
The Multistage Antimalarial Compound Calxinin Perturbates P. falciparum Ca2+ Homeostasis by Targeting a Unique Ion Channel

 ,
,  ,
,  , , , , , ,
, , , , , ,  and
and 
Abstract
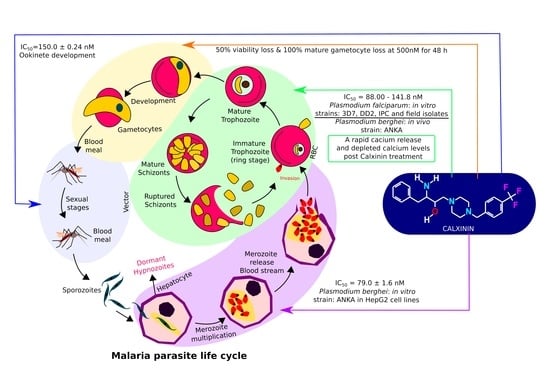
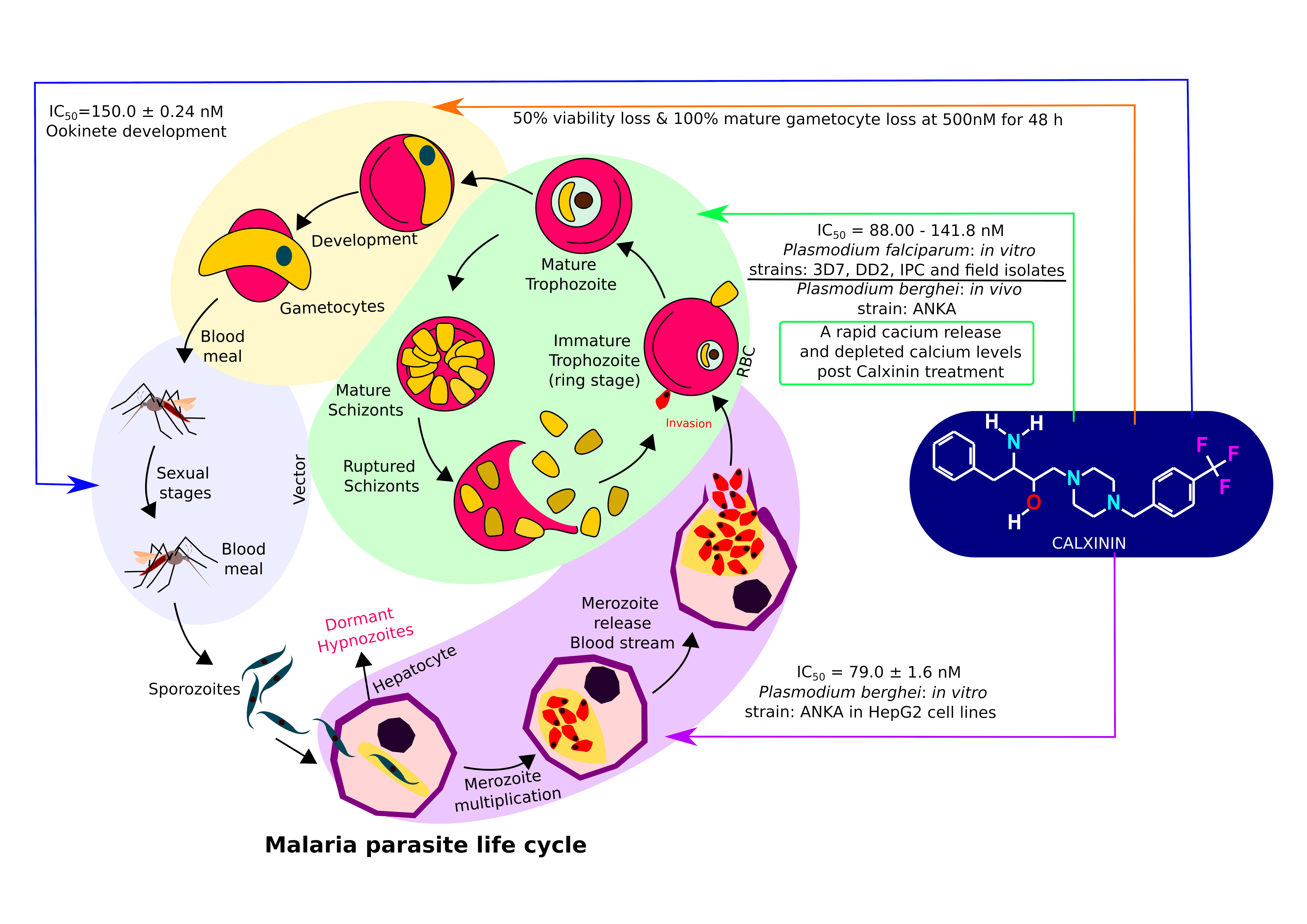
:
1. Introduction
2. Methods
2.1. Synthesis and Purification of Calxinin
2.1.2. Tert-butyl-(3-hydroxy-1-phenyl-4-(4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)butan-2-yl)carbamate (2)
2.1.3. 3-Amino-4-phenyl-1-(4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)butan-2-ol (Calxinin)
2.2. Maintenance of Reference Pf Parasites and Testing
2.2.1. Pf Reference Strains
2.2.2. Preparation of Drug Dilutions and Test Plates
2.2.3. In Vitro Antiparasitic Sensitivity Testing
2.2.4. Clinical Train Testing
Clinical Samples and Study Participants
Antiplasmodial Activity Testing in Pf Field Strain
2.2.5. Gametocyte Production
2.2.6. In Vitro Gametocyte Growth Inhibition Assay
2.2.7. Calxinin Resistant Mutant Selection
2.3. Testing the Efficacy of Calxinin in Pb Mouse/Mosquito Malaria Models
2.3.1. In Vitro Model
2.3.2. In Vivo Models
Erythrocytic Stage
Liver Stage Infection in Mice, Calxinin Treatment, and Survival Assay
2.3.3. Ex Vivo Models
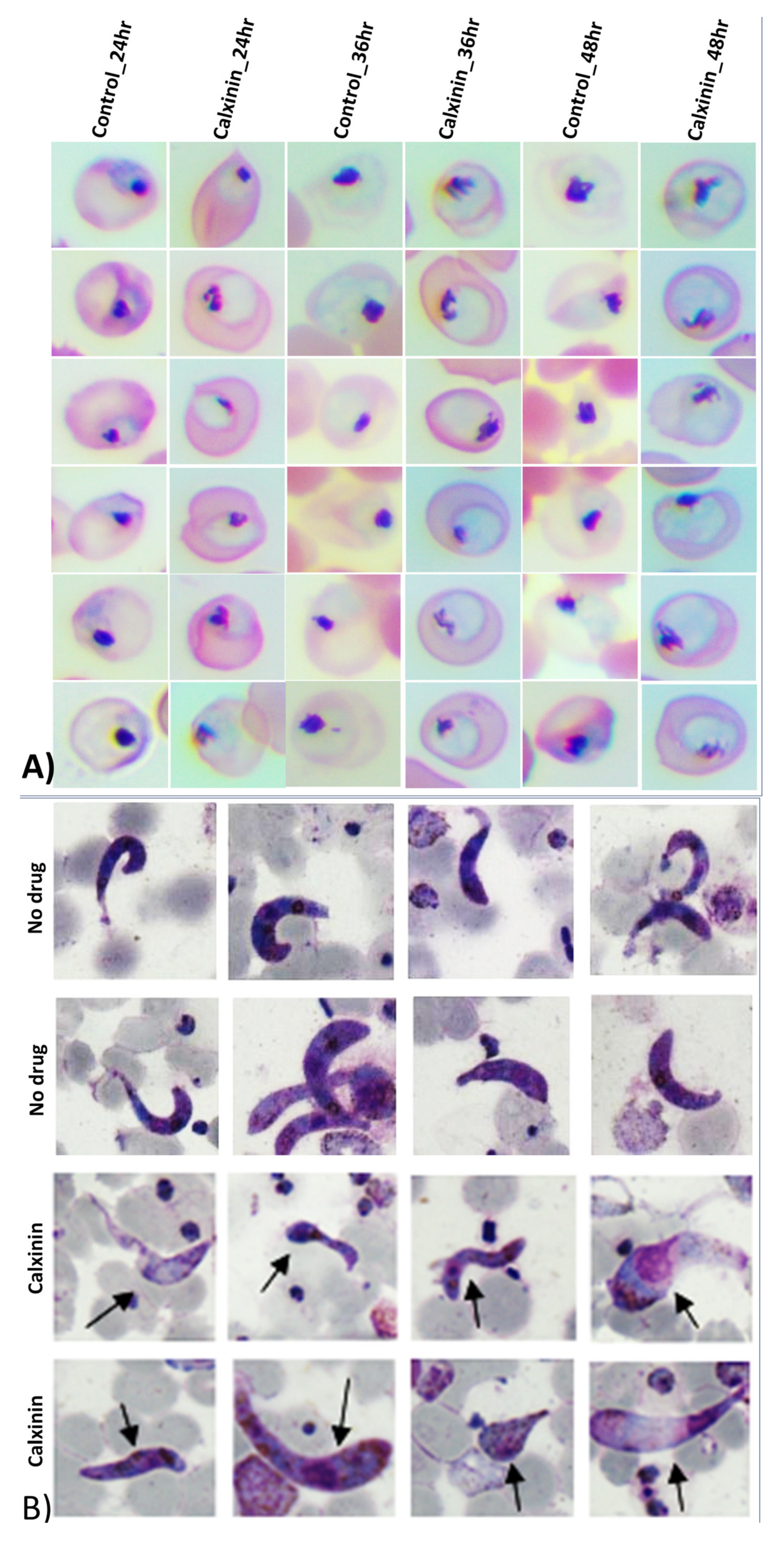
The Effect of Calxinin in an In Vitro Ookinete Inhibition Assay
2.4. Human Host Cross-Reactivity
2.4.1. Calxinin Cytotoxicity Testing in Human Cells
2.4.2. Patch-Clamp (T-Type)
2.4.3. Evaluation of the Hemostatic Effects of Calxinin
2.5. Calxinin Target Prediction In Silico
2.5.1. Target Search
2.5.2. Phylogenetic Analysis of the Putative Calxinin Target
2.5.3. In Silico Validations of the Putative Calxinin Target
2.6. Target Confirmatory Assays
2.6.1. Confocal Live Imaging
2.6.2. Super-Resolution Live Imaging
2.6.3. Electron Microscopic Evaluation of Calxinin’s Effect on the Subcellular Localization of Pf Ca2+
2.7. Data Analyses
2.8. Image Analysis
2.9. Statistical Analysis
3. Results
3.1. Rational Antimalarial Design and Synthesis
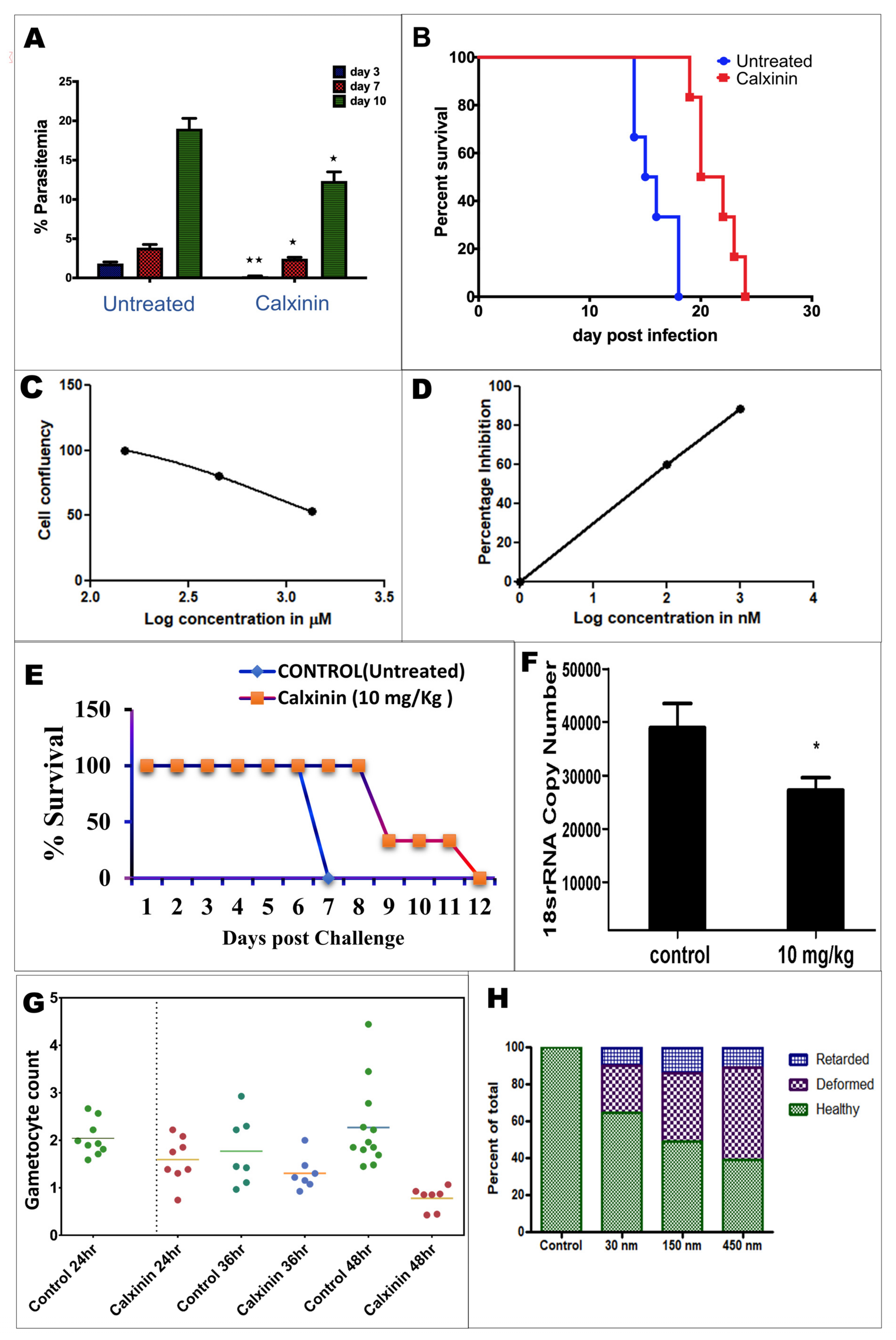
3.2. Plasmodium Falciparum Antiparasitic Activity Breakpoints
3.2.1. Blood Stage Antimalarial Activity of Calxinin in the Reference Strain
3.2.2. Effect of Calxinin on the ART-Resistant Field Strain IPC 4912
3.2.3. Calxinin Activity in Pf Field Isolates from a Malaria-Endemic Region
3.2.4. Activity of Calxinin against Gametocytes
3.3. Calxinin Antiparasitic Activity in the Plasmodium berghei Mouse Model Breakpoints
3.3.1. Efficacy of Single-Dose Calxinin on In Vivo Blood-Stage P. berghei in Mice
3.3.2. Activity of Calxinin against Parasite Liver Stages In Vitro and In Vivo
3.3.3. Plasmodium berghei Ex Vivo Model Antiparasitic Activity Breakpoints; In Vitro Ookinete Inhibition Assay for Calxinin
3.3.4. Resistant Variant Selection
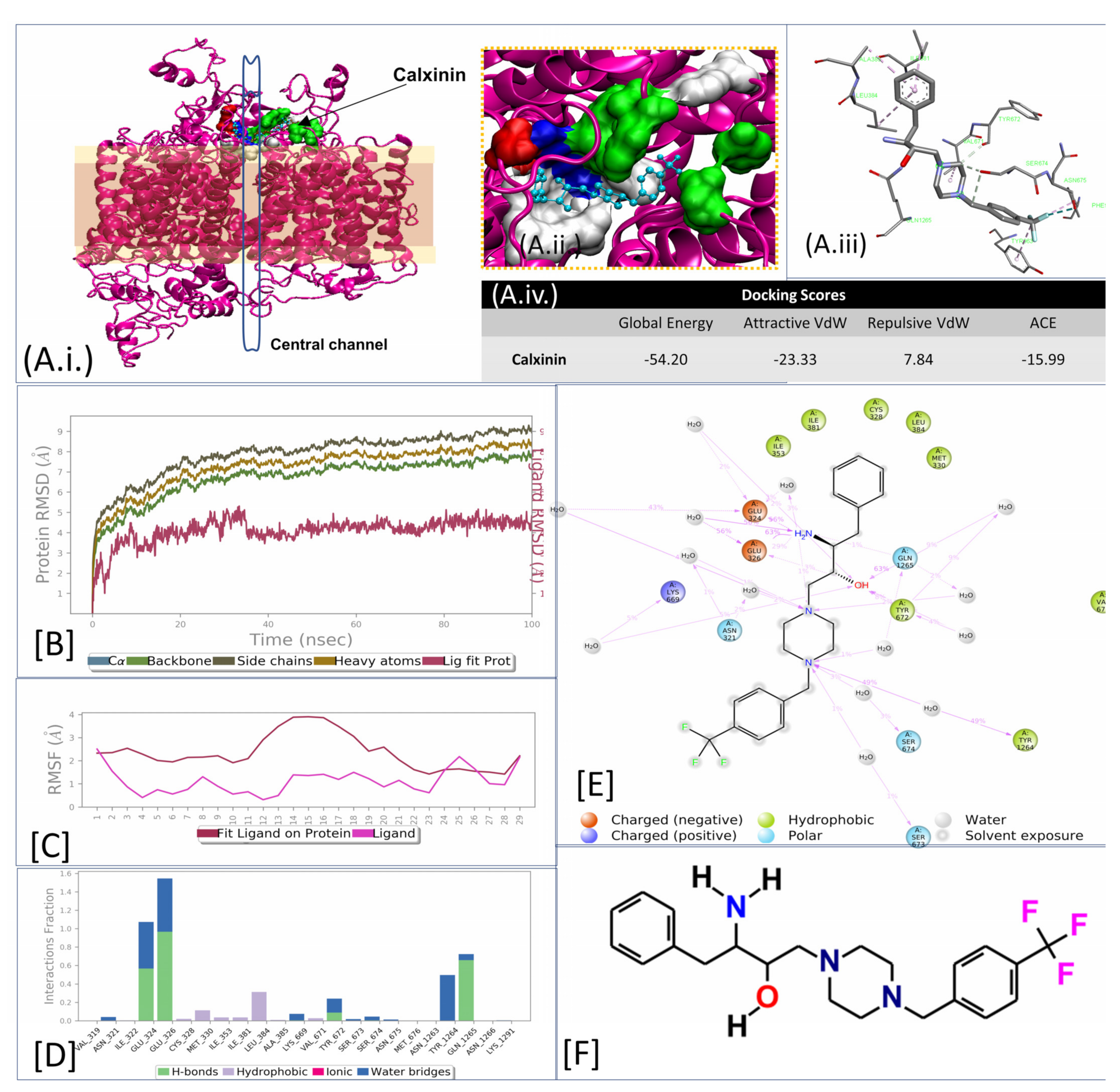
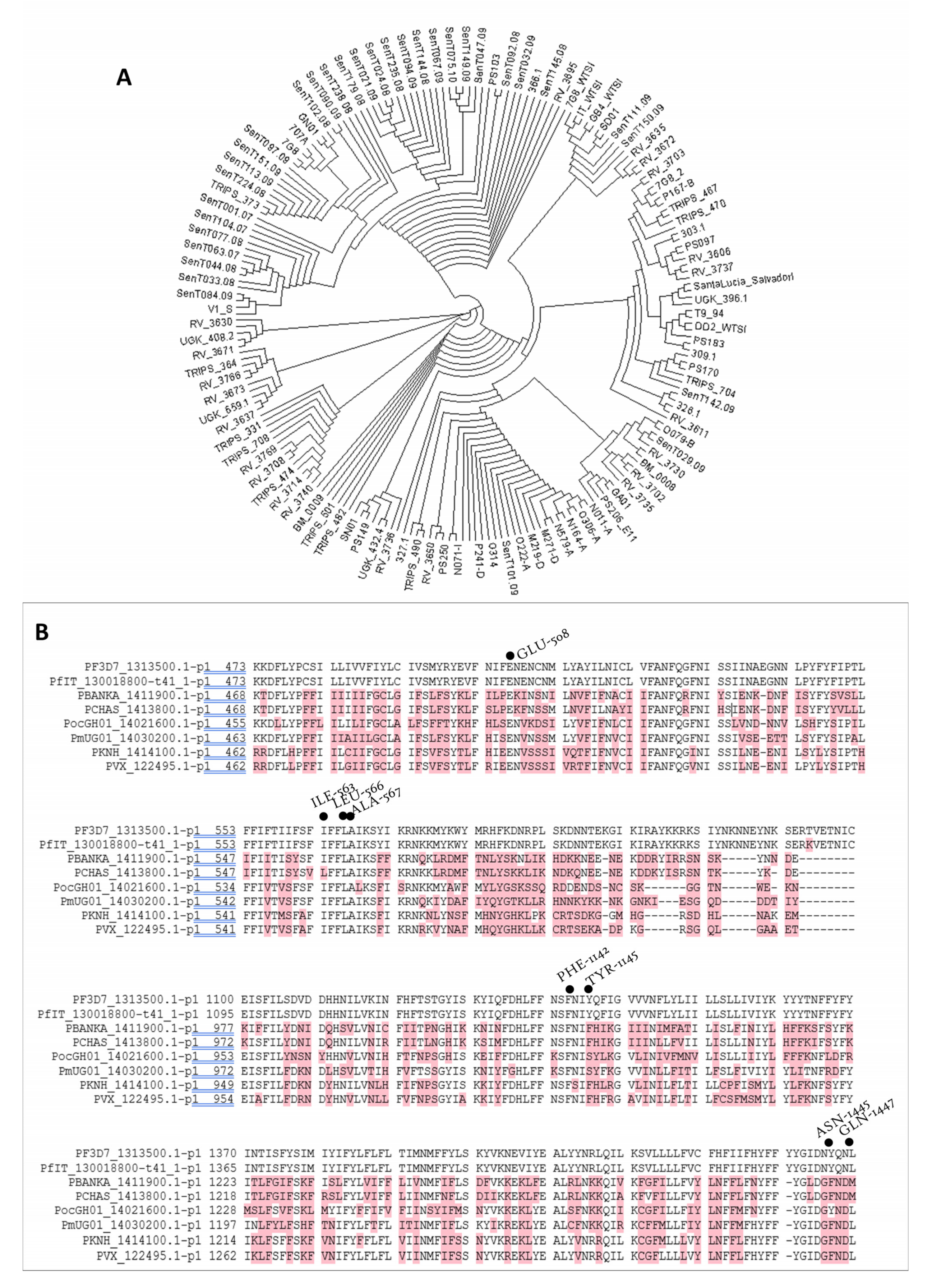
3.3.5. In Silico Target Elucidation
3.3.6. PF3D7_1313500 Genetic Polymorphism
3.3.7. Target Validations through the In Silico Approach
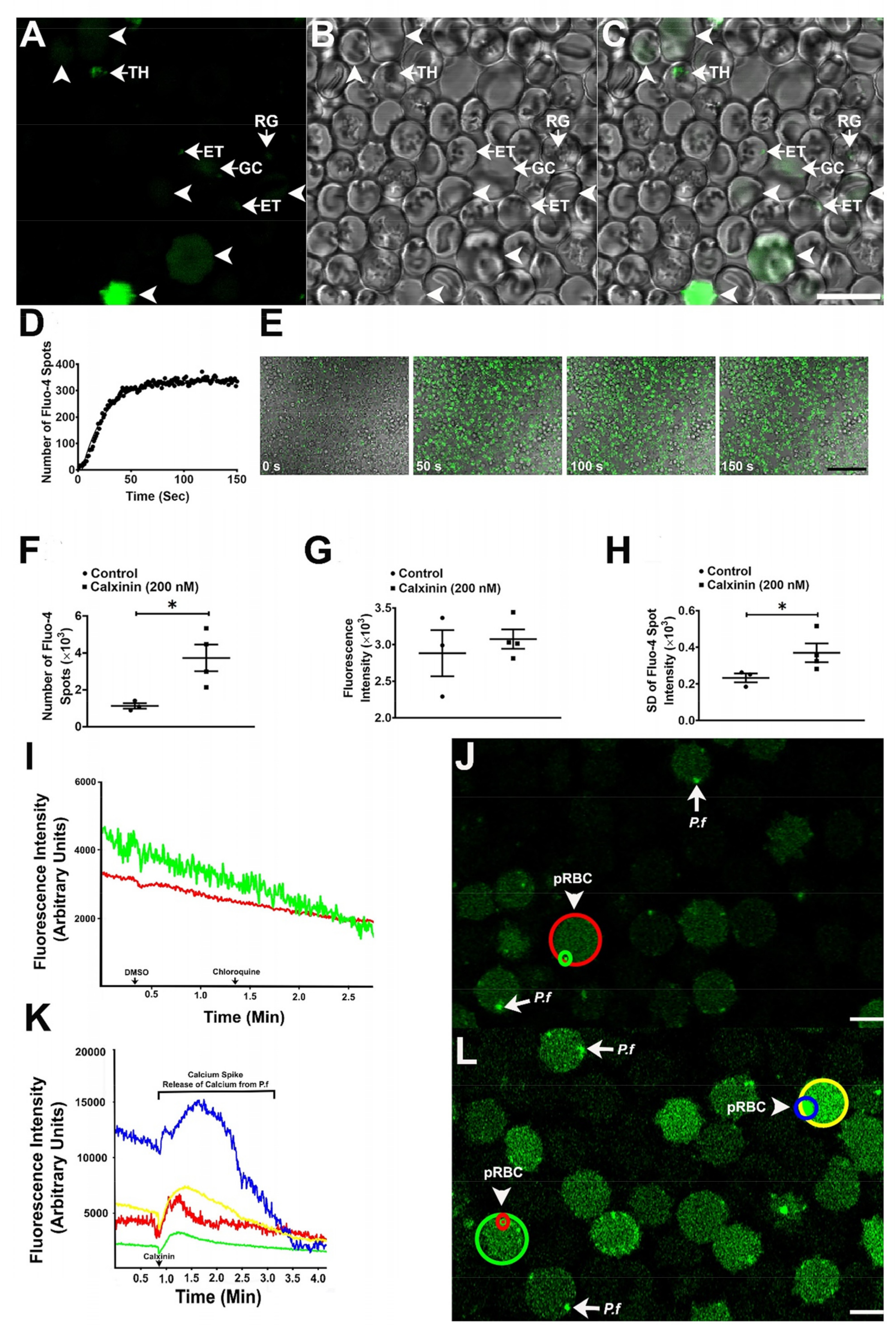
3.3.8. Calxinin’s Effect on Pf Intracellular Ca2+ Levels
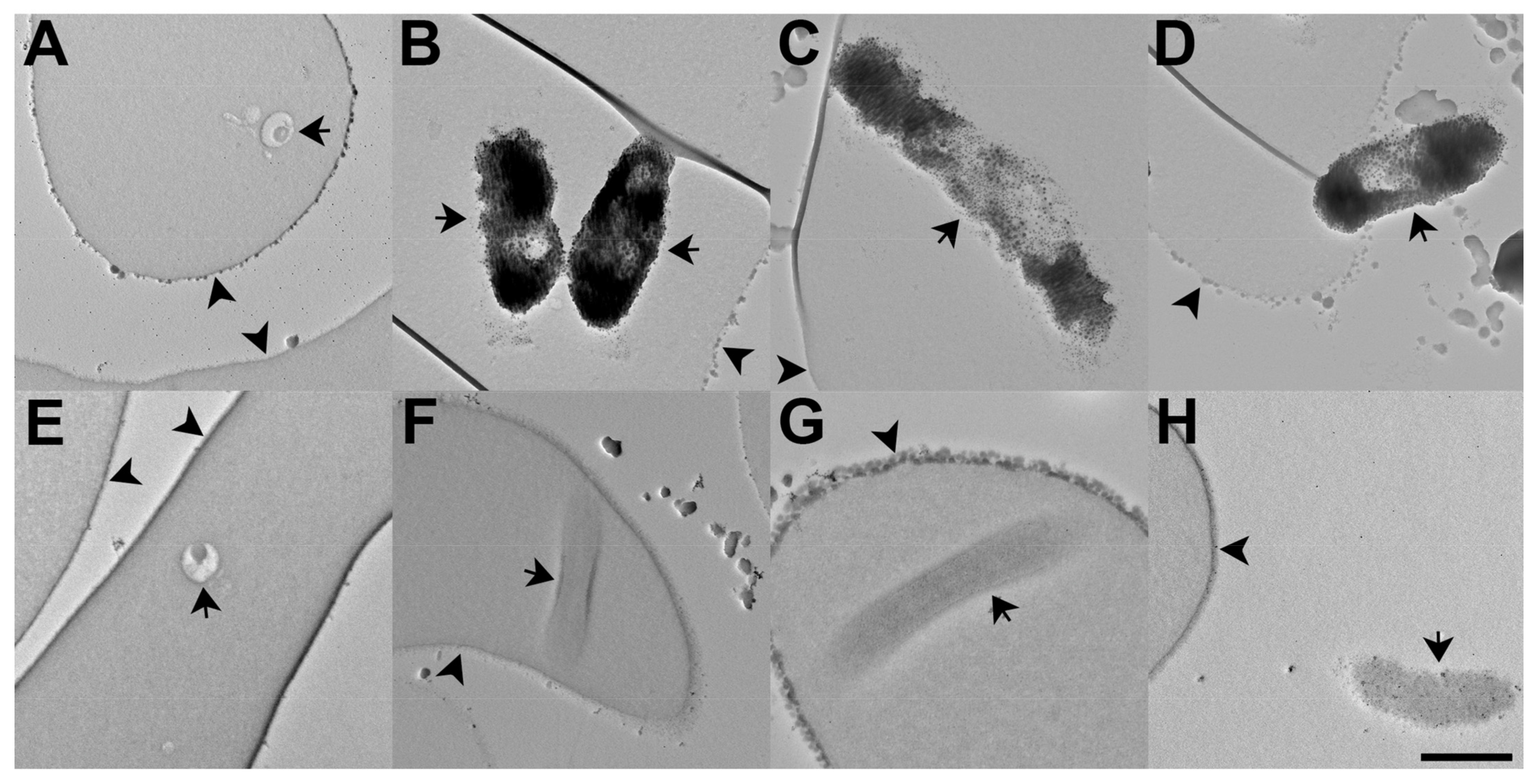
3.3.9. Determination of Intracellular Pf Ca2+ Levels through DAB Photoconversion TEM
3.3.10. Measuring Ca2+ Levels through Fluo4-AM Staining and Super-Resolution Live Microscopy
3.4. Host Toxicity and Host Calcium Homeostasis Interference
3.4.1. Cytotoxicity Effects of Calxinin on Human Primary and Cell Lines
3.4.2. Effect of Calxinin on Human Calcium Channel
3.4.3. Effects of Calxinin on Blood Hemostatic Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- WHO. World Malaria Report 2020: 20 Years of Global Progress and Challenges; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- WHO. Malaria Eradication: Benefits, Future Scenarios and Feasibility; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Achieng, A.O.; Rawat, M.; Ogutu, B.; Guyah, B.; Ong’echa, J.M.; Perkins, D.J.; Kempaiah, P. Antimalarials: Molecular Drug Targets and Mechanism of Action. Curr. Top. Med. Chem. 2017, 17, 2114–2128. [Google Scholar] [CrossRef] [PubMed]
- Bwire, G.M.; Ngasala, B.; Mikomangwa, W.P.; Kilonzi, M.; Kamuhabwa, A.A. Detection of Mutations Associated with Artemisinin Resistance at K13-Propeller Gene and a near Complete Return of Chloroquine Susceptible Falciparum Malaria in Southeast of Tanzania. Sci. Rep. 2020, 10, 3500. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Saha, B.; Hati, A.K.; Roy, S. Evidence of Artemisinin-Resistant Plasmodium Falciparum Malaria in Eastern India. N. Engl. J. Med. 2018, 379, 1962–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashley, E.A.; Phyo, A.P. Drugs in Development for Malaria. Drugs 2018, 78, 861–879. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, F.A.; Boonhok, R.; Cabrera, M.; Mbenda, H.G.N.; Wang, M.; Min, H.; Liang, X.; Qin, J.; Zhu, X.; Miao, J.; et al. Role of Plasmodium Falciparum Kelch 13 Protein Mutations in P. Falciparum Populations from Northeastern Myanmar in Mediating Artemisinin Resistance. mBio 2020, 11, e01134-19. [Google Scholar] [CrossRef] [Green Version]
- Witmer, K.; Dahalan, F.A.; Delves, M.J.; Yahiya, S.; Watson, O.J.; Straschil, U.; Chiwcharoen, D.; Sornboon, B.; Pukrittayakamee, S.; Pearson, R.D.; et al. Artemisinin-Resistant Malaria Parasites Show Enhanced Transmission to Mosquitoes under Drug Pressure. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Uwimana, A.; Legrand, E.; Stokes, B.H.; Ndikumana, J.-L.M.; Warsame, M.; Umulisa, N.; Ngamije, D.; Munyaneza, T.; Mazarati, J.-B.; Munguti, K.; et al. Emergence and Clonal Expansion of in Vitro Artemisinin-Resistant Plasmodium Falciparum Kelch13 R561H Mutant Parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. [Google Scholar] [CrossRef]
- Bihan, A.L.; Kanter, R.D.; Angulo-Barturen, I.; Binkert, C.; Boss, C.; Brun, R.; Brunner, R.; Buchmann, S.; Burrows, J.; Dechering, K.J.; et al. Characterization of Novel Antimalarial Compound ACT-451840: Preclinical Assessment of Activity and Dose–Efficacy Modeling. PLoS Med. 2016, 13, e1002138. [Google Scholar] [CrossRef] [Green Version]
- Schuerman, L. RTS, S Malaria Vaccine Could Provide Major Public Health Benefits. Lancet 2019, 394, 735–736. [Google Scholar] [CrossRef]
- Gupta, Y.; Gupta, N.; Singh, S.; Wu, L.; Chhikara, B.S.; Rawat, M.; Rathi, B. Multistage Inhibitors of the Malaria Parasite: Emerging Hope for Chemoprotection and Malaria Eradication. Med. Res. Rev. 2018, 38, 1511–1535. [Google Scholar] [CrossRef]
- Singh, A.K.; Rajendran, V.; Singh, S.; Kumar, P.; Kumar, Y.; Singh, A.; Miller, W.; Potemkin, V.; Poonam; Grishina, M.; et al. Antiplasmodial Activity of Hydroxyethylamine Analogs: Synthesis, Biological Activity and Structure Activity Relationship of Plasmepsin Inhibitors. Bioorg. Med. Chem. 2018, 26, 3837–3844. [Google Scholar] [CrossRef]
- Singh, A.K.; Rathore, S.; Tang, Y.; Goldfarb, N.E.; Dunn, B.M.; Rajendran, V.; Ghosh, P.C.; Singh, N.; Latha, N.; Singh, B.K.; et al. Hydroxyethylamine Based Phthalimides as New Class of Plasmepsin Hits: Design, Synthesis and Antimalarial Evaluation. PLoS ONE 2015, 10, e0139347. [Google Scholar] [CrossRef] [Green Version]
- Raynaud, F.I. Drug Development. In Metabolomics for Biomedical Research; Elsevier: Amsterdam, The Netherlands, 2020; pp. 159–199. [Google Scholar]
- Kgokong, J.L.; Smith, P.P.; Matsabisa, G.M. 1, 2, 4-Triazino-[5,6b] Indole Derivatives: Effects of the Trifluoromethyl Group on in Vitro Antimalarial Activity. Bioorganic Med. Chem. 2005, 13, 2935–2942. [Google Scholar] [CrossRef]
- Magueur, G.; Crousse, B.; Charneau, S.; Grellier, P.; Bégué, J.-P.; Bonnet-Delpon, D. Fluoroartemisinin: Trifluoromethyl Analogues of Artemether and Artesunate. J. Med. Chem. 2004, 47, 2694–2699. [Google Scholar] [CrossRef]
- Molestina, R.E. BEI Resources: A Biological Resource Center for Parasitologists. Trends Parasitol. 2010, 26, 559. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Rajendran, V.; He, J.; Singh, A.K.; Achieng, A.O.; Kumari, V.; Pant, A.; Nasamu, A.S.; Pandit, M.; Singh, J.; et al. Fast-Acting Small Molecules Targeting Malarial Aspartyl Proteases, Plasmepsins, Inhibit Malaria Infection at Multiple Life Stages. ACS Infect. Dis. 2018, 5, 184–198. [Google Scholar] [CrossRef]
- Kumar, P.; Achieng, A.O.; Rajendran, V.; Ghosh, P.C.; Singh, B.K.; Rawat, M.; Perkins, D.J.; Kempaiah, P.; Rathi, B. Synergistic Blending of High-Valued Heterocycles Inhibits Growth of Plasmodium Falciparum in Culture and P. berghei Infection in Mouse Model. Sci. Rep. 2017, 7, 6724. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium Falciparum Erythrocytic Stages in Culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef]
- Dery, V.; Duah, N.O.; Ayanful-Torgby, R.; Matrevi, S.A.; Anto, F.; Quashie, N.B. An Improved SYBR Green-1-Based Fluorescence Method for the Routine Monitoring of Plasmodium Falciparum Resistance to Anti-Malarial Drugs. Malar. J. 2015, 14, 481. [Google Scholar] [CrossRef] [Green Version]
- Cheruiyot, A.C.; Auschwitz, J.M.; Lee, P.J.; Yeda, R.A.; Okello, C.O.; Leed, S.E.; Talwar, M.; Murthy, T.; Gaona, H.W.; Hickman, M.R.; et al. Assessment of the Worldwide Antimalarial Resistance Network Standardized Procedure for in Vitro Malaria Drug Sensitivity Testing Using SYBR Green Assay for Field Samples with Various Initial Parasitemia Levels. Antimicrob. Agents Chemother. 2016, 60, 2417–2424. [Google Scholar] [CrossRef] [Green Version]
- Akala, H.M.; Eyase, F.L.; Cheruiyot, A.C.; Omondi, A.A.; Ogutu, B.R.; Waters, N.C.; Johnson, J.D.; Polhemus, M.E.; Schnabel, D.C.; Walsh, D.S. Antimalarial Drug Sensitivity Profile of Western Kenya Plasmodium Falciparum Field Isolates Determined by a SYBR Green I in Vitro Assay and Molecular Analysis. Am. J. Trop. Med. Hyg. 2011, 85, 34–41. [Google Scholar] [CrossRef]
- Smilkstein, M.; Sriwilaijaroen, N.; Kelly, J.X.; Wilairat, P.; Riscoe, M. Simple and Inexpensive Fluorescence-Based Technique for High-Throughput Antimalarial Drug Screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.D.; Dennull, R.A.; Gerena, L.; Lopez-Sanchez, M.; Roncal, N.E.; Waters, N.C. Assessment and Continued Validation of the Malaria SYBR Green I-Based Fluorescence Assay for Use in Malaria Drug Screening. Antimicrob. Agents Chemother. 2007, 51, 1926–1933. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Gupta, Y.; Bansal, M.; Singh, S.; Pathak, P.; Shahbaaz, M.; Mathur, R.; Singh, J.; Kashif, M.; Grishina, M.; et al. Multistage Antiplasmodial Activity of Hydroxyethylamine Compounds, in Vitro and in Vivo Evaluations. RSC Adv. 2020, 10, 35516–35530. [Google Scholar] [CrossRef]
- Njokah, M.J.; Kang’ethe, J.N.; Kinyua, J.; Kariuki, D.; Kimani, F.T. In Vitro Selection of Plasmodium Falciparum Pfcrt and Pfmdr1 Variants by Artemisinin. Malar. J. 2016, 15, 381. [Google Scholar] [CrossRef] [Green Version]
- Oduola, A.M.; Milhous, W.K.; Weatherly, N.F.; Bowdre, J.H.; Desjardins, R.E. Plasmodium Falciparum: Induction of Resistance to Mefloquine in Cloned Strains by Continuous Drug Exposure in Vitro. Exp. Parasitol. 1988, 67, 354–360. [Google Scholar] [CrossRef]
- Ma, C.; Harrison, P.; Wang, L.; Coppel, R.L. Automated Estimation of Parasitaemia of Plasmodium Yoelii-Infected Mice by Digital Image Analysis of Giemsa-Stained Thin Blood Smears. Malar. J. 2010, 9, 348. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the AlamarBlue Assay. Cold Spring Harb. Protoc. 2018, 2018, pdb-prot095489. [Google Scholar] [CrossRef]
- Tringham, E.; Powell, K.L.; Cain, S.M.; Kuplast, K.; Mezeyova, J.; Weerapura, M.; Eduljee, C.; Jiang, X.; Smith, P.; Morrison, J.-L.; et al. T-Type Calcium Channel Blockers That Attenuate Thalamic Burst Firing and Suppress Absence Seizures. Sci. Transl. Med. 2012, 4, 121ra19. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Daud, A.N.; Cribbs, L.L.; Lacerda, A.E.; Pereverzev, A.; Klöckner, U.; Schneider, T.; Perez-Reyes, E. Cloning and Expression of a Novel Member of the Low Voltage-Activated T-Type Calcium Channel Family. J. Neurosci. 1999, 19, 1912–1921. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awale, M.; Reymond, J.L. The Polypharmacology Browser: A Web-Based Multi-Fingerprint Target Prediction Tool Using ChEMBL Bioactivity Data. J. Cheminformatics 2017, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awale, M.; Reymond, J.-L. Web-Based Tools for Polypharmacology Prediction. In Systems Chemical Biology; Springer: New York, NY, USA, 2019; pp. 255–272. [Google Scholar]
- Lee, A.; Kim, D. CRDS: Consensus Reverse Docking System for Target Fishing. Bioinformatics 2020, 36, 959–960. [Google Scholar] [CrossRef] [PubMed]
- Gotsbacher, M.P.; Cho, S.M.; Kim, N.H.; Liu, F.; Kwon, H.J.; Karuso, P. Reverse Chemical Proteomics Identifies an Unanticipated Human Target of the Antimalarial Artesunate. ACS Chem. Biol. 2019, 14, 636–643. [Google Scholar] [CrossRef]
- Cheemadan, S.; Ramadoss, R.; Bozdech, Z. Role of Calcium Signaling in the Transcriptional Regulation of the Apicoplast Genome of Plasmodium Falciparum. BioMed Res. Int. 2014, 2014, 869401. [Google Scholar] [CrossRef] [Green Version]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O.S.; et al. PlasmoDB: A Functional Genomic Database for Malaria Parasites. Nucleic Acids Res. 2008, 37, D539–D543. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y. KEGG Mapper for Inferring Cellular Functions from Protein Sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Prole, D.L.; Taylor, C.W. Identification of Intracellular and Plasma Membrane Calcium Channel Homologues in Pathogenic Parasites. PLoS ONE 2011, 6, e26218. [Google Scholar] [CrossRef]
- Adams, J.H.; Fraser, M.J.; Balu, B.; Shoue, D.A. High Efficiency Transformation of Plasmodium Falciparum by the Lepidopteran Transposon, PiggyBac. U.S. Patent No. 7,932,088, 26 April 2011. [Google Scholar]
- Thomas, P.; Sedillo, J.; Oberstaller, J.; Li, S.; Zhang, M.; Singh, N.; Wang, C.C.; Udenze, K.; Jiang, R.H.; Adams, J.H. Phenotypic Screens Identify Parasite Genetic Factors Associated with Malarial Fever Response in Plasmodium Falciparum PiggyBac Mutants. Msphere 2016, 1, e00273-16. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2015, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, J.; Roy, A.; Zhang, Y. Automated Protein Structure Modeling in CASP9 by I-TASSER Pipeline Combined with QUARK-Based Ab Initio Folding and FG-MD-Based Structure Refinement. Proteins Struct. Funct. Bioinform. 2011, 79, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 2010, 5, 725. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.; Jones, P.; Mitchell, A.; Apweiler, R.; Attwood, T.K.; Bateman, A.; Bernard, T.; Binns, D.; Bork, P.; Burge, S.; et al. InterPro in 2011: New Developments in the Family and Domain Prediction Database. Nucleic Acids Res. 2012, 40, D306–D312. [Google Scholar] [CrossRef] [Green Version]
- Mulder, N.J.; Apweiler, R. The InterPro Database and Tools for Protein Domain Analysis. Curr. Protoc. Bioinform. 2003, 2, 2–7. [Google Scholar] [CrossRef]
- Roy, A.; Yang, J.; Zhang, Y. COFACTOR: An Accurate Comparative Algorithm for Structure-Based Protein Function Annotation. Nucleic Acids Res. 2012, 40, W471–W477. [Google Scholar] [CrossRef] [Green Version]
- Cunha, M.R.; Bhardwaj, R.; Carrel, A.L.; Lindinger, S.; Romanin, C.; Parise-Filho, R.; Hediger, M.A.; Reymond, J.-L. Natural Product Inspired Optimization of a Selective TRPV6 Calcium Channel Inhibitor. RSC Med. Chem. 2020, 11, 1032–1040. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega. Curr. Protoc. Bioinform. 2014, 48, 3–13. [Google Scholar] [CrossRef]
- Gupta, Y.; Goicoechea, S.; Pearce, C.M.; Mathur, R.; Romero, J.G.; Kwofie, S.K.; Weyenberg, M.C.; Daravath, B.; Sharma, N.; Akala, H.M.; et al. The Emerging Paradigm of Calcium Homeostasis as a New Therapeutic Target for Protozoan Parasites. Med. Res. Rev. 2021, 42, 56–82. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated Genome Annotation and Pathway Identification Using the KEGG Orthology (KO) as a Controlled Vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Warrenfeltz, S.; Basenko, E.Y.; Crouch, K.; Harb, O.S.; Kissinger, J.C.; Roos, D.S.; Shanmugasundram, A.; Silva-Franco, F. EuPathDB: The Eukaryotic Pathogen Genomics Database Resource. In Eukaryotic Genomic Databases; Springer: New York, NY, USA, 2018; pp. 69–113. [Google Scholar]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny. Fr: Robust Phylogenetic Analysis for the Non-Specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef]
- Albà, M. Making Alignments Prettier. Genome Biol. 2000, 1, reports2047. [Google Scholar] [CrossRef]
- Schrödinger, L. Desmond Molecular Dynamics System. In DE Shaw Research, New York, Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2020. [Google Scholar]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Zipprer, E.M.; Neggers, M.; Kushwaha, A.; Rayavara, K.; Desai, S.A. A Kinetic Fluorescence Assay Reveals Unusual Features of Ca++ Uptake in Plasmodium Falciparum-Infected Erythrocytes. Malar. J. 2014, 13, 184. [Google Scholar] [CrossRef] [Green Version]
- Poletto, V.; Galimberti, V.; Guerra, G.; Rosti, V.; Moccia, F.; Biggiogera, M. Fine Structural Detection of Calcium Ions by Photoconversion. Eur. J. Histochem. EJH 2016, 60, 2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, C.L.; El-Safi, S.H.; Wynn, T.A.; Satti, M.M.; Kordofani, A.M.; Hashim, F.A.; Hag-Ali, M.; Neva, F.A.; Nutman, T.B.; Sacks, D.L. In Vivo Cytokine Profiles in Patients with Kala-Azar. Marked Elevation of Both Interleukin-10 and Interferon-Gamma. J. Clin. Investig. 1993, 91, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Oduola, A.M.; Sowunmi, A.; Milhous, W.K.; Brewer, T.G.; Kyle, D.E.; Gerena, L.; Rossan, R.N.; Salako, L.A.; Schuster, B.G. In Vitro and in Vivo Reversal of Chloroquine Resistance in Plasmodium Falciparum with Promethazine. Am. J. Trop. Med. Hyg. 1998, 58, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, C.; Chaudhary, M.; Oliveira, R.N.D.; Borbas, A.; Kempaiah, P.; Singh, P.; Rathi, B. Fluorinated Scaffolds for Antimalarial Drug Discovery. Expert Opin. Drug Discov. 2020, 15, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Meninno, S.; Lattanzi, A. Organocatalytic Asymmetric Reactions of Epoxides: Recent Progress. Chem.-A Eur. J. 2016, 22, 3632–3642. [Google Scholar] [CrossRef]
- Yeda, R.; Ingasia, L.A.; Cheruiyot, A.C.; Okudo, C.; Chebon, L.J.; Cheruiyot, J.; Akala, H.M.; Kamau, E. The Genotypic and Phenotypic Stability of Plasmodium Falciparum Field Isolates in Continuous In Vitro Culture. PLoS ONE 2016, 11, e0143565. [Google Scholar] [CrossRef]
- Ingasia, L.A.; Cheruiyot, J.; Okoth, S.A.; Andagalu, B.; Kamau, E. Genetic Variability and Population Structure of Plasmodium Falciparum Parasite Populations from Different Malaria Ecological Regions of Kenya. Infect. Genet. Evol. 2016, 39, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Achieng, A.O.; Muiruri, P.; Ingasia, L.A.; Opot, B.H.; Juma, D.W.; Yeda, R.; Ngalah, B.S.; Ogutu, B.R.; Andagalu, B.; Akala, H.M.; et al. Temporal Trends in Prevalence of Plasmodium Falciparum Molecular Markers Selected for by Artemether–Lumefantrine Treatment in Pre-ACT and Post-ACT Parasites in Western Kenya. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Mbaisi, A.; Liyala, P.; Eyase, F.; Achilla, R.; Akala, H.; Wangui, J.; Mwangi, J.; Osuna, F.; Alam, U.; Smoak, B.L.; et al. Drug Susceptibility and Genetic Evaluation of Plasmodium Falciparum Isolates Obtained in Four Distinct Geographical Regions of Kenya. Antimicrob. Agents Chemother. 2004, 48, 3598–3601. [Google Scholar] [CrossRef] [Green Version]
- Savini, H.; Bogreau, H.; Bertaux, L.; Bouchiba, H.; Kraemer, P.; Parzy, D.; Garnotel, E.; Rogier, C.; Simon, F.; Pradines, B. First Case of Emergence of Atovaquone-Proguanil Resistance in Plasmodium Falciparum during Treatment in a Traveler in Comoros. Antimicrob. Agents Chemother. 2008, 52, 2283–2284. [Google Scholar] [CrossRef] [Green Version]
- Gee, K.R.; Brown, K.; Chen, W.U.; Bishop-Stewart, J.; Gray, D.; Johnson, I. Chemical and Physiological Characterization of Fluo-4 Ca2+-Indicator Dyes. Cell Calcium 2000, 27, 97–106. [Google Scholar] [CrossRef]
- Perkins, D.J.; Were, T.; Davenport, G.C.; Kempaiah, P.; Hittner, J.B.; Ong’echa, J.M. Severe Malarial Anemia: Innate Immunity and Pathogenesis. Int. J. Biol. Sci. 2011, 7, 1427–1442. [Google Scholar] [CrossRef] [Green Version]
- Kempaiah, P.; Anyona, S.B.; Raballah, E.; Davenport, G.C.; Were, T.; Hittner, J.B.; Ong’echa, J.M.; Perkins, D.J. Reduced Interferon (IFN)-Alpha Conditioned by IFNA2 (-173) and IFNA8 (-884) Haplotypes Is Associated with Enhanced Susceptibility to Severe Malarial Anemia and Longitudinal All-Cause Mortality. Hum. Genet. 2012, 131, 1375–1391. [Google Scholar] [CrossRef] [Green Version]
- Kempaiah, P.; Dokladny, K.; Karim, Z.; Raballah, E.; Ong’echa, J.M.; Moseley, P.L.; Perkins, D.J. Reduced Hsp70 and Glutamine in Pediatric Severe Malaria Anemia: Role of Hemozoin in Suppressing Hsp70 and NF-KappaB Activation. Mol. Med. 2016, 22, 570–584. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Mharakurwa, S.; Ndiaye, D.; Rathod, P.K.; Rosenthal, P.J. Antimalarial Drug Resistance: Literature Review and Activities and Findings of the ICEMR Network. Am. J. Trop. Med. Hyg. 2015, 93, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Kashif, M.; Singh, V.; Fontinha, D.; Mukherjee, B.; Kumar, D.; Singh, S.; Prudencio, M.; Singh, A.P.; Rathi, B. Novel Antiplasmodial Compounds Leveraged with Multistage Potency against the Parasite Plasmodium Falciparum: In Vitro and In Vivo Evaluations and Pharmacokinetic Studies. J. Med. Chem. 2021, 64, 8666–8683. [Google Scholar] [CrossRef]
- Recacha, R.; Leitans, J.; Akopjana, I.; Aprupe, L.; Trapencieris, P.; Jaudzems, K.; Jirgensons, A.; Tars, K. Structures of Plasmepsin II from Plasmodium Falciparum in Complex with Two Hydroxyethylamine-Based Inhibitors. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 1531–1539. [Google Scholar] [CrossRef] [Green Version]
- Souza, M.C.; Padua, T.A.; Torres, N.D.; Costa, M.F.D.S.; Facchinetti, V.; Gomes, C.R.B.; Souza, M.V.N.; Henriques, M.D.G. Study of the Antimalarial Properties of Hydroxyethylamine Derivatives Using Green Fluorescent Protein Transformed Plasmodium Berghei. Memórias Do Instituto Oswaldo Cruz 2015, 110, 560–565. [Google Scholar] [CrossRef]
- Cunico, W.; Ferreira, M.D.L.G.; Ferreira, T.G.; Penido, C.; Henriques, M.G.; Krettli, L.G.; Varotti, F.P.; Krettli, A.U. Synthesis and Antimalarial Activity of Novel Hydroxyethylamines, Potential Aspartyl Protease Inhibitors. Lett. Drug Des. Discov. 2008, 5, 178–181. [Google Scholar] [CrossRef]
- Singh, A.K.; Rajendran, V.; Pant, A.; Ghosh, P.C.; Singh, N.; Latha, N.; Garg, S.; Pandey, K.C.; Singh, B.K.; Rathi, B. Design, Synthesis and Biological Evaluation of Functionalized Phthalimides: A New Class of Antimalarials and Inhibitors of Falcipain-2, a Major Hemoglobinase of Malaria Parasite. Bioorganic Med. Chem. 2015, 23, 1817–1827. [Google Scholar] [CrossRef]
- Volloch, V.; Rits, S. Results of Beta Secretase-Inhibitor Clinical Trials Support Amyloid Precursor Protein-Independent Generation of Beta Amyloid in Sporadic Alzheimer’s Disease. Med. Sci. 2018, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Jin, B.; Lee, H.M.; Kim, S.K. Conformational Analysis of Genotoxic Benzo [a] Pyrene-7, 8-Dione-Duplex DNA Adducts Using a Molecular Dynamics Method. J. Biomol. Struct. Dyn. 2010, 27, 457–464. [Google Scholar] [CrossRef]
- Daydé-Cazals, B.; Fauvel, B.; Singer, M.; Feneyrolles, C.; Bestgen, B.; Gassiot, F.; Spenlinhauer, A.; Warnault, P.; Hijfte, N.V.; Borjini, N.; et al. Rational Design, Synthesis, and Biological Evaluation of 7-Azaindole Derivatives as Potent Focused Multi-Targeted Kinase Inhibitors. J. Med. Chem. 2016, 59, 3886–3905. [Google Scholar] [CrossRef]
- Bhagavathula, A.S.; Elnour, A.A.; Shehab, A. Alternatives to Currently Used Antimalarial Drugs: In Search of a Magic Bullet. Infect. Dis. Poverty 2016, 5, 103. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Starr, C.G.; Troendle, E.; Wiedman, G.; Wimley, W.C.; Ulmschneider, J.P.; Ulmschneider, M.B. Simulation-Guided Rational de Novo Design of a Small Pore-Forming Antimicrobial Peptide. J. Am. Chem. Soc. 2019, 141, 4839–4848. [Google Scholar] [CrossRef]
- Lin, X.; Huang, X.-P.; Chen, G.; Whaley, R.; Peng, S.; Wang, Y.; Zhang, G.; Wang, S.X.; Wang, S.; Roth, B.L.; et al. Life beyond Kinases: Structure-Based Discovery of Sorafenib as Nanomolar Antagonist of 5-HT Receptors. J. Med. Chem. 2012, 55, 5749–5759. [Google Scholar] [CrossRef] [Green Version]
- Chitrala, K.N.; Yang, X.; Busbee, B.; Singh, N.P.; Bonati, L.; Xing, Y.; Nagarkatti, P.; Nagarkatti, M. Computational Prediction and in Vitro Validation of VEGFR1 as a Novel Protein Target for 2, 3, 7, 8-Tetrachlorodibenzo-p-Dioxin. Sci. Rep. 2019, 9, 6810. [Google Scholar] [CrossRef]
- Huang, J.; Chen, F.; Zhong, Z.; Tan, H.Y.; Wang, N.; Liu, Y.; Fang, X.; Yang, T.; Feng, Y. Interpreting the Pharmacological Mechanisms of Huachansu Capsules on Hepatocellular Carcinoma through Combining Network Pharmacology and Experimental Evaluation. Front. Pharmacol. 2020, 11, 414. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Feng, Z.-P.; Zhang, L.; Pajouhesh, H.; Ding, Y.; Belardetti, F.; Pajouhesh, H.; Dolphin, D.; Mitscher, L.A.; Snutch, T.P. Scaffold-Based Design and Synthesis of Potent N-Type Calcium Channel Blockers. Bioorganic Med. Chem. Lett. 2009, 19, 6467–6472. [Google Scholar] [CrossRef] [PubMed]
- Tutumlu, G.; Dogan, B.; Avsar, T.; Orhan, M.D.; Calis, S.; Durdagi, S. Integrating Ligand and Target-Driven Based Virtual Screening Approaches With in Vitro Human Cell Line Models and Time-Resolved Fluorescence Resonance Energy Transfer Assay to Identify Novel Hit Compounds Against BCL-2. Front. Chem. 2020, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Sharma, D.; Soni, R.; Khatoon, N.; Sharma, B.; Bhatt, T.K. Plasmodium Falciparum Apicoplast and Its Transcriptional Regulation through Calcium Signaling. J. Microbiol. 2017, 55, 231–236. [Google Scholar] [CrossRef]
- Milton, M.E.; Nelson, S.W. Replication and Maintenance of the Plasmodium Falciparum Apicoplast Genome. Mol. Biochem. Parasitol. 2016, 208, 56–64. [Google Scholar] [CrossRef]
- Yellowley, C.E.; Hancox, J.C.; Donahue, H.J. Effects of Cell Swelling on Intracellular Calcium and Membrane Currents in Bovine Articular Chondrocytes. J. Cell. Biochem. 2002, 86, 290–301. [Google Scholar] [CrossRef]
- Straimer, J.; Gnädig, N.F.; Witkowski, B.; Amaratunga, C.; Duru, V.; Ramadani, A.P.; Dacheux, M.; Khim, N.; Zhang, L.; Lam, S.; et al. K13-Propeller Mutations Confer Artemisinin Resistance in Plasmodium Falciparum Clinical Isolates. Science 2015, 347, 428–431. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.; Shivappagowdar, A.; Hada, R.S.; Ayana, R.; Bathula, C.; Sen, S.; Kalia, I.; Pati, S.; Singh, A.P.; Singh, S. Plasmodium Perforin-like Protein Pores on the Host Cell Membrane Contribute in Its Multistage Growth and Erythrocyte Senescence. Front. Cell. Infect. Microbiol. 2020, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.; Agarwal, S.; Kumar, S.; Yazdani, S.S.; Chitnis, C.E.; Singh, S. Calcium-Dependent Permeabilization of Erythrocytes by a Perforin-like Protein during Egress of Malaria Parasites. Nat. Commun. 2013, 4, 1736. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.Y.; Choi, H.C. Nifedipine Inhibits Vascular Smooth Muscle Cell Proliferation and Reactive Oxygen Species Production through AMP-Activated Protein Kinase Signaling Pathway. Vasc. Pharmacol. 2012, 56, 1–8. [Google Scholar] [CrossRef]
- Antoine, T.; Fisher, N.; Amewu, R.; O’Neill, P.M.; Ward, S.A.; Biagini, G.A. Rapid Kill of Malaria Parasites by Artemisinin and Semi-Synthetic Endoperoxides Involves ROS-Dependent Depolarization of the Membrane Potential. J. Antimicrob. Chemother. 2014, 69, 1005–1016. [Google Scholar] [CrossRef]
- Schrevel, J.; Asfaux-Foucher, G.; Hopkins, J.; Robert, V.; Bourgouin, C.; Prensier, G.; Bannister, L. Vesicle Trafficking during Sporozoite Development in Plasmodium Berghei: Ultrastructural Evidence for a Novel Trafficking Mechanism. Parasitology 2008, 135, 1–12. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
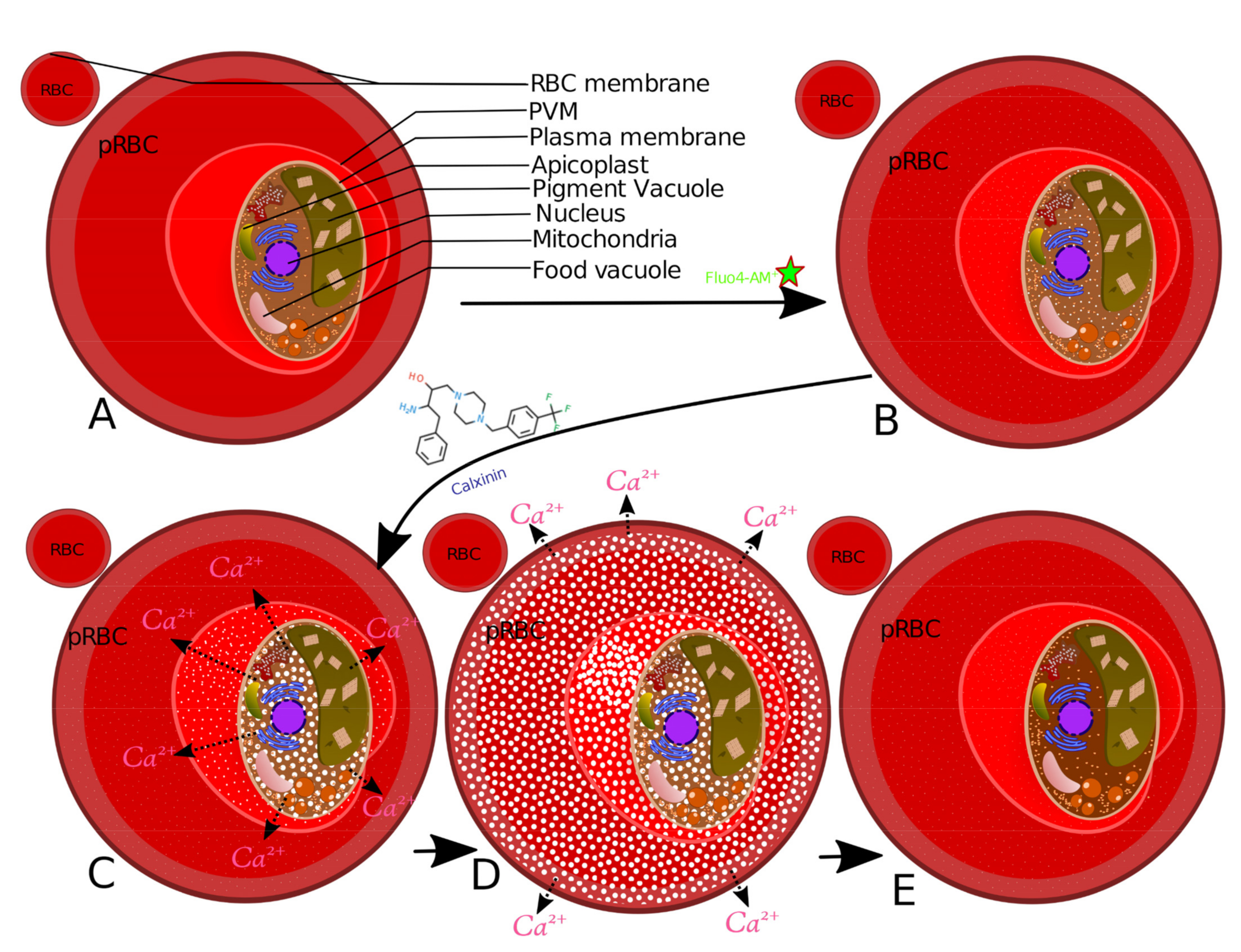{kind=link}
| Strain | Stage | Host | Efficacy | |
|---|---|---|---|---|
| Pf3D7 | In vitro | Erythrocytic | Human RBCs | IC50 = 90.0 ± 1.9 nM |
| PfDD2 | In vitro | Erythrocytic | Human RBCs | IC50 = 88.0 ±1.1 nM |
| PfIPC | In vitro | Erythrocytic | Human RBCs | IC50 = 93.0 ± 4.9 nM |
| Pf3D7 | In vitro | Gametocytic | Human RBCs | 50% viability loss and 100% mature gametocyte loss at 500 nM for 48 h |
| PbANKA | In vivo | Erythrocytic | Mice | Single 50mg/kg dose = 27.4% reduction in parasitemia |
| PbANKA | In vitro | Liver stages | HepG2 | 79.0 ± 1.6 nM |
| PbANKA | Ex vivo | Ookinete | Mice | Ookinete development IC50 = 150.0 ± 0.24 nM |
| Pf field strains | In vitro | Erythrocytic | Human RBCs | IC50 = 135.0 ± 6.7 nM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, Y.; Sharma, N.; Singh, S.; Romero, J.G.; Rajendran, V.; Mogire, R.M.; Kashif, M.; Beach, J.; Jeske, W.; Poonam; et al. The Multistage Antimalarial Compound Calxinin Perturbates P. falciparum Ca2+ Homeostasis by Targeting a Unique Ion Channel. Pharmaceutics 2022, 14, 1371. https://doi.org/10.3390/pharmaceutics14071371
Gupta Y, Sharma N, Singh S, Romero JG, Rajendran V, Mogire RM, Kashif M, Beach J, Jeske W, Poonam, et al. The Multistage Antimalarial Compound Calxinin Perturbates P. falciparum Ca2+ Homeostasis by Targeting a Unique Ion Channel. Pharmaceutics. 2022; 14(7):1371. https://doi.org/10.3390/pharmaceutics14071371
Chicago/Turabian StyleGupta, Yash, Neha Sharma, Snigdha Singh, Jesus G. Romero, Vinoth Rajendran, Reagan M. Mogire, Mohammad Kashif, Jordan Beach, Walter Jeske, Poonam, and et al. 2022. "The Multistage Antimalarial Compound Calxinin Perturbates P. falciparum Ca2+ Homeostasis by Targeting a Unique Ion Channel" Pharmaceutics 14, no. 7: 1371. https://doi.org/10.3390/pharmaceutics14071371