
PD-L1-Targeted Co-Delivery of Two Chemotherapeutics for Efficient Suppression of Skin Cancer Growth

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
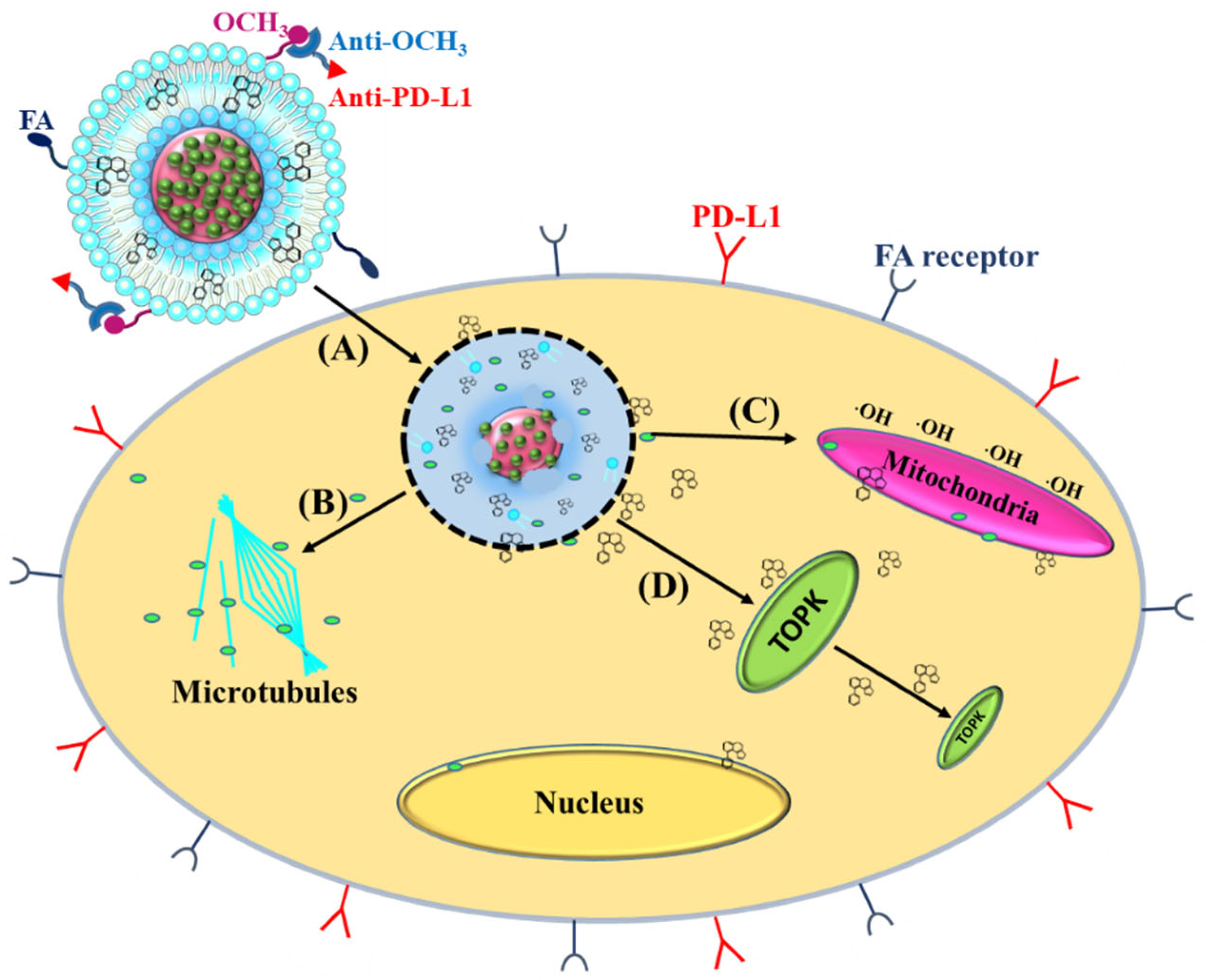{kind=link}
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Fabrication and Characterisation of LCP-Based Nanoparticles
2.3. Determination of the Number of Folic Acid and Bi-Anti-PD-L1 Conjugates
2.4. Quantification of Surface PD-L1 Expression
2.5. Cellular Uptake of Bi-Anti-PD-L1-Conjugated LCP NPs
2.6. Cytotoxicity Studies
2.7. Analysis of Apoptosis Induction, Reactive Oxygen Species (ROS) Generation and VEGF Secretion
2.8. Wound Healing and Transwell Invasion Assays
2.9. In Vivo Tumour Growth Inhibition
2.10. Statistical Analysis
3. Results and Discussion
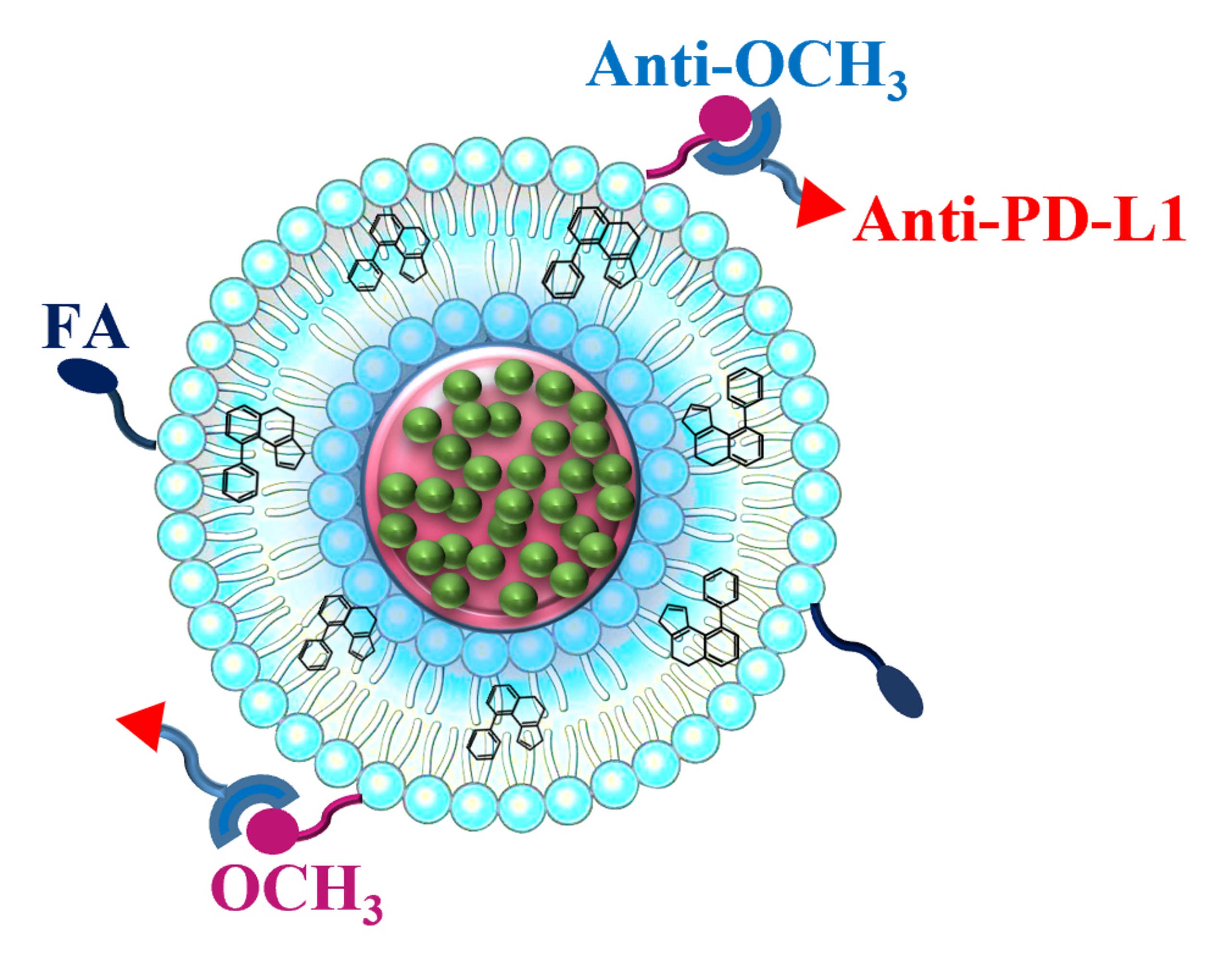
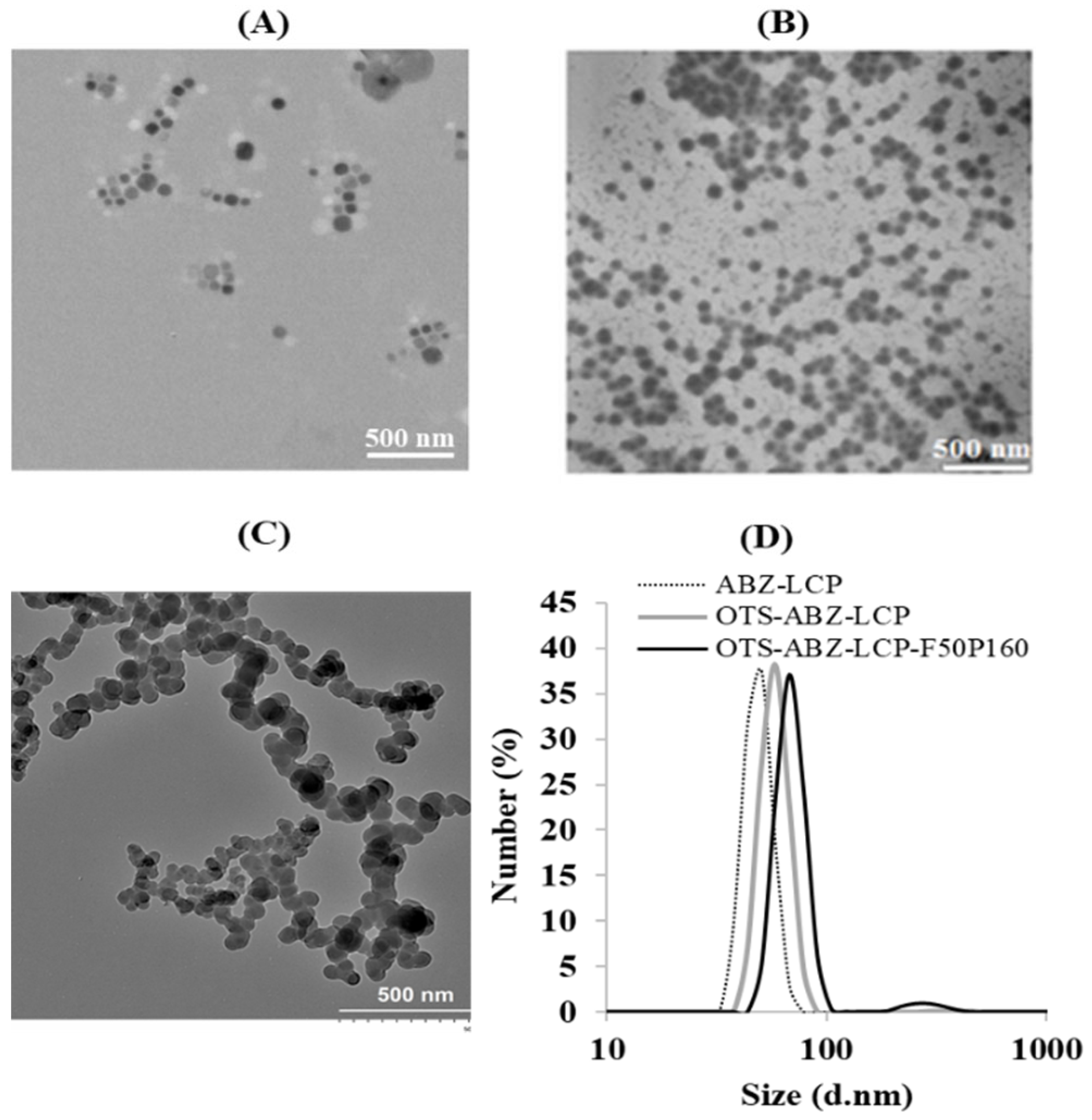
3.1. Physicochemical Properties of LCP-Based Nanoparticles
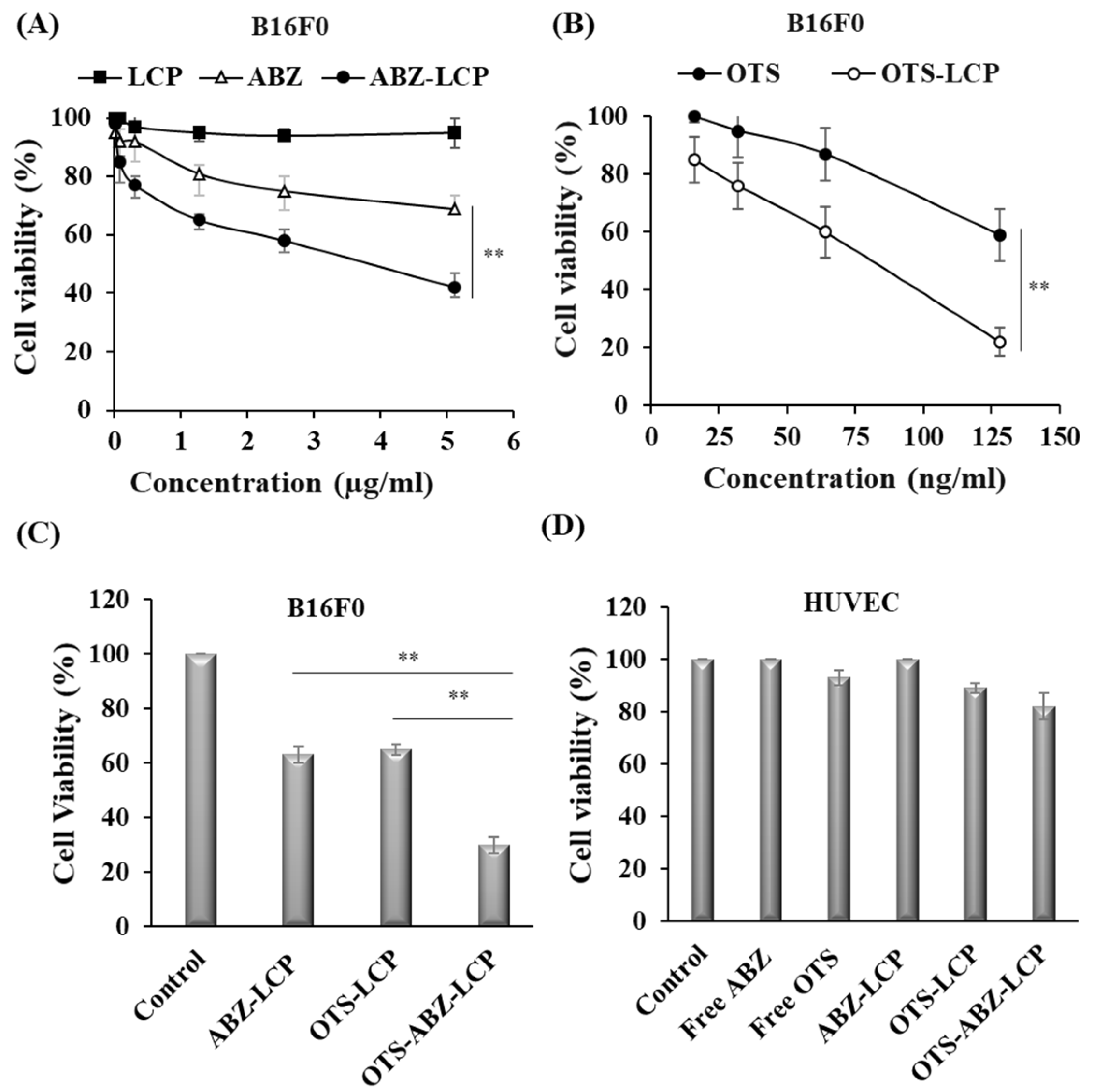
3.2. Cytotoxicity of LCP-Based Nanoparticles against Skin Cancer Cells
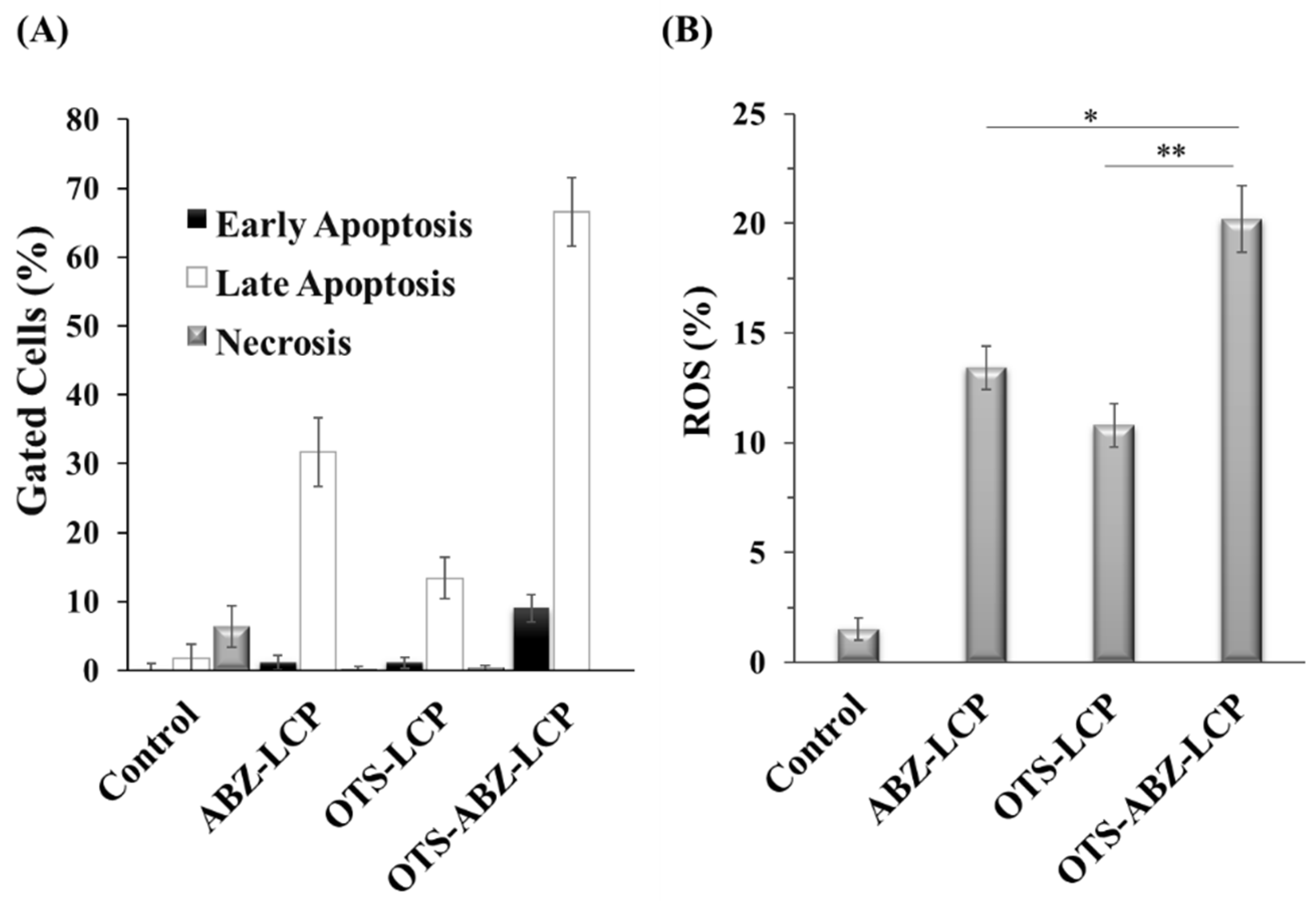
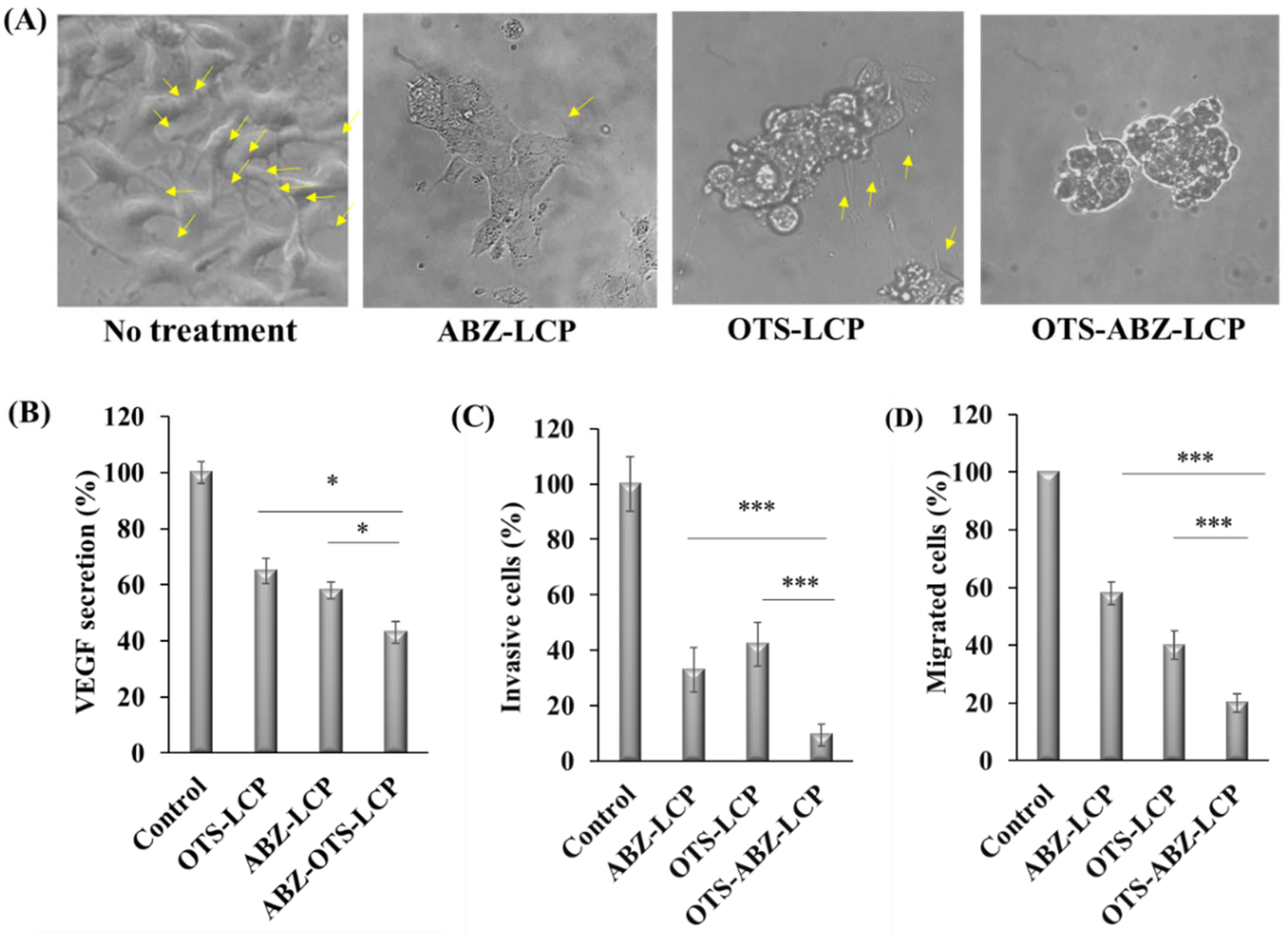
3.3. ROS-Induced Apoptosis, Inhibition of Tumour Progression and Dissemination
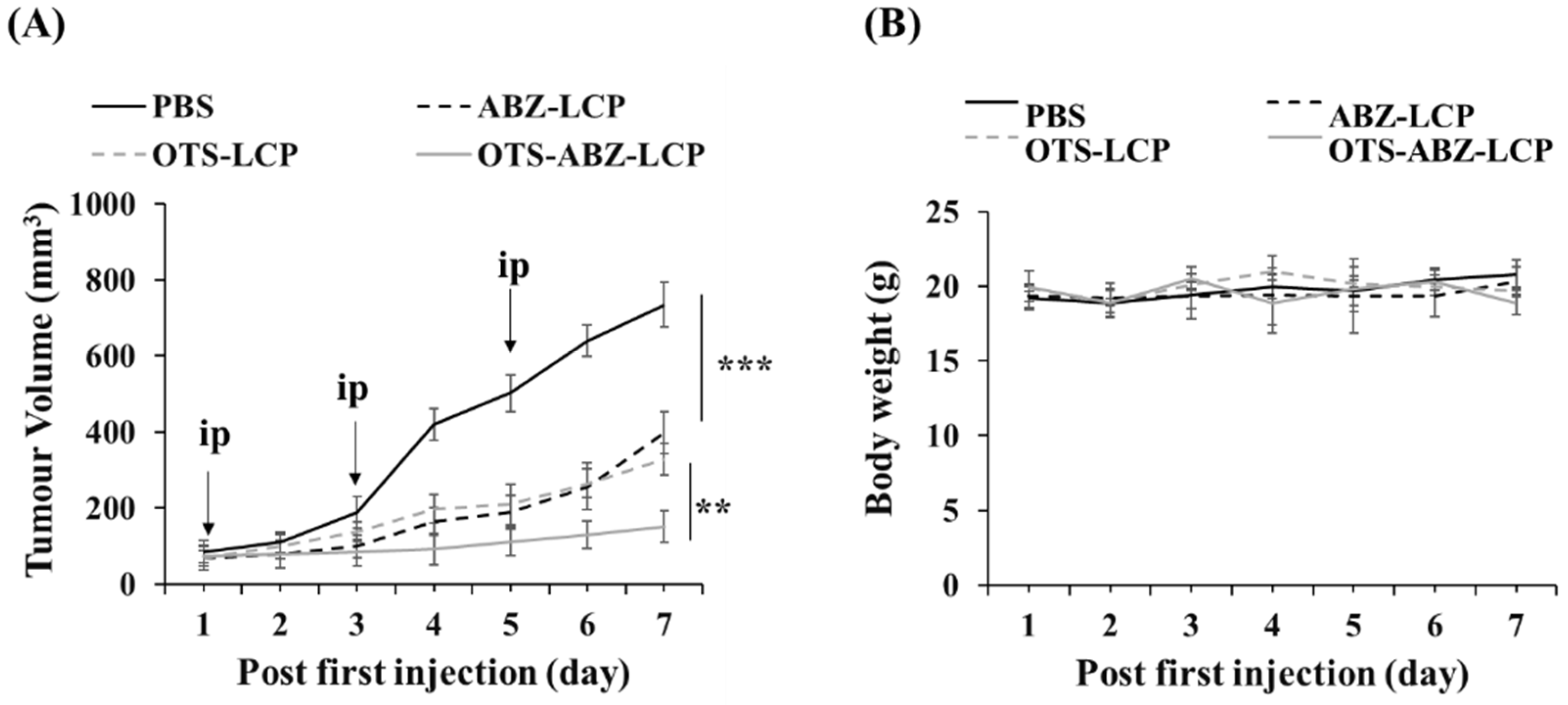
3.4. Efficient In Vivo Combination Chemotherapy
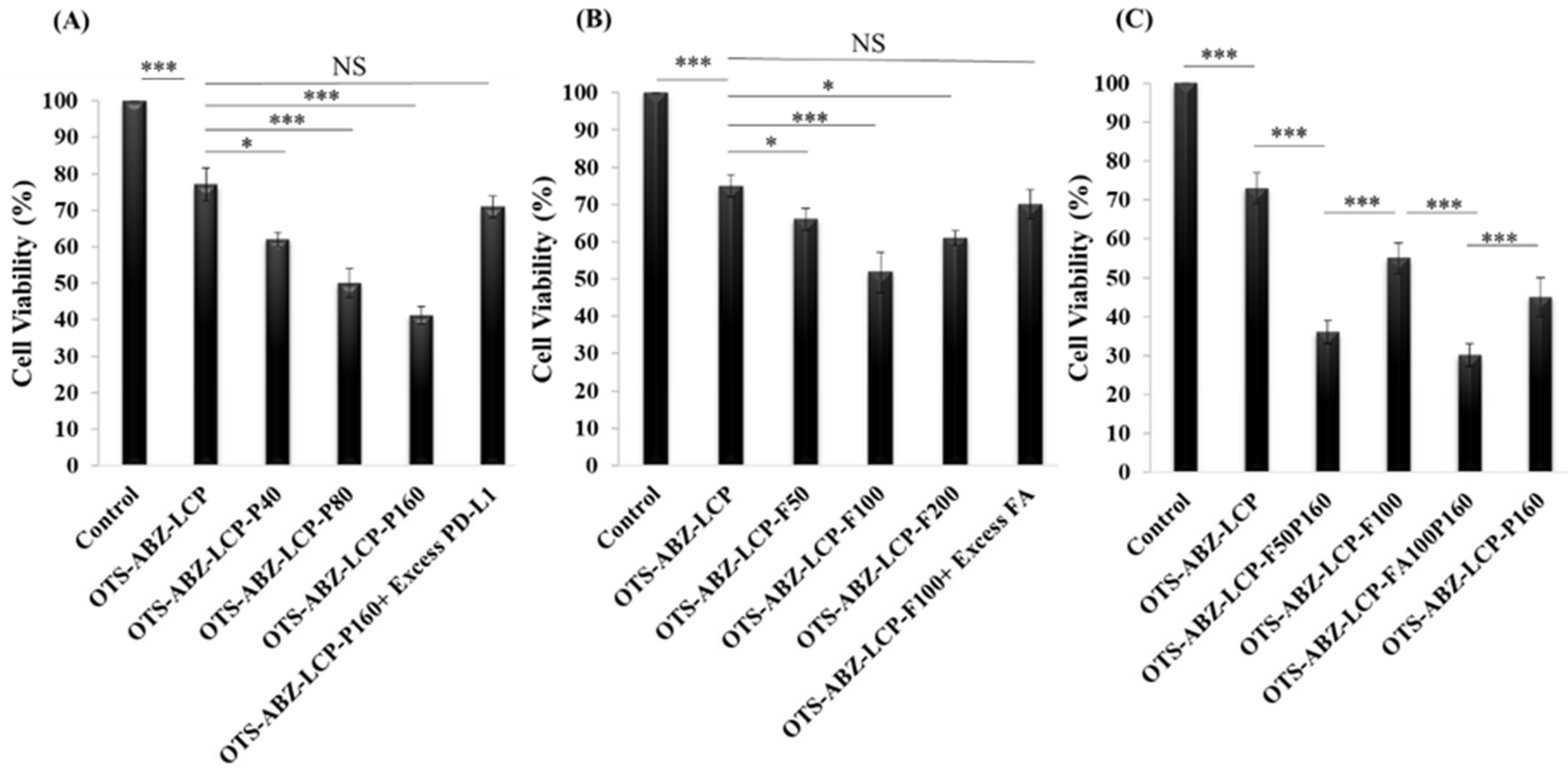
3.5. Dual Targeting Further Enhances Cytotoxicity against Skin Cancer Cells
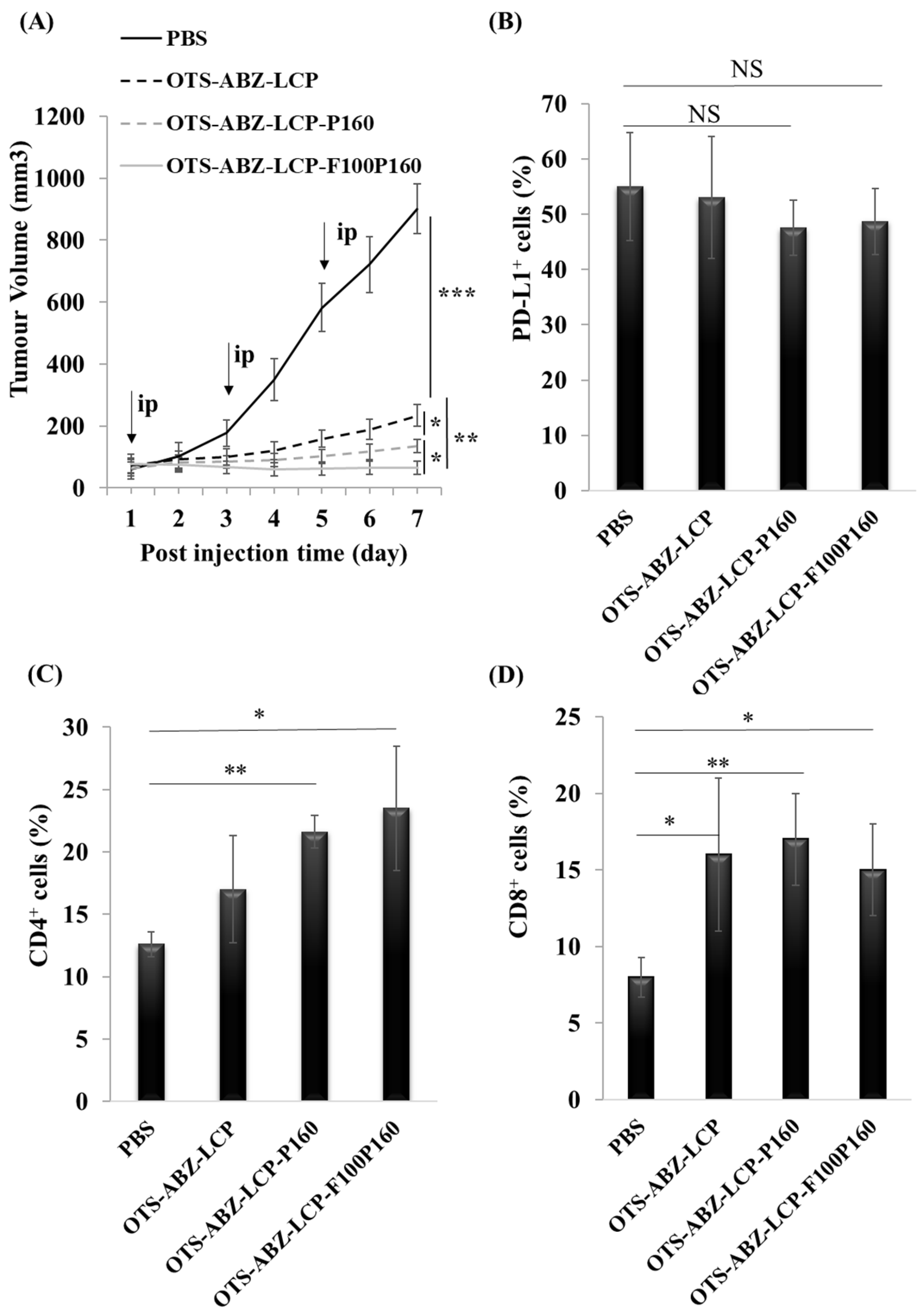
3.6. Dual Targeting Completely Inhibits Skin Tumour Growth
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 4 February 2020).
- Carlotto, A.; Hogsett, V.L.; Maiorini, E.M.; Razulis, J.G.; Sonis, S.T. The Economic Burden of Toxicities Associated with Cancer Treatment: Review of the Literature and Analysis of Nausea and Vomiting, Diarrhoea, Oral Mucositis and Fatigue. Pharmacoeconomics 2013, 31, 753–766. [Google Scholar] [CrossRef]
- Zhang, Y.; Bush, X.; Yan, B.; Chen, J.A. Gemcitabine nanoparticles promote antitumor immunity against melanoma. Biomaterials 2019, 189, 48–59. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.-C.; Tseng, Y.-C.; Mozumdar, S.; Huang, L. Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J. Control. Release 2010, 142, 416–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habraken, W.; Habibovic, P.; Epple, M.; Bohner, M. Calcium phosphates in biomedical applications: Materials for the future? Mater. Today 2016, 19, 69–87. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- Olton, D.; Li, J.; Wilson, M.E.; Rogers, T.; Close, J.; Huang, L.; Kumta, P.N.; Sfeir, C. Nanostructured calcium phosphates (NanoCaPs) for non-viral gene delivery: Influence of the synthesis parameters on transfection efficiency. Biomaterials 2007, 28, 1267–1279. [Google Scholar] [CrossRef]
- Wu, Y.; Gu, W.; Xu, Z.P. Enhanced combination cancer therapy using lipid-calcium carbonate/phosphate nanoparticles as a targeted delivery platform. Nanomedicine 2019, 14, 77–92. [Google Scholar] [CrossRef]
- Wu, Y.; Gu, W.; Tang, J.; Xu, Z.P. Devising new lipid-coated calcium phosphate/carbonate hybrid nanoparticles for controlled release in endosomes for efficient gene delivery. J. Mater. Chem. B 2017, 5, 7194–7203. [Google Scholar] [CrossRef]
- Movahedi, F.; Wu, Y.; Gu, W.; Xu, Z.P. Nanostructuring a widely used anti-worm drug into the lipid-coated calcium phosphate matrix for enhanced skin tumour treatment. ACS Appl. Bio Mater. 2020, 3, 4230–4238. [Google Scholar] [CrossRef]
- Chu, S.W.L.; Badar, S.; Morris, D.L.; Pourgholami, M.H. Potent inhibition of tubulin polymerisation and proliferation of paclitaxel-resistant 1A9PTX22 human ovarian cancer cells by albendazole. Anticancer Res. 2009, 29, 3791–3796. [Google Scholar]
- Sun, H.; Zhang, L.; Shi, C.; Hu, P.; Yan, W.; Wang, Z.; Duan, Q.; Lu, F.; Qin, L.; Lu, T.; et al. TOPK is highly expressed in circulating tumor cells, enabling metastasis of prostate cancer. Oncotarget 2015, 6, 12392–12404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, K.J.; Ashton, T.M.; Prevo, R.; Pirovano, G.; Higgins, G.S. T-LAK cell-originated protein kinase (TOPK): An emerging target for cancer-specific therapeutics. Cell Death Dis. 2018, 9, 1089. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Li, B.; Howard, C.B.; Mahler, S.M.; Thurecht, K.J.; Wu, Y.; Huang, L.; Xu, Z.P. Multifunctional lipid-coated calcium phosphate nanoplatforms for complete inhibition of large triple negative breast cancer via targeted combined therapy. Biomaterials 2019, 216, 119232. [Google Scholar] [CrossRef] [PubMed]
- Patrinely, J.R.; Dewan, A.K.; Johnson, D.B. The Role of Anti-PD-1/PD-L1 in the Treatment of Skin Cancer. BioDrugs 2020, 34, 495–503. [Google Scholar] [CrossRef]
- Zwicke, G.L.; Mansoori, G.A.; Jeffery, C.J. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012, 3, 18496. [Google Scholar] [CrossRef]
- Oh, S.A.; Wu, D.-C.; Cheung, J.; Navarro, A.; Xiong, H.; Cubas, R.; Totpal, K.; Chiu, H.; Wu, Y.; Comps-Agrar, L.; et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer 2020, 1, 681–691. [Google Scholar] [CrossRef]
- Passariello, M.; D’Alise, A.M.; Esposito, A.; Vetrei, C.; Froechlich, G.; Scarselli, E.; Nicosia, A.; De Lorenzo, C. Novel Human Anti-PD-L1 mAbs Inhibit Immune-Independent Tumor Cell Growth and PD-L1 Associated Intracellular Signalling. Sci. Rep. 2019, 9, 13125. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, F.; Liu, S.; Jiang, S. Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. J. Control. Release 2016, 244, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Fernández, M.; Javaid, F.; Chudasama, V. Advances in targeting the folate receptor in the treatment/imaging of cancers. Chem. Sci. 2018, 9, 790–810. [Google Scholar] [CrossRef] [Green Version]
- Raftery, L.J.; Grewal, Y.S.; Howard, C.B.; Jones, M.L.; Shiddiky, M.J.A.; Carrascosa, L.G.; Thurecht, K.J.; Mahler, S.M.; Trau, M. Biosensing made easy with PEG-targeted bi-specific antibodies. Chem. Commun. 2016, 52, 5730–5733. [Google Scholar] [CrossRef]
- Tang, J.; Li, L.; Howard, C.B.; Mahler, S.M.; Huang, L.; Xu, Z.P. Preparation of optimized lipid-coated calcium phosphate nanoparticles for enhanced in vitro gene delivery to breast cancer cells. J. Mater. Chem. B 2015, 3, 6805–6812. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-Y.; Chen, M.; Chen, L.-C.; Ho, Y.-S.; Ho, H.-O.; Lin, S.-Y.; Chuang, K.-H.; Sheu, M.-T. Bispecific antibodies (anti-mPEG/anti-HER2) for active tumor targeting of docetaxel (DTX)-loaded mPEGylated nanocarriers to enhance the chemotherapeutic efficacy of HER2-overexpressing tumors. Drug Deliv. 2018, 25, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Kalac, M.; Scotto, L.; Marchi, E.; Amengual, J.; Seshan, V.E.; Bhagat, G.; Ulahannan, N.; Leshchenko, V.V.; Temkin, A.M.; Parekh, S.; et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood 2011, 118, 5506–5516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Howard, C.B.; Mahler, S.M.; Thurecht, K.J.; Huang, L.; Xu, Z.P. Enhanced delivery of siRNA to triple negative breast cancer cells in vitro and in vivo through functionalizing lipid-coated calcium phosphate nanoparticles with dual target ligands. Nanoscale 2018, 10, 4258–4266. [Google Scholar] [CrossRef]
- Xu, S.; Cui, F.; Huang, D.; Zhang, D.; Zhu, A.; Sun, X.; Cao, Y.; Ding, S.; Wang, Y.; Gao, E.; et al. PD-L1 monoclonal antibody-conjugated nanoparticles enhance drug delivery level and chemotherapy efficacy in gastric cancer cells. Int. J. Nanomed. 2018, 14, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Xiao, K.; Li, Y.; Luo, J.; Lee, J.S.; Xiao, W.; Gonik, A.M.; Agarwal, R.G.; Lam, K.S. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials 2011, 32, 3435–3446. [Google Scholar] [CrossRef] [Green Version]
- Castro, L.S.E.W.; Kviecinski, M.R.; Ourique, F.; Parisotto, E.B.; Grienevicius, V.M.A.S.; Correia, J.F.G.; Wilhelm Filho, D.; Pedrosa, R.C. Albendazole as a promising molecule for tumor control. Redox Biol. 2016, 10, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Sugimori, M.; Hayakawa, Y.; Tamura, R.; Kuroda, S. The combined efficacy of OTS964 and temozolomide for reducing the size of power-law coded heterogeneous glioma stem cell populations. Oncotarget 2019, 10, 2397–2415. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, Y.; Park, J.-H.; Miyamoto, T.; Yamamoto, S.; Hisada, S.; Alachkar, H.; Nakamura, Y. TOPK inhibitor induces complete tumor regression in xenograft models of human cancer through inhibition of cytokinesis. Sci. Transl. Med. 2014, 6, 259ra145. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Yan Cai, Z.; Lu, Y.; Wang, L.; Morris, D.L. Albendazole: A potent inhibitor of vascular endothelial growth factor and malignant ascites formation in OVCAR-3 tumor-bearing nude mice. Clin. Cancer Res. 2006, 12, 1928–1935. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Thapa, R.K.; Yong, C.S.; Kim, J.O. Nanoparticle-based combination drug delivery systems for synergistic cancer treatment. J. Pharm. Investig. 2016, 46, 325–339. [Google Scholar] [CrossRef]
- Li, T.; Ito, A.; Pengcuo, R.; Sako, Y.; Chen, X.; Qiu, D.; Xiao, N.; Craig, P.S. Post-treatment follow-up study of abdominal cystic echinococcosis in Tibetan communities of Northwest Sichuan province, China. PLoS Negl. Trop. Dis. 2011, 5, e1364. [Google Scholar] [CrossRef] [PubMed]
- Altinoǧlu, E.I.; Adair, J.H. Calcium phosphate nanocomposite particles: A safer and more effective alternative to conventional chemotherapy? Futur. Oncol. 2009, 5, 279–281. [Google Scholar] [CrossRef]
- Locatelli, C.; Pedrosa, R.C.; De Bem, A.F.; Creczynski-Pasa, T.B.; Cordova, C.A.S.; Wilhelm-Filho, D. A comparative study of albendazole and mebendazole-induced, time-dependent oxidative stress. Redox Rep. 2004, 9, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Ye, N.; Dai, D.; Chen, Y.; Li, C.; Sun, Y. The protective role of the TOPK/PBK pathway in myocardial ischemia/reperfusion and H2O2-induced injury in H9C2 cardiomyocytes. Int. J. Mol. Sci. 2016, 17, 267. [Google Scholar] [CrossRef] [Green Version]
- Vega, L.R.; Solomon, F. Microtubule function in morphological differentiation: Growth zones and growth cones. Cell 1997, 89, 825–828. [Google Scholar] [CrossRef] [Green Version]
- Eccles, S.A.; Box, C.; Court, W. Cell migration/invasion assays and their application in cancer drug discovery. Biotechnol. Annu. Rev. 2005, 11, 391–421. [Google Scholar]
- Ghasemi, F.; Black, M.; Vizeacoumar, F.; Pinto, N.; Ruicci, K.M.; Le, C.C.S.H.; Lowerison, M.R.; Leong, H.S.; Yoo, J.; Fung, K.; et al. Repurposing Albendazole: New potential as a chemotherapeutic agent with preferential activity against HPV-negative head and neck squamous cell cancer. Oncotarget 2017, 8, 71512–71519. [Google Scholar] [CrossRef] [Green Version]
- Seol, M.A.; Park, J.H.; Jeong, J.H.; Lyu, J.; Han, S.Y.; Oh, S.M. Role of TOPK in lipopolysaccharide-induced breast cancer cell migration and invasion. Oncotarget 2017, 8, 40190–40203. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Liang, J.; Wu, A.; Chen, Y.; Zhao, P.; Lin, T.; Zhang, M.; Xu, Q.; Wang, J.; Huang, Y. Co-Delivery of Trichosanthin and Albendazole by Nano-Self-Assembly for Overcoming Tumor Multidrug-Resistance and Metastasis. ACS Appl. Mater. Interfaces 2017, 9, 26648–26664. [Google Scholar] [CrossRef] [PubMed]
- Saifar, M.G.P.; Williams, L.D.; Sobczyk, M.A.; Michaels, S.J.; Sherman, M.R. Selectivity of binding of PEGs and PEG-like oligomers to anti-PEG antibodies induced by methoxyPEG-proteins. Mol. Immunol. 2014, 57, 236–346. [Google Scholar] [CrossRef] [PubMed]
- Bellone, M.; Calcinotto, A.; Filipazzi, P.; De Milito, A.; Fais, S.; Rivoltini, L. The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors. Oncoimmunology 2013, 2, e22058. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Movahedi, F.; Liu, J.; Sun, B.; Cao, P.; Sun, L.; Howard, C.; Gu, W.; Xu, Z.P. PD-L1-Targeted Co-Delivery of Two Chemotherapeutics for Efficient Suppression of Skin Cancer Growth. Pharmaceutics 2022, 14, 1488. https://doi.org/10.3390/pharmaceutics14071488
Movahedi F, Liu J, Sun B, Cao P, Sun L, Howard C, Gu W, Xu ZP. PD-L1-Targeted Co-Delivery of Two Chemotherapeutics for Efficient Suppression of Skin Cancer Growth. Pharmaceutics. 2022; 14(7):1488. https://doi.org/10.3390/pharmaceutics14071488
Chicago/Turabian StyleMovahedi, Fatemeh, Jie Liu, Bing Sun, Pei Cao, Luyao Sun, Christopher Howard, Wenyi Gu, and Zhi Ping Xu. 2022. "PD-L1-Targeted Co-Delivery of Two Chemotherapeutics for Efficient Suppression of Skin Cancer Growth" Pharmaceutics 14, no. 7: 1488. https://doi.org/10.3390/pharmaceutics14071488