Alshehri, B.M.; Banawas, S.; Bin Dukhyil, A.A.; Rub, A. Targeting sterol alpha-14 demethylase of Leishmania donovani to fight against leishmaniasis. J. Cell. Biochem. 2021, 122, 1037–1047. [Google Scholar] [CrossRef]
- Hammill, J.T.; Sviripa, V.M.; Kril, L.M.; Ortiz, D.; Fargo, C.M.; Kim, H.S.; Chen, Y.; Rector, J.; Rice, A.L.; Domagalska, M.A.; et al. Amino-Substituted 3-Aryl- and 3-Heteroarylquinolines as Potential Antileishmanial Agents. J. Med. Chem. 2021, 64, 12152–12162. [Google Scholar] [CrossRef]
- Chanquia, S.N.; Larregui, F.; Puente, V.; Labriola, C.; Lombardo, E.; Liñares, G.G. Synthesis and biological evaluation of new quinoline derivatives as antileishmanial and antitrypanosomal agents. Bioorg. Chem. 2019, 83, 526–534. [Google Scholar] [CrossRef]
- Bompart, D.; Núñez-Durán, J.; Rodríguez, D.; Kouznetsov, V.V.; Gómez, C.M.M.; Sojo, F.; Arvelo, F.; Visbal, G.; Alvarez, A.; Serrano-Martín, X.; et al. Bioorganic & Medicinal Chemistry Anti-leishmanial evaluation of C2-aryl quinolines: Mechanistic insight on bioenergetics and sterol biosynthetic pathway of Leishmania braziliensis. Bioorganic Med. Chem. 2013, 21, 4426–4431. [Google Scholar] [CrossRef]
- Serrano-Martín, X.; Payares, G.; De Lucca, M.; Martinez, J.C.; Mendoza-León, A.; Benaim, G. Amiodarone and Miltefosine Act Synergistically against Leishmania mexicana and Can Induce Parasitological Cure in a Murine Model of Cutaneous Leishmaniasis. Antimicrob. Agents Chemother. 2009, 53, 5108–5113. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Wilson, M.E. Dynamics of sterol synthesis during development of Leishmania spp. parasites to their virulent form. Parasites Vectors 2016, 9, 200. [Google Scholar] [CrossRef] [Green Version]
- Dinesh, N.; Kaur, P.K.; Swamy, K.K.; Singh, S. Mianserin, an antidepressant kills Leishmania donovani by depleting ergosterol levels. Exp. Parasitol. 2014, 144, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dinesh, N.; Kaur, P.K.; Shamiulla, B. Ketanserin, an antidepressant, exerts its antileishmanial action via inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) enzyme of Leishmania donovani. Parasitol. Res. 2014, 113, 2161–2168. [Google Scholar] [CrossRef]
- Zeiman, E.; Greenblatt, C.L.; Elgavish, S.; Khozin-Goldberg, I.; Golenser, J. Mode of Action of Fenarimol against Leishmania spp. J. Parasitol. 2008, 94, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.A. Exploring the Pivotal Immunomodulatory and Anti-Inflammatory Potentials of Glycyrrhizic and Glycyrrhetinic Acids. Mediat. Inflamm. 2021, 2021, 6699560. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Majumder, S.; Majumdar, S.B.; Choudhuri, S.K.; Roy, S.; Majumdar, S. Co-administration of glycyrrhizic acid with the antileishmanial drug sodium antimony gluconate (SAG) cures SAG-resistant visceral leishmaniasis. Int. J. Antimicrob. Agents 2015, 45, 268–277. [Google Scholar] [CrossRef]
- Dinesh, N.; Neelagiri, S.; Kumar, V.; Singh, S. Glycyrrhizic acid attenuates growth of Leishmania donovani by depleting ergosterol levels. Exp. Parasitol. 2017, 176, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ansari, Y.; Dikhit, M.R.; Sahoo, G.C.; Das, P. Comparative modeling of HGPRT enzyme of L. donovani and binding affinities of different analogs of GMP. Int. J. Biol. Macromol. 2012, 50, 637–649. [Google Scholar] [CrossRef]
- Figueroa-Villar, J.D.; Sales, E.M. The importance of nucleoside hydrolase enzyme (NH) in studies to treatment of Leishmania: A review. Chem. Interact. 2017, 263, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.-A.; Zheng, Z.; Wen, X.; Manivannan, S.; Pastor, A.; Kaiser, M.; Brun, R.; Snyder, F.F.; Back, T.G. Synthesis and activity of nucleoside-based antiprotozoan compounds. Bioorganic Med. Chem. 2017, 25, 2091–2104. [Google Scholar] [CrossRef] [PubMed]
- Serafim, T.D.; Figueiredo, A.B.; Costa, P.A.C.; Marques-Da-Silva, E.; Goncalves, R.H.; De Moura, S.A.L.; Gontijo, N.; Da Silva, S.M.; Michalick, M.S.M.; Meyer-Fernandes, J.R.; et al. Leishmania Metacyclogenesis Is Promoted in the Absence of Purines. PLoS Negl. Trop. Dis. 2012, 6, e1833. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Yates, P.A.; Soysa, R.; Alfaro, J.F.; Yang, F.; Burnum-Johnson, K.; Petyuk, V.; Weitz, K.K.; Camp, D.G.; Smith, R.; et al. Metabolic Reprogramming during Purine Stress in the Protozoan Pathogen Leishmania donovani. PLoS Pathog. 2014, 10, e1003938. [Google Scholar] [CrossRef] [Green Version]
- Azzouz, S.; Lawton, P. In vitro effects of purine and pyrimidine analogues on Leishmania donovani and Leishmania infantum promastigotes and intracellular amastigotes. Acta Parasitol. 2017, 62, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Bouton, J.; Ferreira de Almeida Fiuza, L.; Santos, C.C.; Mazzarella, M.A.; Soeiro, M.D.N.C.; Maes, L.; Karalic, I.; Caljon, G.; Van Calenbergh, S. Revisiting Pyrazolo[3,4-d]pyrimidine Nucleosides as Anti-Trypanosoma cruzi and Antileishmanial Agents. J. Med. Chem. 2021, 64, 4206–4238. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, D.; Sanchez, M.A.; Pierce, S.; Herrmann, T.; Kimblin, N.; Bouwer, H.G.A.; Landfear, S.M. Molecular genetic analysis of purine nucleobase transport in Leishmania major. Mol. Microbiol. 2007, 64, 1228–1243. [Google Scholar] [CrossRef]
- Bichiou, H.; Bouabid, C.; Rabhi, I.; Guizani-Tabbane, L. Transcription Factors Interplay Orchestrates the Immune-Metabolic Response of Leishmania Infected Macrophages. Front. Cell. Infect. Microbiol. 2021, 11, 660415. [Google Scholar] [CrossRef]
- Bogdan, C. Macrophages as host, effector and immunoregulatory cells in leishmaniasis: Impact of tissue micro-environment and metabolism. Cytokine X 2020, 2, 100041. [Google Scholar] [CrossRef]
- Zhang, W.-W.; McCall, L.-I.; Matlashewski, G. Role of Cytosolic Glyceraldehyde-3-Phosphate Dehydrogenase in Visceral Organ Infection by Leishmania donovani. Eukaryot. Cell 2013, 12, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Balmer, E.A.; Faso, C. The Road Less Traveled? Unconventional Protein Secretion at Parasite–Host Interfaces. Front. Cell Dev. Biol. 2021, 9, 662711. [Google Scholar] [CrossRef] [PubMed]
- Yousef, B.A.; Elwaseela, T.H.; Ali, T.A.; Mohammed, F.E.; Mohammed, W.O.; Alobaid, M.; Dirar, A.I. Anti-malarial drugs as potential inhibitors of leishmania glycolytic enzymes: Development of new anti-leishmanial agents. Pharmacol. Clin. Pharm. Res. 2020, 5, 77. [Google Scholar] [CrossRef]
- Silva, L.A.; Vinaud, M.C.; Castro, A.M.; Cravo, P.V.L.; Bezerra, J.C.B. In SilicoSearch of Energy Metabolism Inhibitors for Alternative Leishmaniasis Treatments. BioMed Res. Int. 2015, 2015, 965725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanra, S.; Juin, S.K.; Jawed, J.J.; Ghosh, S.; Dutta, S.; Nabi, S.A.; Dash, J.; Dasgupta, D.; Majumdar, S.; Banerjee, R. In vivo experiments demonstrate the potent antileishmanial efficacy of repurposed suramin in visceral leishmaniasis. PLoS Negl. Trop. Dis. 2020, 14, e0008575. [Google Scholar] [CrossRef] [PubMed]
- von der Ahe, D.; Huehnchen, P.; Balkaya, M.; Peruzzaro, S.; Endres, M.; Boehmerle, W. Suramin-Induced Neurotoxicity: Preclinical Models and Neuroprotective Strategies. Molecules 2018, 23, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albulescu, I.C.; van Hoolwerff, M.; Wolters, L.A.; Bottaro, E.; Nastruzzi, C.; Yang, S.C.; Tsay, S.-C.; Hwu, J.R.; Snijder, E.J.; van Hemert, M.J. Suramin inhibits chikungunya virus replication through multiple mechanisms. Antivir. Res. 2015, 121, 39–46. [Google Scholar] [CrossRef]
- Joshi, S.; Yadav, N.K.; Rawat, K.; Tripathi, C.D.P.; Jaiswal, A.K.; Khare, P.; Tandon, R.; Baharia, R.K.; Das, S.; Gupta, R.; et al. Comparative Analysis of Cellular Immune Responses in Treated Leishmania Patients and Hamsters against Recombinant Th1 Stimulatory Proteins of Leishmania donovani. Front. Microbiol. 2016, 7, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Samant, M.; Misra, P.; Khare, P.; Sisodia, B.; Shasany, A.K.; Dube, A. Th1-stimulatory polyproteins of soluble Leishmania donovani promastigotes ranging from 89.9 to 97.1kDa offers long-lasting protection against experimental visceral leishmaniasis. Vaccine 2008, 26, 5700–5711. [Google Scholar] [CrossRef]
- Gupta, R.; Kumar, V.; Kushawaha, P.K.; Tripathi, C.P.; Joshi, S.; Sahasrabuddhe, A.A.; Mitra, K.; Sundar, S.; Siddiqi, M.I.; Dube, A. Characterization of Glycolytic Enzymes-rAldolase and rEnolase of Leishmania donovani, Identified as Th1 Stimulatory Proteins, for Their Immunogenicity and Immunoprophylactic Efficacies against Experimental Visceral Leishmaniasis. PLoS ONE 2014, 9, e86073. [Google Scholar] [CrossRef] [Green Version]
- Khare, P.; Jaiswal, A.K.; Tripathi, C.D.P.; Sundar, S.; Dube, A. Immunoprotective responses of T helper type 1 stimulatory protein-S-adenosyl-L-homocysteine hydrolase against experimental visceral leishmaniasis. Clin. Exp. Immunol. 2016, 185, 165–179. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, M.S.; Junqueira, C.; Trigueiro, R.C.; Shams-Eldin, H.; Macedo, C.S.; Araújo, P.R.; Gomes, D.A.; Martinelli, P.M.; Kimmel, J.; Stahl, P.; et al. Identification and Functional Analysis of Trypanosoma cruzi Genes That Encode Proteins of the Glycosylphosphatidylinositol Biosynthetic Pathway. PLoS Negl. Trop. Dis. 2013, 7, e2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmahallawy, E.; Alkhaldi, A. Insights into Leishmania Molecules and Their Potential Contribution to the Virulence of the Parasite. Veter. Sci. 2021, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- de Assis, R.R.; Ibraim, I.C.; Nogueira, P.M.; Soares, R.P.; Turco, S.J. Glycoconjugates in New World species of Leishmania: Polymorphisms in lipophosphoglycan and glycoinositolphospholipids and interaction with hosts. Biochim. Biophys. Acta-Gen. Subj. 2012, 1820, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- de Macedo, C.S.; Shams-Eldin, H.; Smith, T.K.; Schwarz, R.T.; Azzouz, N. Inhibitors of glycosyl-phosphatidylinositol anchor biosynthesis. Biochimie 2003, 85, 465–472. [Google Scholar] [CrossRef]
- Masterson, W.J.; Ferguson, M.A. Phenylmethanesulphonyl fluoride inhibits GPI anchor biosynthesis in the African trypanosome. EMBO J. 1991, 10, 2041–2045. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.K. Amphomycin inhibits mannosylphosphoryldolichol synthesis by forming a complex with dolichylmo-nophosphate. J. Biol. Chem. 1989, 264, 2024–2028. [Google Scholar] [CrossRef]
- McDowell, W.; Schwarz, R.T. Specificity of GDP-Man: Dolichyl-phosphate mannosyltransferase for the guanosine diphosphate esters of mannose analogues containing deoxy and deoxyfluoro substituents. FEBS Lett. 1989, 243, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Shinde, S.; Mol, M.; Jamdar, V.; Singh, S. Molecular modeling and molecular dynamics simulations of GPI 14 in Leishmania major: Insight into the catalytic site for active site directed drug design. J. Theor. Biol. 2014, 351, 37–46. [Google Scholar] [CrossRef]
- Doering, T.L.; Raper, J.; Buxbaum, L.U.; Adams, S.P.; Gordon, J.I.; Hart, G.W.; Englund, P.T. An Analog of Myristic Acid with Selective Toxicity for African Trypanosomes. Science 1991, 252, 1851–1854. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.L.; Lu, T.; Werbovetz, K.A.; Gokel, G.W.; Hart, G.W.; Gordon, J.I.; Englund, P.T. Toxicity of myristic acid analogs toward African trypanosomes. Proc. Natl. Acad. Sci. USA 1994, 91, 9735–9739. [Google Scholar] [CrossRef] [Green Version]
- Shulpekova, Y.; Nechaev, V.; Kardasheva, S.; Sedova, A.; Kurbatova, A.; Bueverova, E.; Kopylov, A.; Malsagova, K.; Dlamini, J.; Ivashkin, V. The Concept of Folic Acid in Health and Disease. Molecules 2021, 26, 3731. [Google Scholar] [CrossRef]
- Dias, M.V.B.; Santos, J.C.; Libreros-Zúñiga, G.A.; Ribeiro, J.A.; Chavez-Pacheco, S.M. Folate biosynthesis pathway: Mechanisms and insights into drug design for infectious diseases. Futur. Med. Chem. 2018, 10, 935–959. [Google Scholar] [CrossRef]
- Kelly, F.D.; Yates, P.A.; Landfear, S.M. Nutrient sensing in Leishmania: Flagellum and cytosol. Mol. Microbiol. 2021, 115, 849–859. [Google Scholar] [CrossRef]
- Hendrickx, S.; Caljon, G.; Maes, L. Need for sustainable approaches in antileishmanial drug discovery. Parasitol. Res. 2019, 118, 2743–2752. [Google Scholar] [CrossRef]
- Sharma, V.K.; Bharatam, P.V. Identification of Selective Inhibitors of LdDHFR Enzyme Using Pharmacoinformatic Methods. J. Comput. Biol. 2021, 28, 43–59. [Google Scholar] [CrossRef]
- Das Neves, G.M.; Kagami, L.P.; Gonçalves, I.L.; Eifler-Lima, V.L. Targeting pteridine reductase 1 and dihydrofolate reductase: The old is a new trend for leishmaniasis drug discovery. Futur. Med. Chem. 2019, 11, 2107–2130. [Google Scholar] [CrossRef]
- Tomašić, T.; Zidar, N.; Šink, R.; Kovač, A.; Blanot, D.; Contreras-Martel, C.; Dessen, A.; Müller-Premru, M.; Zega, A.; Gobec, S.; et al. Structure-Based Design of a New Series of d-Glutamic Acid Based Inhibitors of Bacterial UDP-N-acetylmuramoyl-l-alanine: D-glutamate Ligase (MurD). J. Med. Chem. 2011, 54, 4600–4610. [Google Scholar] [CrossRef]
- Bibi, M.; Qureshi, N.A.; Sadiq, A.; Farooq, U.; Hassan, A.; Shaheen, N.; Asghar, I.; Umer, D.; Ullah, A.; Khan, F.A.; et al. Exploring the ability of dihydropyrimidine-5-carboxamide and 5-benzyl-2,4-diaminopyrimidine-based analogues for the selective inhibition of L. major dihydrofolate reductase. Eur. J. Med. Chem. 2021, 210, 112986. [Google Scholar] [CrossRef]
- Vickers, T.J.; Beverley, S.M. Folate metabolic pathways in Leishmania. Essays Biochem. 2011, 51, 63–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, F.H.A.; Froes, T.Q.; da Silva, S.G.; de Souza, E.I.M.; Vital-Fujii, D.G.; Trossini, G.H.G.; da Rocha Pita, S.S.; Castilho, M.S. An integrated approach towards the discovery of novel non-nucleoside Leishmania major pteridine reductase 1 inhibitors. Eur. J. Med. Chem. 2017, 132, 322–332. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Leprohon, P.; Ouellette, M. Combined gene deletion of dihydrofolate reductase-thymidylate synthase and pteridine reductase in Leishmania infantum. PLoS Negl. Trop. Dis. 2021, 15, e0009377. [Google Scholar] [CrossRef]
- Patil, S.R.; Asrondkar, A.; Patil, V.; Sangshetti, J.N.; Khan, F.A.K.; Damale, M.G.; Patil, R.H.; Bobade, A.S.; Shinde, D.B. Antileishmanial potential of fused 5-(pyrazin-2-yl)-4H-1,2,4-triazole-3-thiols: Synthesis, biological evaluations and computational studies. Bioorg. Med. Chem. Lett. 2017, 27, 3845–3850. [Google Scholar] [CrossRef]
- Aggarwal, R.; Sumran, G. An insight on medicinal attributes of 1,2,4-triazoles. Eur. J. Med. Chem. 2020, 205, 112652. [Google Scholar] [CrossRef] [PubMed]
- Temraz, M.G.; Elzahhar, P.A.; Bekhit, A.E.-D.A.; Bekhit, A.A.; Labib, H.F.; Belal, A.S. Anti-leishmanial click modifiable thiosemicarbazones: Design, synthesis, biological evaluation and in silico studies. Eur. J. Med. Chem. 2018, 151, 585–600. [Google Scholar] [CrossRef]
- Kapil, S.; Singh, P.K.; Kashyap, A.; Silakari, O. Structure based designing of benzimidazole/benzoxazole derivatives as anti-leishmanial agents. SAR QSAR Environ. Res. 2019, 30, 919–933. [Google Scholar] [CrossRef]
- Herrmann, F.C.; Sivakumar, N.; Jose, J.; Costi, M.P.; Pozzi, C.; Schmidt, T.J. In Silico Identification and In Vitro Evaluation of Natural Inhibitiors of Leishmania major Pteridine Reductase I. Molecules 2017, 22, 2166. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, B.V.F.; Teles, A.L.B.; da Silva, S.G.; Brito, C.C.B.; de Freitas, H.F.; Pires, A.B.L.; Froes, T.Q.; Castilho, M.S. Dual and selective inhibitors of pteridine reductase 1 (PTR1) and dihydrofolate reductase-thymidylate synthase (DHFR-TS) from Leishmania chagasi. J. Enzym. Inhib. Med. Chem. 2019, 34, 1439–1450. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Chauhan, K.; Shivahare, R.; Vishwakarma, P.; Suthar, M.; Sharma, A.; Gupta, S.; Saxena, J.K.; Lal, J.; Chandra, P.; et al. Discovery of a New Class of Natural Product-Inspired Quinazolinone Hybrid as Potent Antileishmanial agents. J. Med. Chem. 2013, 56, 4374–4392. [Google Scholar] [CrossRef]
- Muhammad, M.T.; Ghouri, N.; Khan, K.M.; Arshia; Choudhary, M.I.; Perveen, S. Synthesis of Thiocarbohydrazones and Evaluation of their in vitro Antileishmanial Activity. Med. Chem. 2018, 14, 725–732. [Google Scholar] [CrossRef]
- Sahu, A.; Kumar, D.; Agrawal, R.K. Antileishmanial Drug Discovery: Synthetic Methods, Chemical Characteristics, and Biological Potential of Quinazolines and its Derivatives. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2017, 16, 3–32. [Google Scholar] [CrossRef]
- Romero, A.H.; Rodríguez, N.; Oviedo, H. 2-Aryl-quinazolin-4(3H)-ones as an inhibitor of leishmania folate pathway: In vitro biological evaluation, mechanism studies and molecular docking. Bioorganic Chem. 2019, 83, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Fiorillo, A.; Genovese, I.; Colotti, G. Polyamine-trypanothione pathway: An update. Futur. Med. Chem. 2017, 9, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Saccoliti, F.; Di Santo, R.; Costi, R. Recent Advancement in the Search of Innovative Antiprotozoal Agents Targeting Trypanothione Metabolism. ChemMedChem 2020, 15, 2420–2435. [Google Scholar] [CrossRef]
- Kraeva, N.; Horáková, E.; Kostygov, A.Y.; Kořený, L.; Butenko, A.; Yurchenko, V.; Lukeš, J. Catalase in Leishmaniinae: With me or against me? Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2017, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Khare, P.; Jaiswal, A.K.; Tripathi, C.D.P.; Joshi, S.; Sundar, S.; Dube, A. Efficacy of Leishmania donovani trypanothione reductase, identified as a potent Th1 stimulatory protein, for its immunogenicity and prophylactic potential against experimental visceral leishmaniasis. Parasitol. Res. 2014, 113, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A.; Carrillo, C.; Comini, M. The Thiol-polyamine Metabolism of Trypanosoma cruzi: Molecular Targets and Drug Repurposing Strategies. Curr. Med. Chem. 2019, 26, 6614–6635. [Google Scholar] [CrossRef]
- Battista, T.; Colotti, G.; Ilari, A.; Fiorillo, A. Targeting Trypanothione Reductase, a Key Enzyme in the Redox Trypanosomatid Metabolism, to Develop New Drugs against Leishmaniasis and Trypanosomiases. Molecules 2020, 25, 1924. [Google Scholar] [CrossRef] [Green Version]
- Saccoliti, F.; Angiulli, G.; Pupo, G.; Pescatori, L.; Madia, V.N.; Messore, A.; Colotti, G.; Fiorillo, A.; Scipione, L.; Gramiccia, M.; et al. Inhibition of Leishmania infantum trypanothione reductase by diaryl sulfide derivatives. J. Enzym. Inhib. Med. Chem. 2017, 32, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Vishwakarma, P.; Parmar, N.; Chandrakar, P.; Sharma, T.; Kathuria, M.; Agnihotri, P.K.; Siddiqi, M.I.; Mitra, K.; Kar, S. Ammonium trichloro [1,2-ethanediolato-O,O′]-tellurate cures experimental visceral leishmaniasis by redox modulation of Leishmania donovani trypanothione reductase and inhibiting host integrin linked PI3K/Akt pathway. Cell Mol. Life Sci. 2018, 75, 563–588. [Google Scholar] [CrossRef]
- Revuelto, A.; Ruiz-Santaquiteria, M.; de Lucio, H.; Gamo, A.M.; Carriles, A.A.; Gutiérrez, K.J.; Sánchez-Murcia, P.A.; Hermoso, J.A.; Gago, F.; Camarasa, M.-J.; et al. Pyrrolopyrimidine vs Imidazole-Phenyl-Thiazole Scaffolds in Nonpeptidic Dimerization Inhibitors of Leishmania infantum Trypanothione Reductase. ACS Infect. Dis. 2019, 5, 873–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revuelto, A.; de Lucio, H.; García-Soriano, J.C.; Sánchez-Murcia, P.A.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.-J.; Velázquez, S. Efficient Dimerization Disruption of Leishmania infantum Trypanothione Reductase by Triazole-phenyl-thiazoles. J. Med. Chem. 2021, 64, 6137–6160. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.D.; dos Santos, J.A.; Machado, P.A.; Alves, L.A.; Laque, L.C.; de Souza, V.C.; Coimbra, E.S.; Capriles, P.V.S.Z. Insights about resveratrol analogs against trypanothione reductase of Leishmania braziliensis: Molecular modeling, computational docking and in vitro antileishmanial studies. J. Biomol. Struct. Dyn. 2019, 37, 2960–2969. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, R.; Watowich, S.J.; Flórez, A.; Mesa, C.V.; Robledo, S.M.; Muskus, C. Drug search for leishmaniasis: A virtual screening approach by grid computing. J. Comput. Aided Mol. Des. 2016, 30, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Matadamas-Martínez, F.; Hernández-Campos, A.; Téllez-Valencia, A.; Vázquez-Raygoza, A.; Comparán-Alarcón, S.; Yépez-Mulia, L.; Castillo, R. Leishmania mexicana Trypanothione Reductase Inhibitors: Computational and Biological Studies. Molecules 2019, 24, 3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Aquino, T.M.; França, P.H.B.; Rodrigues, E.E.S.; Nascimento, I.J.; Santos-Júnior, P.F.S.; Aquino, P.G.V.; Santos, M.S.; Queiroz, A.C.; Araújo, M.V.; Alexandre-Moreira, M.S.; et al. Synthesis, Antileishmanial Activity and in silico Studies of Aminoguanidine Hydrazones (AGH) and Thiosemicarbazones (TSC) against Leishmania chagasi Amastigotes. Med. Chem. 2021, 18, 151–169. [Google Scholar] [CrossRef]
- Maamri, S.; Benarous, K.; Yousfi, M. Identification of 3-Methoxycarpachromene and Masticadienonic Acid as New Target Inhibitors against Trypanothione Reductase from Leishmania Infantum Using Molecular Docking and ADMET Prediction. Molecules 2021, 26, 3335. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, P.K.; Chakraborti, S.; Bagchi, A.; Chakraborti, T. Bioassay-based Corchorus capsularis L. leaf-derived β-sitosterol exerts antileishmanial effects against Leishmania donovani by targeting trypanothione reductase. Sci. Rep. 2020, 10, 20440. [Google Scholar] [CrossRef] [PubMed]
- Khare, P.; Rastogi, P.; Gupta, S.; Maurya, R.; Dube, A. In vitro and In vivo Efficacy of a New Herbaceous Indian Plant—Abutilon indicum Against Leishmania donovani Infection. AJPCT 2014, 2, 134–139. [Google Scholar]
- Inacio, J.D.F.; Fonseca, M.S.; Limaverde-Sousa, G.; Tomas, A.M.; Castro, H.; Almeida-Amaral, E.E. Epigallocathechin-O-3-Gallate Inhibits Trypanothione Reductase of Leishmania infantum, Causing Alterations in Redox Balance and Leading to Parasite Death. Front. Cell. Infect. Microbiol. 2021, 11, 640561. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.A. Polyamines in protozoan pathogens. J. Biol. Chem. 2018, 293, 18746–18756. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, S.; Cleveland, J.L. Targeting the polyamine-hypusine circuit for the prevention and treatment of cancer. Amino Acids 2016, 48, 2353. [Google Scholar] [CrossRef] [Green Version]
- Chawla, B.; Kumar, R.R.; Tyagi, N.; Subramanian, G.; Srinivasan, N.; Park, M.H.; Madhubala, R. A Unique Modification of the Eukaryotic Initiation Factor 5A Shows the Presence of the Complete Hypusine Pathway in Leishmania donovani. PLoS ONE 2012, 7, e33138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Zhang, W.; Chen, H.; Zeng, J. Targeting Polyamine Metabolism for Control of Human Viral Diseases. Infect. Drug Resist. 2020, 13, 4335–4346. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Jones, D.C.; Wyllie, S.; Fairlamb, A.H.; Phillips, M.A. Allosteric Activation of Trypanosomatid Deoxyhypusine Synthase by a Catalytically Dead Paralog. J. Biol. Chem. 2013, 288, 15256–15267. [Google Scholar] [CrossRef] [Green Version]
- Goldman-Pinkovich, A.; Balno, C.; Strasser, R.; Zeituni-Molad, M.; Bendelak, K.; Rentsch, D.; Ephros, M.; Wiese, M.; Jardim, A.; Myler, P.J.; et al. An Arginine Deprivation Response Pathway Is Induced in Leishmania during Macrophage Invasion. PLoS Pathog. 2016, 12, e1005494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, N.; Stamper, B.; Elbarbry, F.; Nguyen, V.; Lopez, S.; Kawasaki, Y.; Poormohamadian, R.; Roberts, S. Natural Products That Target the Arginase in Leishmania Parasites Hold Therapeutic Promise. Microorganisms 2021, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- da Silva, M.F.L.; Floeter-Winter, L.M. Arginase in Leishmania. Subcell Biochem. 2013, 74, 103–117. [Google Scholar] [CrossRef]
- Boitz, J.M.; Gilroy, C.A.; Olenyik, T.D.; Paradis, D.; Perdeh, J.; Dearman, K.; Davis, M.J.; Yates, P.A.; Li, Y.; Riscoe, M.K.; et al. Arginase Is Essential for Survival of Leishmania donovani Promastigotes but Not Intracellular Amastigotes. Infect. Immun. 2017, 85, e00554-16. [Google Scholar] [CrossRef] [Green Version]
- Castilho-Martins, E.A.; da Silva, M.F.L.; dos Santos, M.G.; Muxel, S.M.; Floeter-Winter, L.M. Axenic Leishmania amazonensis Promastigotes Sense both the External and Internal Arginine Pool Distinctly Regulating the Two Transporter-Coding Genes. PLoS ONE 2011, 6, e27818. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Kumari, E.; Bhardwaj, R.; Kumar, R.; Dubey, V.K. Molecular events leading to death of Leishmania donovani under spermidine starvation after hypericin treatment. Chem. Biol. Drug Des. 2017, 90, 962–971. [Google Scholar] [CrossRef]
- Patterson, S.; Wyllie, S.; Norval, S.; Stojanovski, L.; Simeons, F.R.; Auer, J.L.; Osuna-Cabello, M.; Read, K.D.; Fairlamb, A.H. The anti-tubercular drug delamanid as a potential oral treatment for visceral leishmaniasis. eLife 2016, 5, e09744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafi, S.; Afrin, F.; Islamuddin, M.; Chouhan, G.; Ali, I.; Naaz, F.; Sharma, K.; Zaman, M.S. β-Nitrostyrenes as Potential Anti-leishmanial Agents. Front. Microbiol. 2016, 7, 1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, U.; Sultana, R.; Shaheen, N.; Hassan, S.F.; Yaqoob, F.; Ahmad, M.J.; Iftikhar, F.; Sultana, N.; Asghar, S.; Yasinzai, M.; et al. Structure based medicinal chemistry-driven strategy to design substituted dihydropyrimidines as potential antileishmanial agents. Eur. J. Med. Chem. 2016, 115, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Verastegui, C.; Llanos-Cuentas, A.; Arevalo, I.; Ward, B.J.; Matlashewski, G. Randomized, Double-Blind Clinical Trial of Topical Imiquimod 5% with Parenteral Meglumine Antimoniate in the Treatment of Cutaneous Leishmaniasis in Peru. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2005, 40, 1395–1403. [Google Scholar] [CrossRef]
- Portas, A.D.S.; Miguel, D.C.; Yokoyama-Yasunaka, J.K.U.; Uliana, S.R.B.; Espósito, B.P. Increasing the activity of copper(II) complexes against Leishmania through lipophilicity and pro-oxidant ability. J. Biol. Inorg. Chem. JBIC A Publ. Soc. Biol. Inorg. Chem. 2012, 17, 107–112. [Google Scholar] [CrossRef]
- Didwania, N.; Shadab; Sabur, A.; Ali, N. Alternative to Chemotherapy—The Unmet Demand against Leishmaniasis. Front. Immunol. 2017, 8, 1779. [Google Scholar] [CrossRef] [Green Version]
- Robles-Loaiza, A.; Pinos-Tamayo, E.; Mendes, B.; Teixeira, C.; Alves, C.; Gomes, P.; Almeida, J. Peptides to Tackle Leishmaniasis: Current Status and Future Directions. Int. J. Mol. Sci. 2021, 22, 4400. [Google Scholar] [CrossRef]
- Chaudhari, V.; Buttar, H.S.; Bagwe-Parab, S.; Tuli, H.S.; Vora, A.; Kaur, G. Therapeutic and Industrial Applications of Curdlan with Overview on Its Recent Patents. Front. Nutr. 2021, 8, 646988. [Google Scholar] [CrossRef]
- Maurya, R.; Bhattacharya, P.; Ismail, N.; Dagur, P.K.; Joshi, A.B.; Razdan, K., Jr.; Ascher, J.; Dey, R.; Nakhasi, H.L. Differential Role of Leptin as an Immunomodulator in Controlling Visceral Leishmaniasis in Normal and Leptin-Deficient Mice. Am. J. Trop. Med. Hyg. 2016, 95, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodziej, H.; Kiderlen, A.F. Antileishmanial activity and immune modulatory effects of tannins and related compounds on Leishmania parasitised RAW 264.7 cells. Phytochemistry 2005, 66, 2056–2071. [Google Scholar] [CrossRef]
- Munder, M.; Mallo, M.; Eichmann, K.; Modolell, M. Murine Macrophages Secrete Interferon γ upon Combined Stimulation with Interleukin (IL)-12 and IL-18: A Novel Pathway of Autocrine Macrophage Activation. J. Exp. Med. 1998, 187, 2103–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, T.R.; Conserva, G.A.A.; Varela, M.T.; Costa-Silva, T.A.; Thevenard, F.; Ponci, V.; Fortuna, A.; Falcão, A.C.; Tempone, A.; Fernandes, J.P.S.; et al. Improving the drug-likeness of inspiring natural products—Evaluation of the antiparasitic activity against Trypanosoma cruzi through semi-synthetic and simplified analogues of licarin A. Sci. Rep. 2020, 10, 5467. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; O’Connor, P.D.; Blaser, A.; Yardley, V.; Maes, L.; Gupta, S.; Launay, D.; Martin, D.; Franzblau, S.G.; Wan, B.; et al. Repositioning Antitubercular 6-Nitro-2,3-dihydroimidazo[2,1-b][1,3]oxazoles for Neglected Tropical Diseases: Structure–Activity Studies on a Preclinical Candidate for Visceral Leishmaniasis. J. Med. Chem. 2016, 59, 2530–2550. [Google Scholar] [CrossRef]
- Wyllie, S.; Brand, S.; Thomas, M.; De Rycker, M.; Chung, C.-W.; Pena, I.; Bingham, R.P.; Bueren-Calabuig, J.A.; Cantizani, J.; Cebrian, D.; et al. Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 9318–9323. [Google Scholar] [CrossRef] [Green Version]
- Gil, Z.; Martinez-Sotillo, N.; Pinto-Martinez, A.; Mejias, F.; Martinez, J.C.; Galindo, I.; Oldfield, E.; Benaim, G. SQ109 inhibits proliferation of Leishmania donovani by disruption of intracellular Ca2+ homeostasis, collapsing the mitochondrial electrochemical potential (ΔΨm) and affecting acidocalcisomes. Parasitol. Res. 2020, 119, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.G.; Tempone, A.G. Activity of the antiarrhythmic drug amiodarone against Leishmania (L.) infantum: An in vitro and in vivo approach. J. Venom. Anim. Toxins Incl. Trop. Dis. 2018, 24, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singodia, D.; Verma, A.; Khare, P.; Dube, A.; Mitra, K.; Mishra, P.R. Investigations on feasibility of in situ development of amphotericin B liposomes for industrial applications. J. Liposome Res. 2011, 22, 8–17. [Google Scholar] [CrossRef]
- Tempone, A.; Perez, D.; Rath, S.; Vilarinho, A.L.; Mortara, R.; De Andrade, H.F. Targeting Leishmania (L.) chagasi amastigotes through macrophage scavenger receptors: The use of drugs entrapped in liposomes containing phosphatidylserine. J. Antimicrob. Chemother. 2004, 54, 60–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tempone, A.G.; Mortara, R.; de Andrade, H.F.; Reimão, J. Therapeutic evaluation of free and liposome-loaded furazolidone in experimental visceral leishmaniasis. Int. J. Antimicrob. Agents 2010, 36, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.G.; da Costa-Silva, T.A.; Tempone, A.G. Histamine H1-receptor antagonists against Leishmania (L.) infantum: An in vitro and in vivo evaluation using phosphatidylserine-liposomes. Acta Trop. 2014, 137, 206–210. [Google Scholar] [CrossRef]
- Gharbi, M.; Darghouth, M.A.; Elati, K.; Al-Hosary, A.A.T.; Ayadi, O.; Salih, D.A.; El Hussein, A.M.; Mhadhbi, M.; Khbou, M.K.; Hassan, S.M.; et al. Current status of tropical theileriosis in Northern Africa: A review of recent epidemiological investigations and implications for control. Transbound. Emerg. Dis. 2020, 67, 8–25. [Google Scholar] [CrossRef] [PubMed]
- Costa-silva, T.A.; Galisteo, J. Nanoliposomal Buparvaquone Immunomodulates Leishmania infantum—Infected Macrophages and Is Highly Effective in a Murine Model. Spine 2017, 61, e02297-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandrioli, R.; Mercolini, L.; Raggi, M.A. Evaluation of the pharmacokinetics, safety and clinical efficacy of sertraline used to treat social anxiety. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Palit, P.; Ali, N. Oral therapy with sertraline, a selective serotonin reuptake inhibitor, shows activity against Leishmania donovani. J. Antimicrob. Chemother. 2008, 61, 1120–1124. [Google Scholar] [CrossRef] [Green Version]
- Lima, M.L.; Abengózar, M.A.; Nácher-Vázquez, M.; Martínez-Alcázar, M.P.; Barbas, C.; Tempone, A.G.; López-Gonzálvez, Á.; Rivas, L. Molecular Basis of the Leishmanicidal Activity of the Antidepressant Sertraline as a Drug Repurposing Candidate. Antimicrob. Agents Chemother. 2018, 62, e01928-18. [Google Scholar] [CrossRef] [Green Version]
- Romanelli, M.M.; Da Costa-Silva, T.A.; Cunha-Junior, E.; Ferreira, D.D.; Guerra, J.M.; Galisteo, A.J.J.; Pinto, E.G.; Barbosa, L.; Torres-Santos, E.C.; Tempone, A.G. Sertraline Delivered in Phosphatidylserine Liposomes Is Effective in an Experimental Model of Visceral Leishmaniasis. Front. Cell. Infect. Microbiol. 2019, 9, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, C.S.; Oliveira-Da-Silva, J.A.; Lage, D.P.; Costa, R.R.; Mendonça, D.V.C.; Martins, V.T.; Reis, T.A.R.; Antinarelli, L.M.R.; Machado, A.S.; Tavares, G.S.V.; et al. Digitoxigenin presents an effective and selective antileishmanial action against Leishmania infantum and is a potential therapeutic agent for visceral leishmaniasis. Parasitol. Res. 2020, 120, 321–335. [Google Scholar] [CrossRef]
- Costa, R.R.; Oliveira-Da-Silva, J.A.; Reis, T.A.R.; Tavares, G.S.V.; Mendonça, D.V.C.; Freitas, C.S.; Lage, D.P.; Martins, V.T.; Antinarelli, L.M.R.; Machado, A.S.; et al. Acarbose presents in vitro and in vivo antileishmanial activity against Leishmania infantum and is a promising therapeutic candidate against visceral leishmaniasis. Med. Microbiol. Immunol. 2021, 210, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.; Mondal, S.; Riikonen, J.; Rantanen, J.; Näkki, S.; Nissinen, T.; Närvänen, A.; Lehto, V.-P. Biogenic nanoporous silicon carrier improves the efficacy of buparvaquone against resistant visceral leishmaniasis. PLoS Negl. Trop. Dis. 2021, 15, e0009533. [Google Scholar] [CrossRef] [PubMed]
- Freitas-Mesquita, A.L.; Meyer-Fernandes, J.R. Stage-Specific Class I Nucleases of Leishmania Play Important Roles in Parasite Infection and Survival. Front. Cell. Infect. Microbiol. 2021, 11, 769933. [Google Scholar] [CrossRef] [PubMed]
- Kushawaha, P.K.; Gupta, R.; Tripathi, C.D.P.; Khare, P.; Jaiswal, A.K.; Sundar, S.; Dube, A. Leishmania donovani Triose Phosphate Isomerase: A Potential Vaccine Target against Visceral Leishmaniasis. PLoS ONE 2012, 7, e45766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chulanetra, M.; Chaicumpa, W. Revisiting the Mechanisms of Immune Evasion Employed by Human Parasites. Front. Cell. Infect. Microbiol. 2021, 11, 639. [Google Scholar] [CrossRef] [PubMed]
- Matte, C.; Duque, G.A.; Descoteaux, A. Leishmania donovani Metacyclic Promastigotes Impair Phagosome Properties in Inflammatory Monocytes. Infect. Immun. 2021, 89, e0000921. [Google Scholar] [CrossRef]
- O’Neal, A.J.; Butler, L.R.; Rolandelli, A.; Gilk, S.D.; Pedra, J.H. Lipid hijacking: A unifying theme in vector-borne diseases. eLife 2020, 9, e61675. [Google Scholar] [CrossRef]
- Bourourou, M.; Gouix, E.; Melis, N.; Friard, J.; Heurteaux, C.; Tauc, M.; Blondeau, N. Inhibition of eIF5A hypusination pathway as a new pharmacological target for stroke therapy. J. Cereb. Blood Flow Metab. 2021, 41, 1080–1090. [Google Scholar] [CrossRef]
- Muxel, S.M.; Aoki, J.I.; Fernandes, J.C.R.; Laranjeira-Silva, M.F.; Zampieri, R.A.; Acuña, S.M.; Müller, K.E.; Vanderlinde, R.H.; Floeter-Winter, L.M.; Muxel, S.M.; et al. Arginine and Polyamines Fate in Leishmania Infection. Front. Microbiol. 2017, 8, 2682. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Kumar, A.; Gupta, P.; Chand, K.; Samant, M.; Maurya, R.; Dube, A. Evaluation of antileishmanial potential of Tinospora sinensis against experimental visceral leishmaniasis. Parasitol. Res. 2008, 102, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, F.M.; Robinson, J.I.; Bluemling, G.R.; Ronet, C.; Fasel, N.; Beverley, S.M. Antiviral screening identifies adenosine analogs targeting the endogenous dsRNA Leishmania RNA virus 1 (LRV1) pathogenicity factor. Proc. Natl. Acad. Sci. USA 2017, 114, E811–E819. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leishmania Species | Pathway | Drug Target | Potent Drug Candidate | Mode of Action | Reference |
|---|---|---|---|---|---|
| L. braziliensis | Sterol Biosynthetic Pathway | Squalene epoxidase | JS87 | annulation of quinoline and oxindole scaffolds | [25] |
| L. infantum | Sterol Biosynthetic Pathway | Squalene epoxidase | spiro[cyclohexanone-oxindoles] | Inhibition of phosphodiesterase and tyrosine kinase | [23] |
| L. donovani | Sterol Biosynthetic Pathway | Squalene epoxidase | spiro[indole-3,3′-pyrrolizidine]-2-one | inhibitor of bisubunit DNA topoisomerase IB | [24] |
| L. braziliensis | Sterol Biosynthetic Pathway | sterol-14-α-demethylase | 6-ethyl-2-phenylquinoline | disruption of mitochondrial electrochemical potential and alkalinization of acidocalcisomes | [32] |
| L. donovani | Sterol Biosynthetic Pathway | HMGR enzyme | Mevastatin | Inhibits HMGR activity | [17] |
| L. donovani | Sterol Biosynthetic Pathway | Sterol alpha-14 demethylase | Avodart | Avodart-induced ROS caused apoptosis-like cell death in the parasites | [29] |
| L. major | Sterol Biosynthetic Pathway | 14-lanosterol demethylase | fenarimol | destabilization of membrane structure by inhibiting 14α sterol demethylase. | [37] |
| L. donovani | Sterol Biosynthetic Pathway | HMGR enzyme | Glycyrrhizic acid | inhibiting the HMGR enzyme | [40] |
| L. donovani | Purine Salvage Pathway | mRNA translation | 5-fluorouracil 4-thiouracil | binds to RNA and inhibits cell development | [46] |
| L. infantum | Purine Salvage Pathway | mRNA translation | pyrazolo [3,4-d] pyrimidine | binds to RNA and inhibits cell development | [47] |
| L. donovani | Glycolytic Pathway | GAPDH | artesunate | targeting parasites’ glycolytic enzymes mainly Glycerol-3-phosphate dehydrogenase | [53] |
| L. donovani | Glycolytic Pathway | GAPDH | quinine | targeting parasites’ glycolytic enzymes mainly Glycerol-3-phosphate dehydrogenase | [53] |
| L. donovani | Glycolytic Pathway | GAPDH | mefloquine | targeting parasites’ glycolytic enzymes mainly Glycerol-3-phosphate dehydrogenase | [53] |
| L. major | Glycolytic Pathway | phosphoglycerate kinase | Suramin | inhibition of cytosolic phosphoglycerate kinase from | [55] |
| L. major | Glycosyl phosphatidyl inositol | mannosyltransferase | N-4-(-5(trifluromethyl)-1-methyl-1H benzo[d]imidazole-2 yl) phenyl) | inhibit mannosylation of glycosyl phosphatidyl Inositol | [69] |
| L. donovani | Folate Biosynthesis Pathway | DHFR | Methotrexate (MTX, 1), | Inhibit DHFR | [75] |
| L. donovani | Folate Biosynthesis Pathway | DHFR | cycloguanil | Inhibit DHFR | [75] |
| L. donovani | Folate Biosynthesis Pathway | DHFR | trimethoprim (TMP, 2) | Inhibit DHFR | [75] |
| L. donovani | Folate Biosynthesis Pathway | DHFR | ZINC57774418 (Z18) | Inhibits DHFR activity | [76] |
| L. donovani | Folate Biosynthesis Pathway | DHFR | ZINC69844431 (Z31) | Inhibits DHFR activity | [76] |
| L. donovani | Folate Biosynthesis Pathway | DHFR | ZINC71746025 (Z25) | Inhibits DHFR activity | [76] |
| L. donovani | Folate Biosynthesis Pathway | DHFR | and D11596 (DB96) | Inhibits DHFR activity | [76] |
| L. donovani | Folate Biosynthesis Pathway | DHFR | 3,4-dihydropyrimidine-2-one | Inhibits DHFR activity | [77] |
| L. donovani | Folate Biosynthesis Pathway | DHFR | 5-(3,5-dimethoxybenzyl) pyrimidine-2,4-diamine | Inhibits DHFR activity | [77] |
| L. major | Folate Biosynthesis Pathway | PTR1 | thiosemicarbazones and 1,2,4-triazoles | Inhibit DHFR and PTR activity | [85] |
| L. donovani | Folate Biosynthesis Pathway | DHFR and PTR1 | 2-(4-((2,4-dichlorobenzyl)oxy)phenyl)-1H-benzo[d]imidazole | dual inhibitors of DHFR-TS and PTR1 | [86] |
| L. donovani | Folate Biosynthesis Pathway | DHFR and PTR1 | 2-(4-((2,4-dichlorobenzyl)oxy)phenyl)-1H-benzo[d]imidazole-1H-benzo[d]oxazole | dual inhibitors of DHFR-TS and PTR1 | [86] |
| L. major | Folate Biosynthesis Pathway | PTR1 | Sophoraflavanone G | Inhibits PTR1 activity | [87] |
| L. chagasi | Folate Biosynthesis Pathway | DHFR and PTR1 | 2,4-diaminoquinazoline | dual inhibitors of DHFR-TS and PTR1 | [88] |
| L. braziliensis, L. Mexicana, and L. amazonensis | Folate Biosynthesis Pathway | PTR1 | 2-arylquinazolin-4(3H) ones | Inhibits PTR1 activity | [92] |
| L. infantum | Trypanothione pathway | TR | [RDS 777] (6-(sec-butoxy)-2-((3-chlorophenyl) thio) pyrimidin-4-amine) | forms hydrogen bonds with the catalytic residues Glu466’, Cys57, and Cys52, limiting trypanothione binding and preventing its reduction | [99] |
| L. donovani | Trypanothione pathway | TR | trichloro [1,2-ethanediolato-O,O’]-tellurate (AS101) | Inhibits TR by forming thiol bonds with cysteine residues of TR., thus inducing ROS mediated apoptosis | [100] |
| L. infantum | Trypanothione pathway | TR | pyrrolopyrimidine | Disrupting the homodimeric interface trypanothione disulfide reductase | [101] |
| L. infantum | Trypanothione pathway | TR | 5-6-5 imidazole-phenyl-thiazole-helix-mimetic scaffolds | Disrupting the homodimeric interface trypanothione disulfide reductase | [101] |
| L. infantum | Trypanothione pathway | TR | Triazole-phenyl-thiazoles | Disrupting the homodimeric interface trypanothione disulfide reductase | [102] |
| L. braziliensis | Trypanothione pathway | TR | resveratrol analogues | Induce ROS by inhibiting TR activity | [103] |
| L. mexicana | Trypanothione pathway | TR | N-(6-quinolinemethyl)-3-pyrazole carboxamide | formation of hydrogen bonds with the active site of TR | [105] |
| L. donovani | Trypanothione pathway | TR | β-sitosterolCCL | Inhibit TR activity | [108] |
| L. infantum | Trypanothione pathway | TR | (-)-Epigallocatechin 3-O-gallate (EGCG) | a competitive inhibitor of the trypanothione substrate. | [110] |
| L. infantum | Trypanothione pathway | TR | 3-Methoxycarpachromene | Inhibits TR activity | [107] |
| L. donovani | Hypusine pathway | spermidine synthase | hypericin | decrease spermidine availability and induce ROS | [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, S.; Sahu, U.; Kumar, A.; Khare, P. Metabolic Pathways of Leishmania Parasite: Source of Pertinent Drug Targets and Potent Drug Candidates. Pharmaceutics 2022, 14, 1590. https://doi.org/10.3390/pharmaceutics14081590
Jain S, Sahu U, Kumar A, Khare P. Metabolic Pathways of Leishmania Parasite: Source of Pertinent Drug Targets and Potent Drug Candidates. Pharmaceutics. 2022; 14(8):1590. https://doi.org/10.3390/pharmaceutics14081590
Chicago/Turabian StyleJain, Surbhi, Utkarsha Sahu, Awanish Kumar, and Prashant Khare. 2022. "Metabolic Pathways of Leishmania Parasite: Source of Pertinent Drug Targets and Potent Drug Candidates" Pharmaceutics 14, no. 8: 1590. https://doi.org/10.3390/pharmaceutics14081590