
Localized Controlled Release of Kynurenic Acid Encapsulated in Synthetic Polymer Reduces Implant—Induced Dermal Fibrosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Fabrication of KynA Loaded PLGA Microspheres
2.3. Characterization of the Fabricated Microspheres
2.4. Measurement of KynA Using HPLC
2.5. Evaluation of the Encapsulation Efficiency
2.6. KynA Release Profile
2.7. Stability of the KynA after Encapsulation
2.8. In Vivo Animal Studies
2.9. In Vivo Release of KynA
2.10. Histological Analysis of Fibrosis
2.11. In Vivo Biological Activity; Quantification of Collagen
2.12. Total Tissue Cellularity
2.13. In Vivo Biological Activity; α-SMA Protein Expression
2.14. In Vivo Biological Activity; MMP-13, Type-1 Collagen, and α-SMA Gene Expression
2.15. Statistical Analysis
3. Results
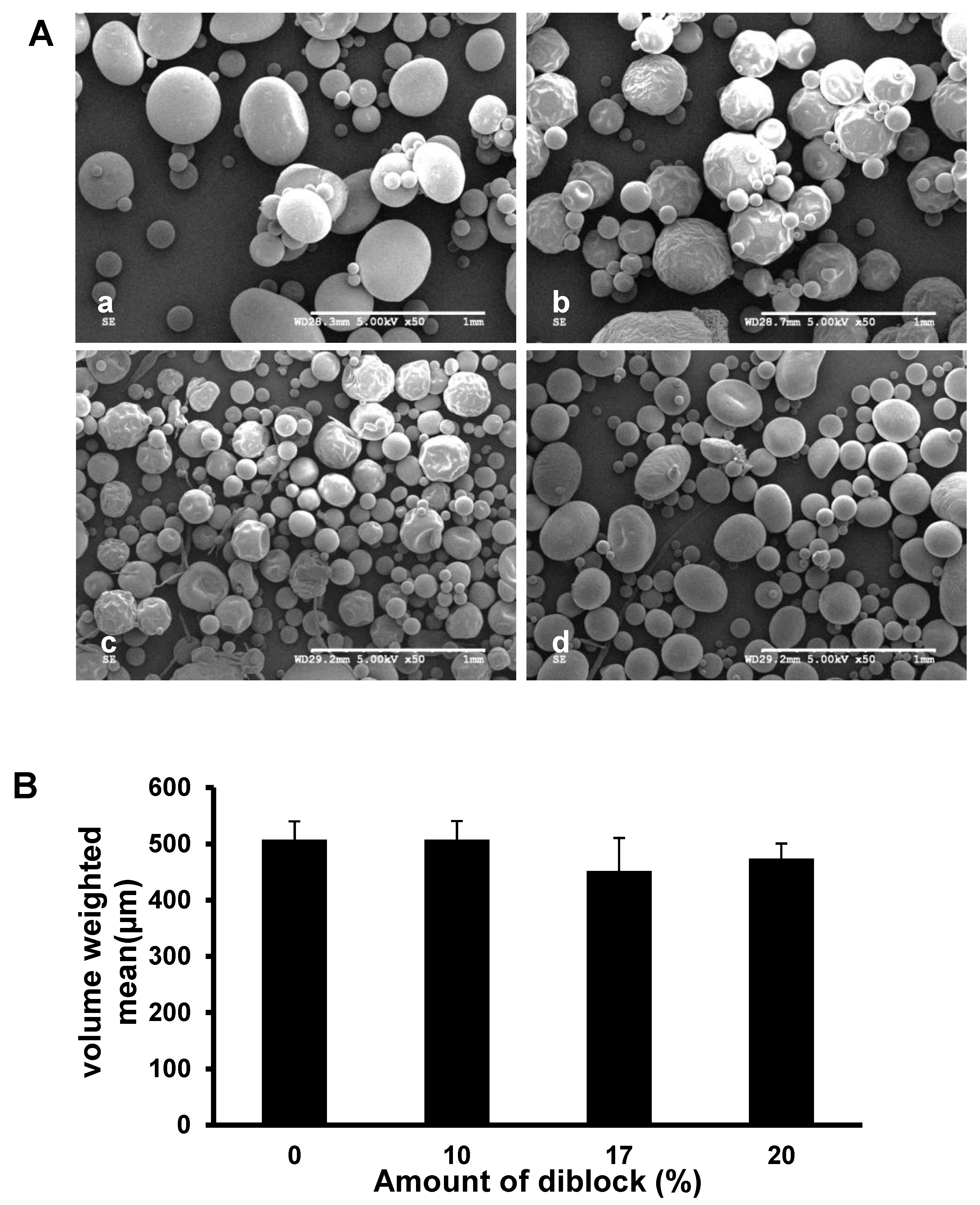
3.1. Morphology and Size Distribution of the Fabricated Microspheres
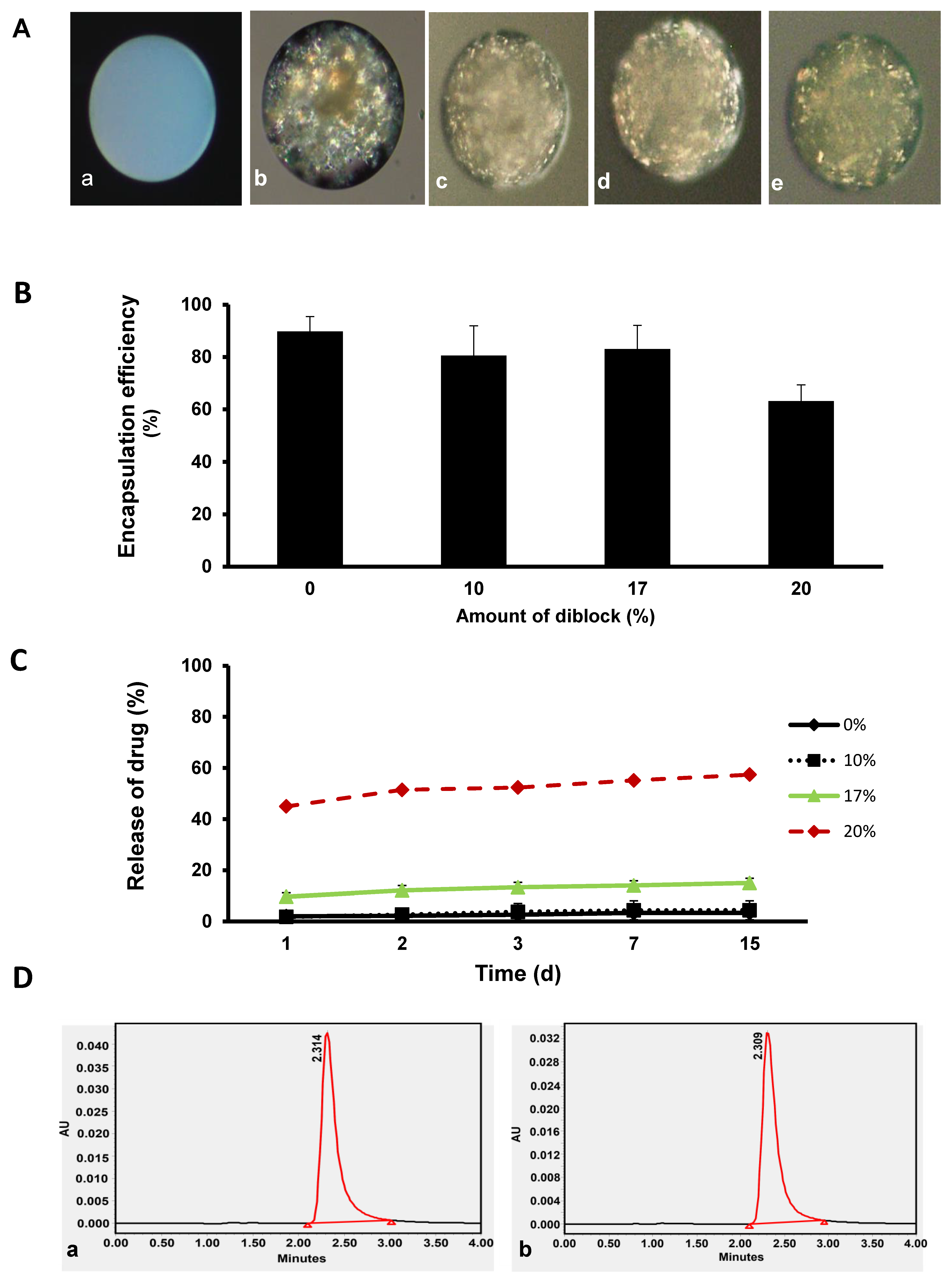
3.2. Encapsulation Efficiency, In Vitro Release Profile, and Stability of KynA
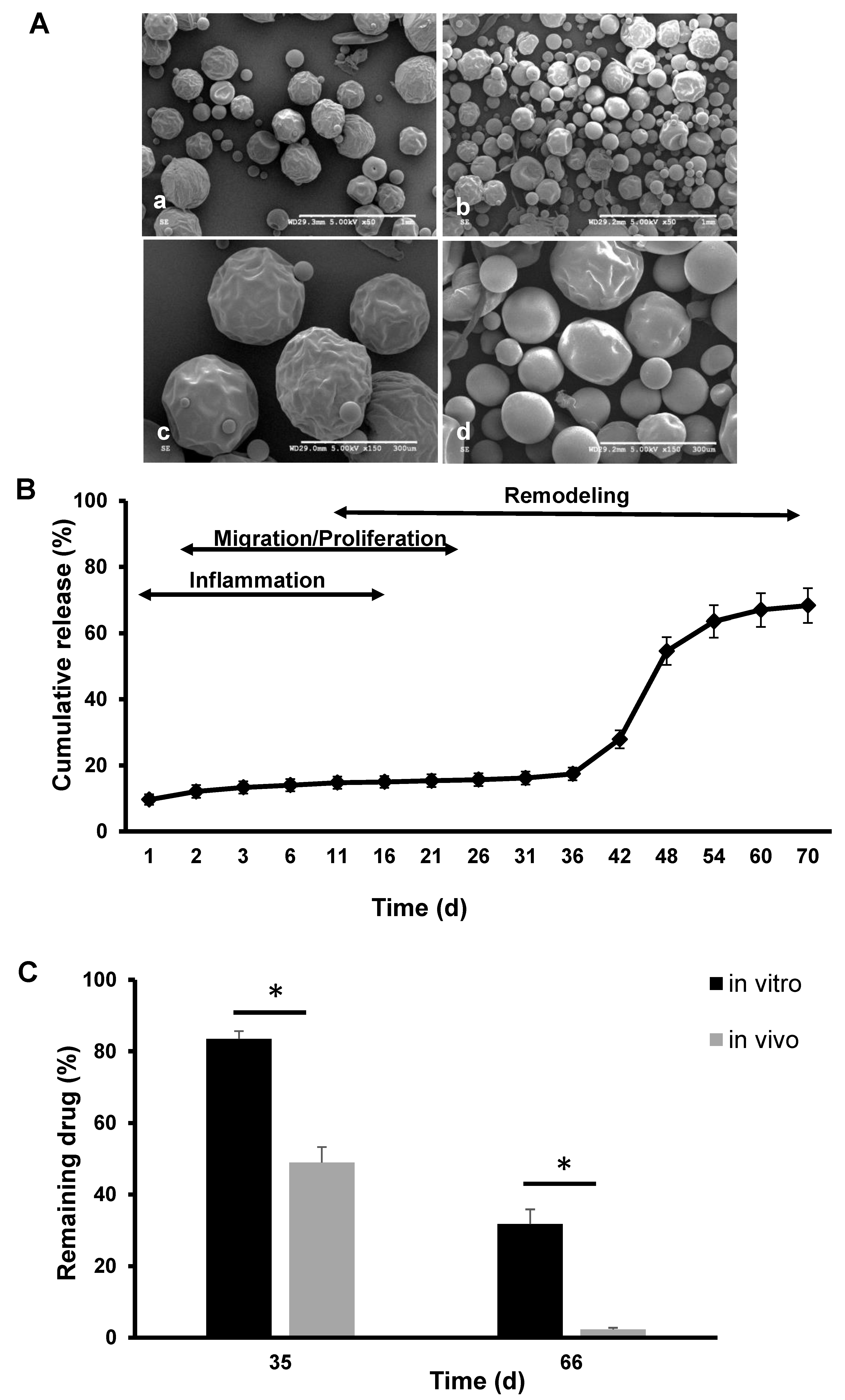
3.3. Morphology of the Polymer Only and Extended-Release Kinetics of KynA Loaded Microspheres In Vitro and In Vivo
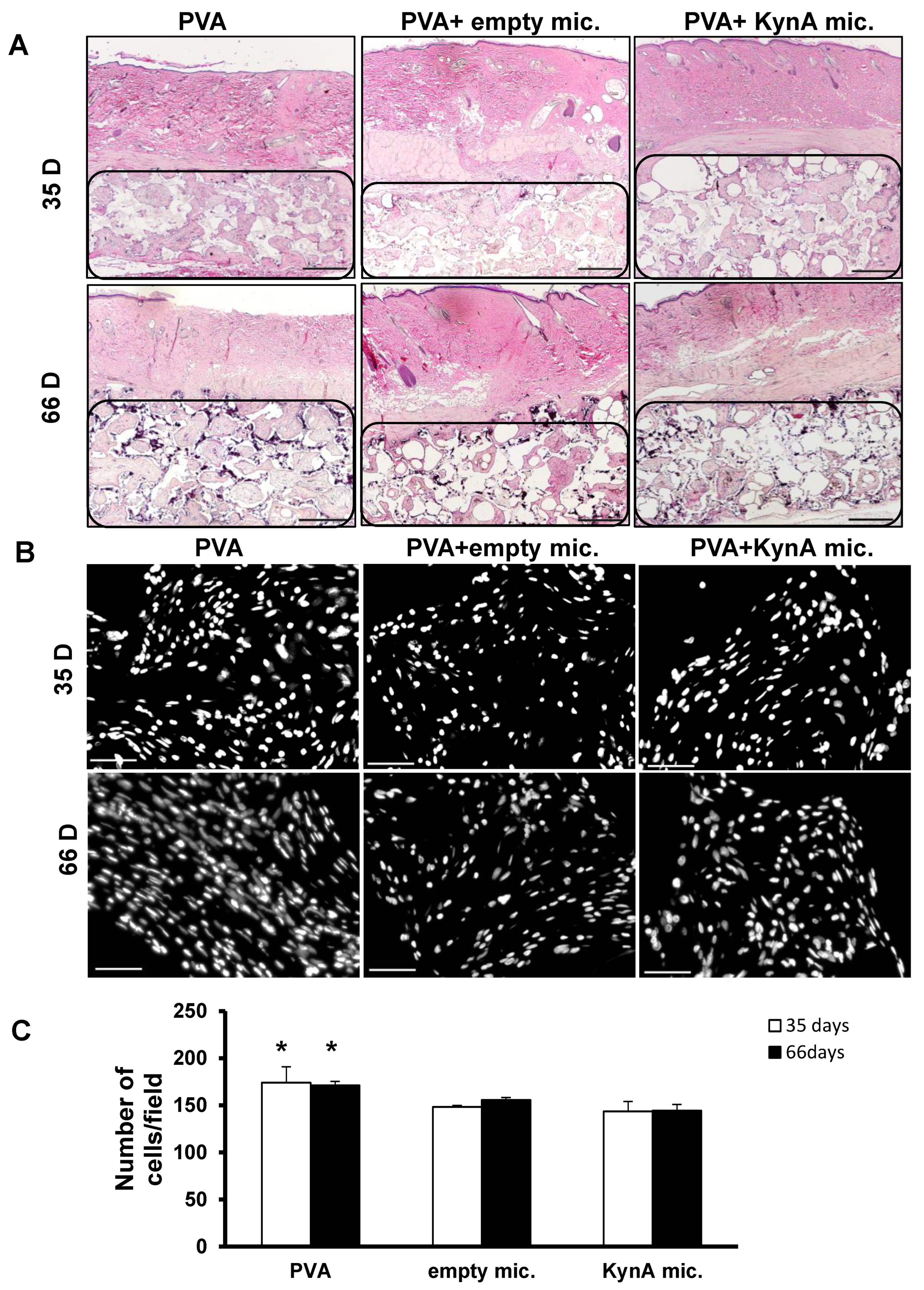
3.4. KynA Microspheres Decrease New Tissue Growth and Total Cellularity inside Implanted PVA Sponges
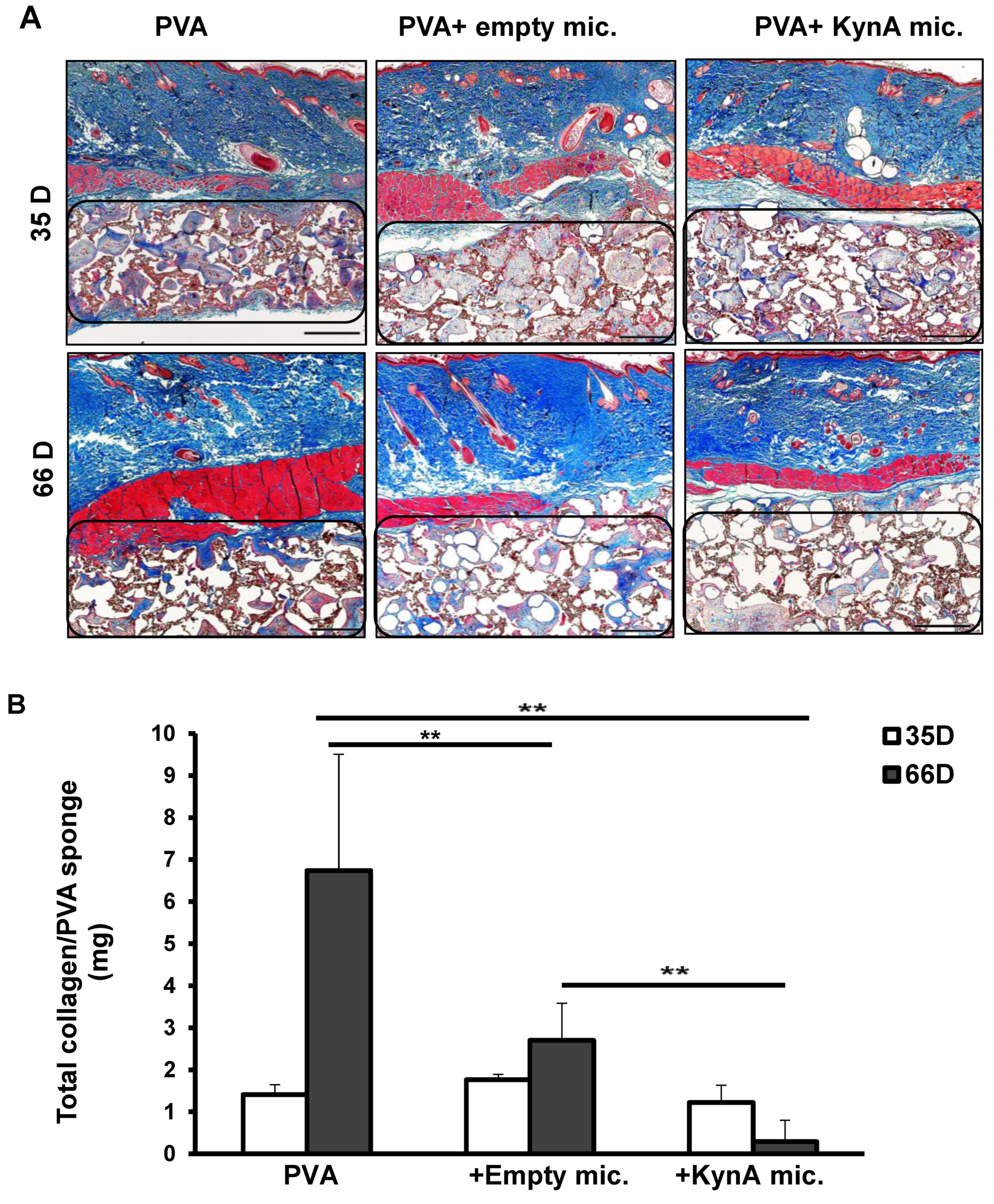
3.5. KynA Microspheres Decrease Collagen Deposition inside and around PVA Sponges
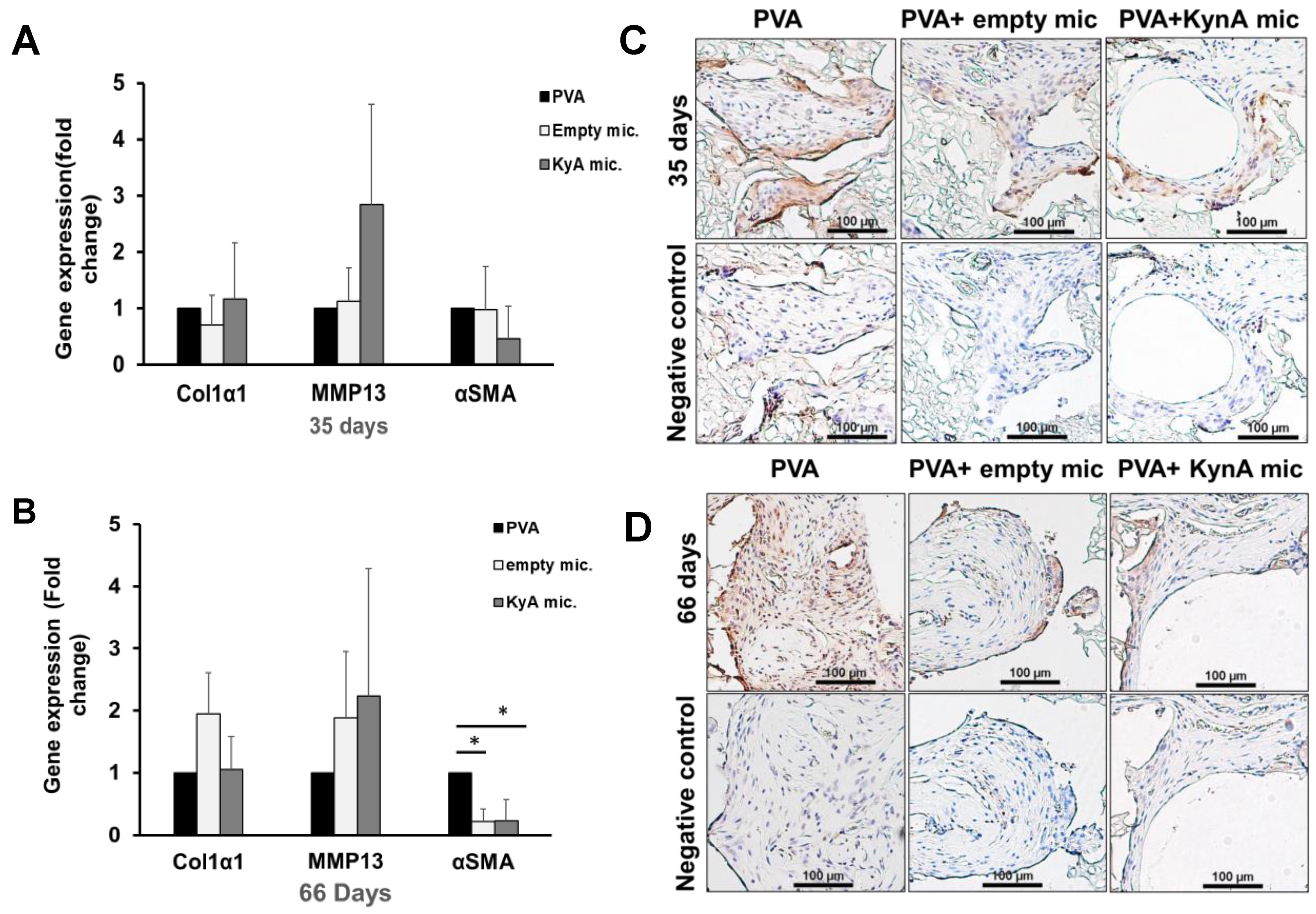
3.6. Col1α1, MMP-13, and α-SMA Expression In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahdavian Delavary, B.; van der Veer, W.M.; Ferreira, J.A.; Niessen, F.B. Formation of hypertrophic scars: Evolution and susceptibility. J. Plast. Surg. Hand Surg. 2012, 46, 95–101. [Google Scholar] [CrossRef]
- Adams, W.P.J. Capsular contracture: What is it? What causes it? How can it be prevented and managed? Clin. Plast. Surg. 2009, 36, 119–126. [Google Scholar] [CrossRef]
- Brüggmann, D.; Tchartchian, G.; Wallwiener, M.; Münstedt, K.; Tinneberg, H.-R.; Hackethal, A. Intra-abdominal adhesions: Definition, origin, significance in surgical practice, and treatment options. Dtsch Arztebl Int. 2010, 107, 769–775. [Google Scholar]
- Menzies, D.; Ellis, H. Intestinal obstruction from adhesions—How big is the problem? Ann. R. Coll. Surg. Engl. 1990, 72, 60–63. [Google Scholar]
- Beyene, R.T.; Kavalukas, S.L.; Barbul, A. Intra-abdominal adhesions: Anatomy, physiology, pathophysiology, and treatment. Curr. Probl. Surg. 2015, 52, 271–319. [Google Scholar] [CrossRef]
- Masopust, V.; Hackel, M.; Netuka, D.; Bradac, O.; Rokyta, R.; Vrabec, M. Postoperative epidural fibrosis. Clin. J. Pain 2009, 25, 600–606. [Google Scholar] [CrossRef]
- Bosscher, H.A.; Heavner, J.E. Incidence and severity of epidural fibrosis after back surgery: An endoscopic study. Pain Pract. 2010, 10, 18–24. [Google Scholar]
- Ellis, H. The clinical significance of adhesions: Focus on intestinal obstruction. Eur. J. Surg. Suppl. 1997, 577, 5–9. [Google Scholar]
- Van Goor, H. Consequences and complications of peritoneal adhesions. Colorectal. Dis. 2007, 9 (Suppl. 2), 25–34. [Google Scholar] [CrossRef]
- Van den Beukel, B.A.; de Ree, R.; van Leuven, S.; Bakkum, E.A.; Strik, C.; van Goor, H.; ten Broek, R.P.G. Surgical treatment of adhesion-related chronic abdominal and pelvic pain after gynaecological and general surgery: A systematic review and meta-analysis. Hum. Reprod. Update 2017, 23, 276–288. [Google Scholar]
- Abdul, N.; Dixon, D.; Walker, A.; Horabin, J.; Smith, N.; Weir, D.J.; Brewster, N.T.; Deehan, D.J.; Mann, D.A.; Borthwick, L.A. Fibrosis is a common outcome following total knee arthroplasty. Sci. Rep. 2015, 5, 16469. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Eaton, J.W. Inflammatory responses to biomaterials. Am. J. Clin. Pathol. 1995, 103, 466–471. [Google Scholar] [CrossRef]
- DiEgidio, P.; Friedman, H.I.; Gourdie, R.G.; Riley, A.E.; Yost, M.J.; Goodwin, R.L. Biomedical implant capsule formation: Lessons learned and the road ahead. Ann. Plast. Surg. 2014, 73, 451–460. [Google Scholar] [CrossRef]
- Ratner, B.D. Reducing capsular thickness and enhancing angiogenesis around implant drug release systems. J. Control Release 2002, 78, 211–218. [Google Scholar] [CrossRef]
- Tresco, P.A.; Winslow, B.D. The challenge of integrating devices into the central nervous system. Crit. Rev. Biomed. Eng. 2011, 39, 29–44. [Google Scholar] [CrossRef]
- Rousche, P.J.; Normann, R.A. Chronic recording capability of the Utah Intracortical Electrode Array in cat sensory cortex. J. Neurosci. Methods 1998, 82, 1–15. [Google Scholar] [CrossRef]
- Amoozgar, B.; Wei, X.; Lee, J.H.; Bloomer, M.; Zhao, Z.; Coh, P.; He, F.; Luan, L.; Xie, C.; Han, Y. A novel flexible microfluidic meshwork to reduce fibrosis in glaucoma surgery. PLoS ONE 2017, 12, e0172556. [Google Scholar] [CrossRef]
- Mylvaganam, S.; Conroy, E.; Williamson, P.R.; Barnes, N.L.; Cutress, R.I.; Gardiner, M.D.; Jain, A.; Skillman, J.M.; Thrush, S.; Whisker, L.J.; et al. Adherence to best practice consensus guidelines for implant-based breast reconstruction: Results from the iBRA national practice questionnaire survey. Eur. J. Surg. Oncol. 2018, 44, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Calobrace, M.B.; Schwartz, M.R.; Zeidler, K.R.; Pittman, T.A.; Cohen, R.; Stevens, W.G. Long-Term Safety of Textured and Smooth Breast Implants. Aesthetic Surg. J. 2017, 38, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Park, M.; Kim, B.H.; Lee, J.E.; Park, H.J.; Lee, S.H.; Park, C.G.; Kim, M.H.; Kim, R.; Kim, E.H.; et al. Acute suppression of TGF-ss with local, sustained release of tranilast against the formation of fibrous capsules around silicone implants. J. Control Release 2015, 200, 125–137. [Google Scholar] [CrossRef]
- Holmdahl, L.; Risberg, B.; E Beck, D.; Burns, J.W.; Chegini, N.; diZerega, G.S.; Ellis, H. Adhesions: Pathogenesis and prevention-panel discussion and summary. Eur. J. Surg. Suppl. 1997, 577, 56–62. [Google Scholar]
- Bellón, J.M.; Buján, J.; Contreras, L.A.; Jurado, F. Use of nonporous polytetrafluoroethylene prosthesis in combination with polypropylene prosthetic abdominal wall implants in prevention of peritoneal adhesions. J. Biomed. Mater. Res. 1997, 38, 197–202. [Google Scholar] [CrossRef]
- Zong, X.; Li, S.; Chen, E.; Garlick, B.; Kim, K.-S.; Fang, D.; Chiu, J.; Zimmerman, T.; Brathwaite, C.; Hsiao, B.S.; et al. Prevention of postsurgery-induced abdominal adhesions by electrospun bioabsorbable nanofibrous poly(lactide-co-glycolide)-based membranes. Ann. Surg. 2004, 240, 910–915. [Google Scholar] [CrossRef]
- Li, J.; Zhu, J.; He, T.; Li, W.; Zhao, Y.; Chen, Z.; Zhang, J.; Wan, H.; Li, R. Prevention of intra-abdominal adhesion using electrospun PEG/PLGA nanofibrous membranes. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 78, 988–997. [Google Scholar] [CrossRef]
- Gauglitz, G.G.; Korting, H.C.; Pavicic, T.; Ruzicka, T.; Jeschke, M.G. Hypertrophic scarring and keloids: Pathomechanisms and current and emerging treatment strategies. Mol. Med. 2011, 17, 113–125. [Google Scholar] [CrossRef]
- Czock, D.; Keller, F.; Rasche, F.M.; Häussler, U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin. Pharm. 2005, 44, 61–98. [Google Scholar] [CrossRef]
- Whittam, A.J.; Maan, Z.; Duscher, D.; Wong, V.W.; Barrera, J.A.; Januszyk, M.; Gurtner, G.C. Challenges and Opportunities in Drug Delivery for Wound Healing. Adv. Wound Care 2016, 5, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Poormasjedi-Meibod, M.-S.; Hartwell, R.; Kilani, R.T.; Ghahary, A. Anti-scarring properties of different tryptophan derivatives. PLoS ONE 2014, 9, e91955. [Google Scholar] [CrossRef]
- Poormasjedi-Meibod, M.-S.; Pakyari, M.; Jackson, J.K.; Salimi Elizei, S.; Ghahary, A. Development of a nanofibrous wound dressing with an antifibrogenic properties in vitro and in vivo model. J. Biomed. Mater. Res. A 2016, 104, 2334–2344. [Google Scholar] [CrossRef]
- Shahriari, M.; Zahiri, M.; Abnous, K.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Enzyme responsive drug delivery systems in cancer treatment. J. Control. Release Off. J. Control. Release Soc. 2019, 308, 172–189. [Google Scholar] [CrossRef]
- Gao, J.; Dutta, K.; Zhuang, J.; Thayumanavan, S. Cellular- and Subcellular-Targeted Delivery Using a Simple All-in-One Polymeric Nanoassembly. Angew Chem. Int. Ed. Engl. 2020, 59, 23466–23470. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.; Asadian-Birjand, M.; Balach, J.; Bergueiro, J.; Miceli, E.; Calderón, M. Stimuli-responsive nanogel composites and their application in nanomedicine. Chem. Soc. Rev. 2015, 44, 6161–6186. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wu, P.; Fernandez, A.; Zhuang, J.; Thayumanavan, S. Cellular AND Gates: Synergistic Recognition to Boost Selective Uptake of Polymeric Nanoassemblies. Angew Chem. Int. Ed. Engl. 2020, 59, 10456–10460. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Preat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Jackson, J.K.; Hung, T.; Letchford, K.; Burt, H.M. The characterization of paclitaxel-loaded microspheres manufactured from blends of poly(lactic-co-glycolic acid) (PLGA) and low molecular weight diblock copolymers. Int. J. Pharm. 2007, 342, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.S.; Jackson, J.; Min, W.; Risovic, V.; Wasan, K.M.; Burt, H.M. Methotrexate loaded poly(L-lactic acid) microspheres for intra-articular delivery of methotrexate to the joint. J. Pharm. Sci. 2004, 93, 943–956. [Google Scholar] [CrossRef]
- Badawy, A.A.; Morgan, C.J. Rapid Isocratic Liquid Chromatographic Separation and Quantification of Tryptophan and Six kynurenine Metabolites in Biological Samples with Ultraviolet and Fluorimetric Detection. Int. J. Tryptophan Res. 2010, 3, 175–186. Available online: http://www.la-press.com/redirect_file.php?fileId=3261&filename=2391-IJTR-Rapid-Isocratic-Liquid-Chromatographic-Separation-and-Quantification-o.pdf&fileType=pdf (accessed on 21 October 2013). [CrossRef] [PubMed] [Green Version]
- Carson, F.L.; Cappellano, C.H. Histotechnology A Self Instructional Text, 4th ed.; ASCP Press: Chicago, IL, USA, 2015. [Google Scholar]
- Peterson, J.T.; Li, H.; Dillon, L.; Bryant, J.W. Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc. Res. 2000, 46, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Sasano, Y.; Zhu, J.-X.; Tsubota, M.; Takahashi, I.; Onodera, K.; Mizoguchi, I.; Kagayama, M. Gene expression of MMP8 and MMP13 during embryonic development of bone and cartilage in the rat mandible and hind limb. J. Histochem. Cytochem. 2002, 50, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, B.M.; Schwendeman, S.P. Characterization of the initial burst release of a model peptide from poly(D,L-lactide-co-glycolide) microspheres. J. Control Release 2002, 82, 289–307. [Google Scholar] [CrossRef]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- Percival, S.L.; McCarty, S.; Hunt, J.A.; Woods, E.J. The effects of pH on wound healing, biofilms, and antimicrobial efficacy. Wound Repair Regen. 2014, 22, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Ogawa, Y.; Yamamoto, M.; Takada, S.; Okada, H.; Shimamoto, T. Controlled-release of leuprolide acetate from polylactic acid or copoly(lactic/glycolic) acid microcapsules: Influence of molecular weight and copolymer ratio of polymer. Chem. Pharm. Bull. 1988, 36, 1502–1507. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, R. Keloid and Hypertrophic Scars Are the Result of Chronic Inflammation in the Reticular Dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, Y.-P.; Huang, H.-L.; Lai, W.-W.; Chung, J.-G.; Yang, J.-H. Antiproliferative effects of lactic acid via the induction of apoptosis and cell cycle arrest in a human keratinocyte cell line (HaCaT). J. Dermatol. Sci. 2009, 54, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nabai, L.; Ghahary, A.; Jackson, J. Localized Controlled Release of Kynurenic Acid Encapsulated in Synthetic Polymer Reduces Implant—Induced Dermal Fibrosis. Pharmaceutics 2022, 14, 1546. https://doi.org/10.3390/pharmaceutics14081546
Nabai L, Ghahary A, Jackson J. Localized Controlled Release of Kynurenic Acid Encapsulated in Synthetic Polymer Reduces Implant—Induced Dermal Fibrosis. Pharmaceutics. 2022; 14(8):1546. https://doi.org/10.3390/pharmaceutics14081546
Chicago/Turabian StyleNabai, Layla, Aziz Ghahary, and John Jackson. 2022. "Localized Controlled Release of Kynurenic Acid Encapsulated in Synthetic Polymer Reduces Implant—Induced Dermal Fibrosis" Pharmaceutics 14, no. 8: 1546. https://doi.org/10.3390/pharmaceutics14081546