
PD-1 Cellular Nanovesicles Carrying Gemcitabine to Inhibit the Proliferation of Triple Negative Breast Cancer Cell

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Regents
2.2. Plasmids
2.3. Cell Lines
2.4. Western Blotting
2.5. Cell Membrane Vesicle Preparation
2.6. Size Distribution, Zeta-Potential Analysis, and Morphology
2.7. Nanovesicle Cell Binding Assay
2.8. Isolation of Peripheral Blood Mononuclear Cell (PBMC) from Human Peripheral Blood
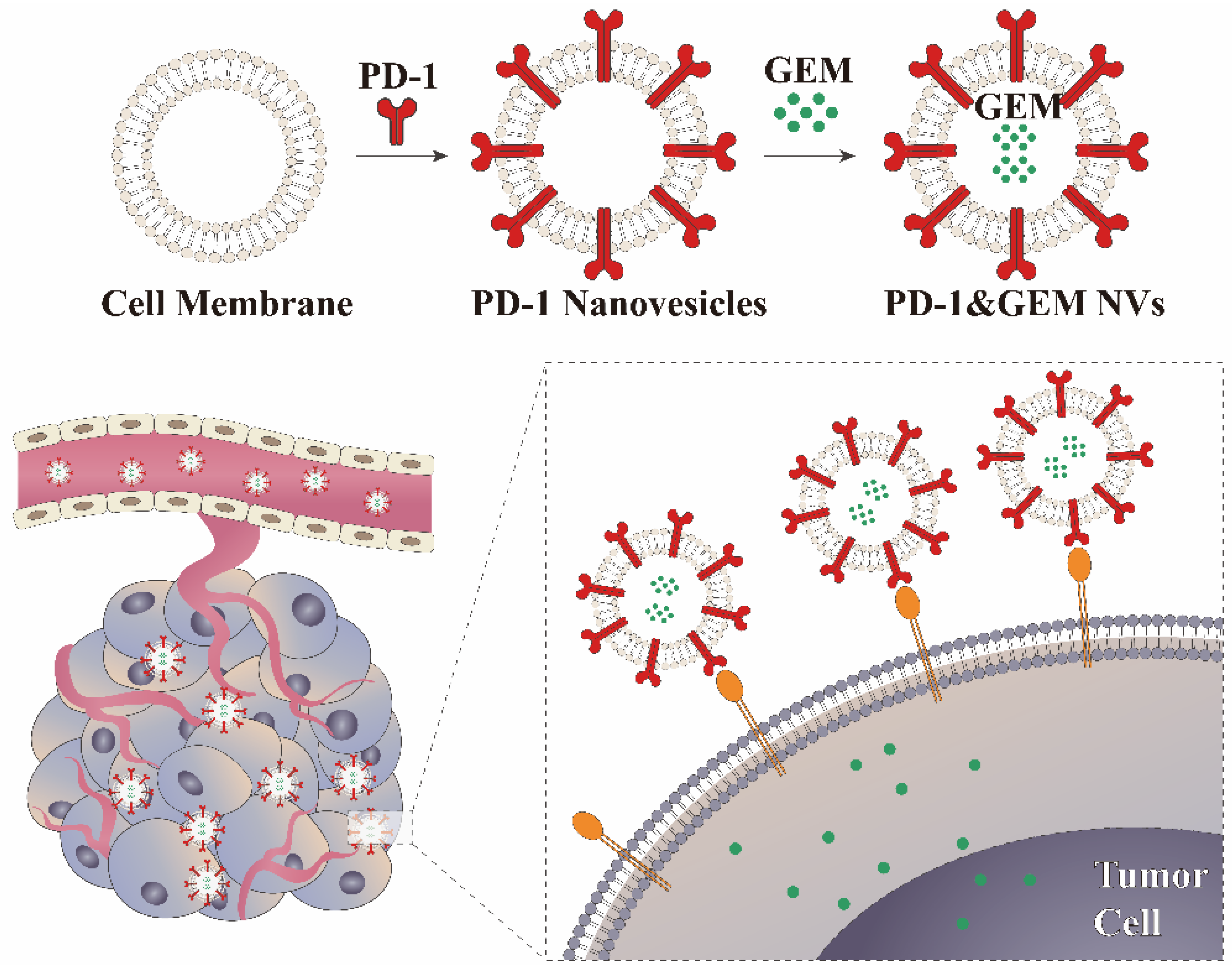
2.9. Production of GEM&PD-1 NVs
2.10. Detection of Encapsulation Ratio
2.11. Carboxyfluorescein Diacetate Succinimidyl Easter (CFSE) Staining
2.12. Biodistribution
2.13. Breast Cancer Mouse Models
2.14. Haematoxylin and Eosin Staining
2.15. Cell Isolation from Spleen, Tumor, and Lymph Node and Flow Cytometry Assay
2.16. Annexin V/Propidium Iodide-Based Cell Apoptosis Assay
2.17. Cell Viability and Colony Formation Assays
2.18. Quantitative Real-Time PCR
2.19. Statistical Analysis
3. Results
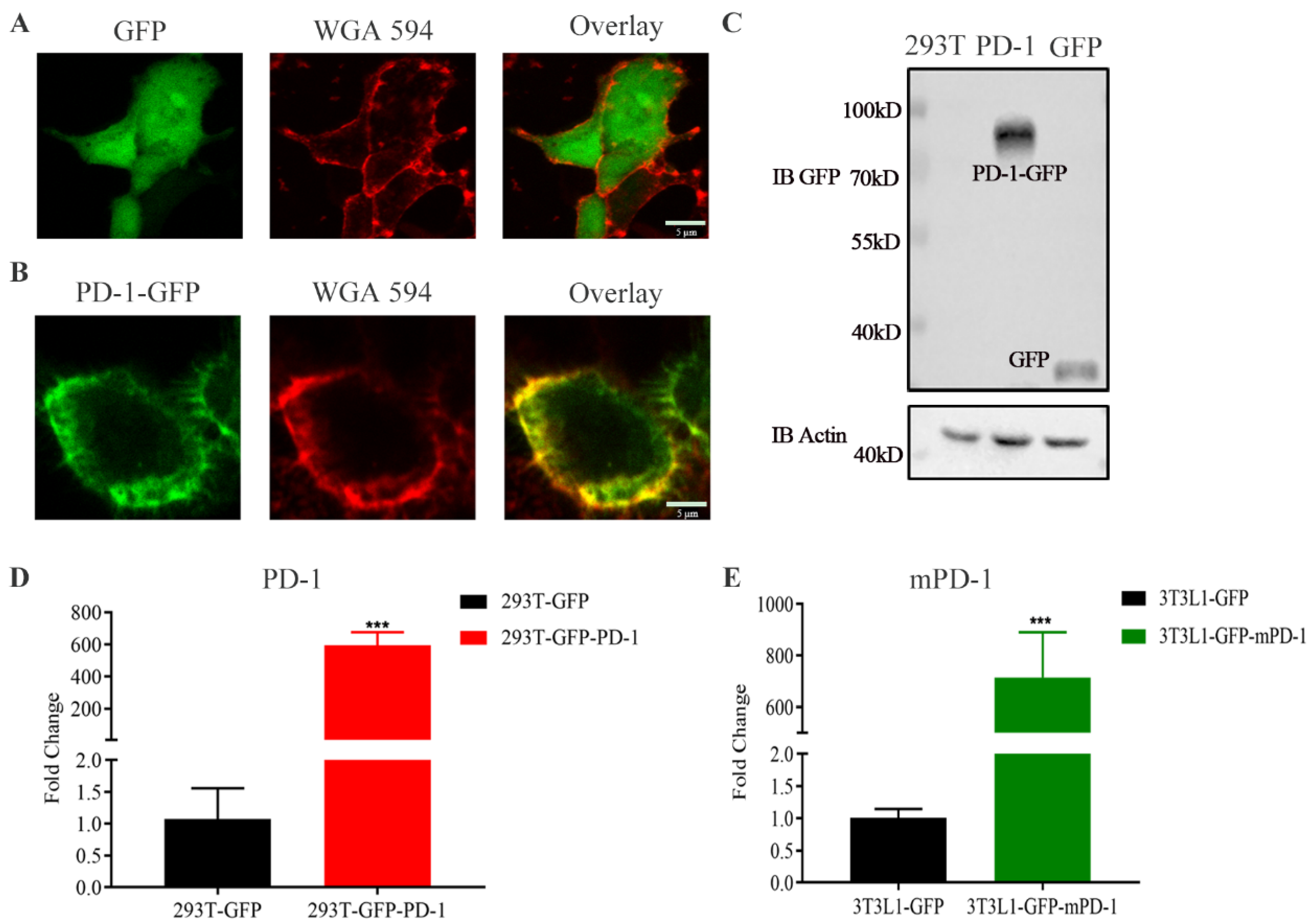
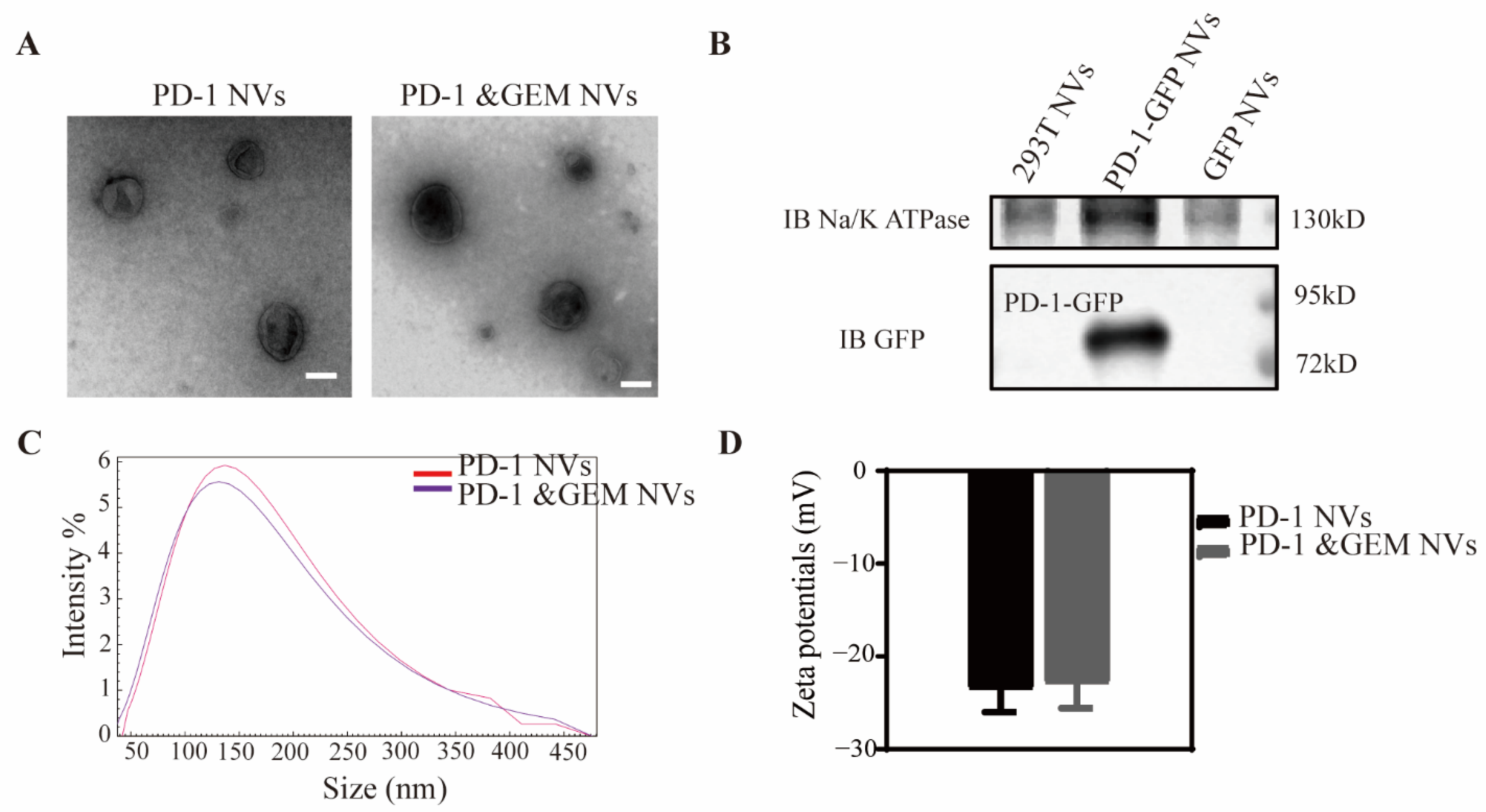
3.1. Construction of Stably Overexpressing PD-1 Cell Lines and Biological Behaviors of PD-1 NVs In Vitro
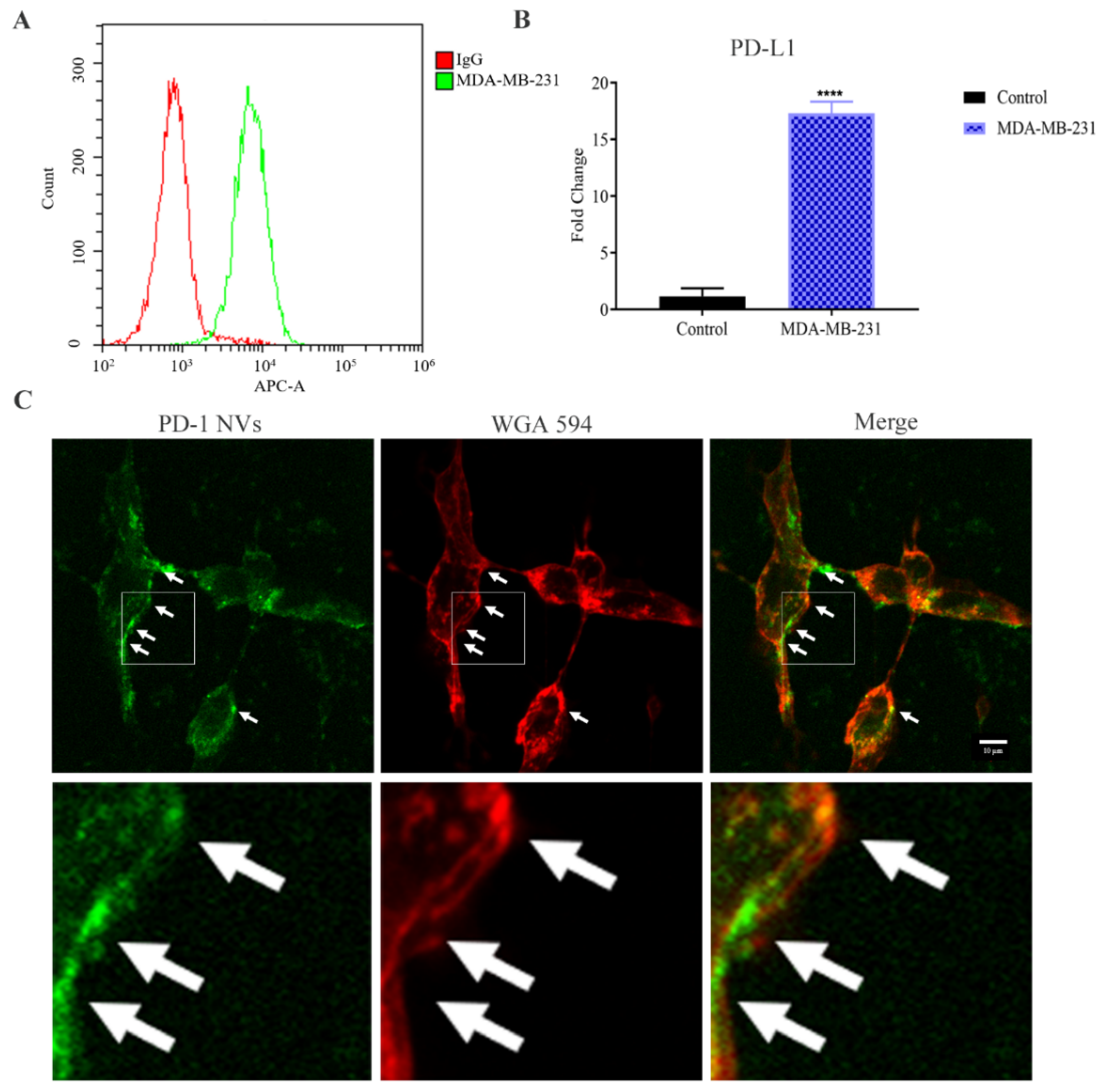
3.2. PD-1-GFP NVs Specifically Bound to PD-L1 on the Surface of MDA-MB-231 Triple-Negative Breast Cancer Cells
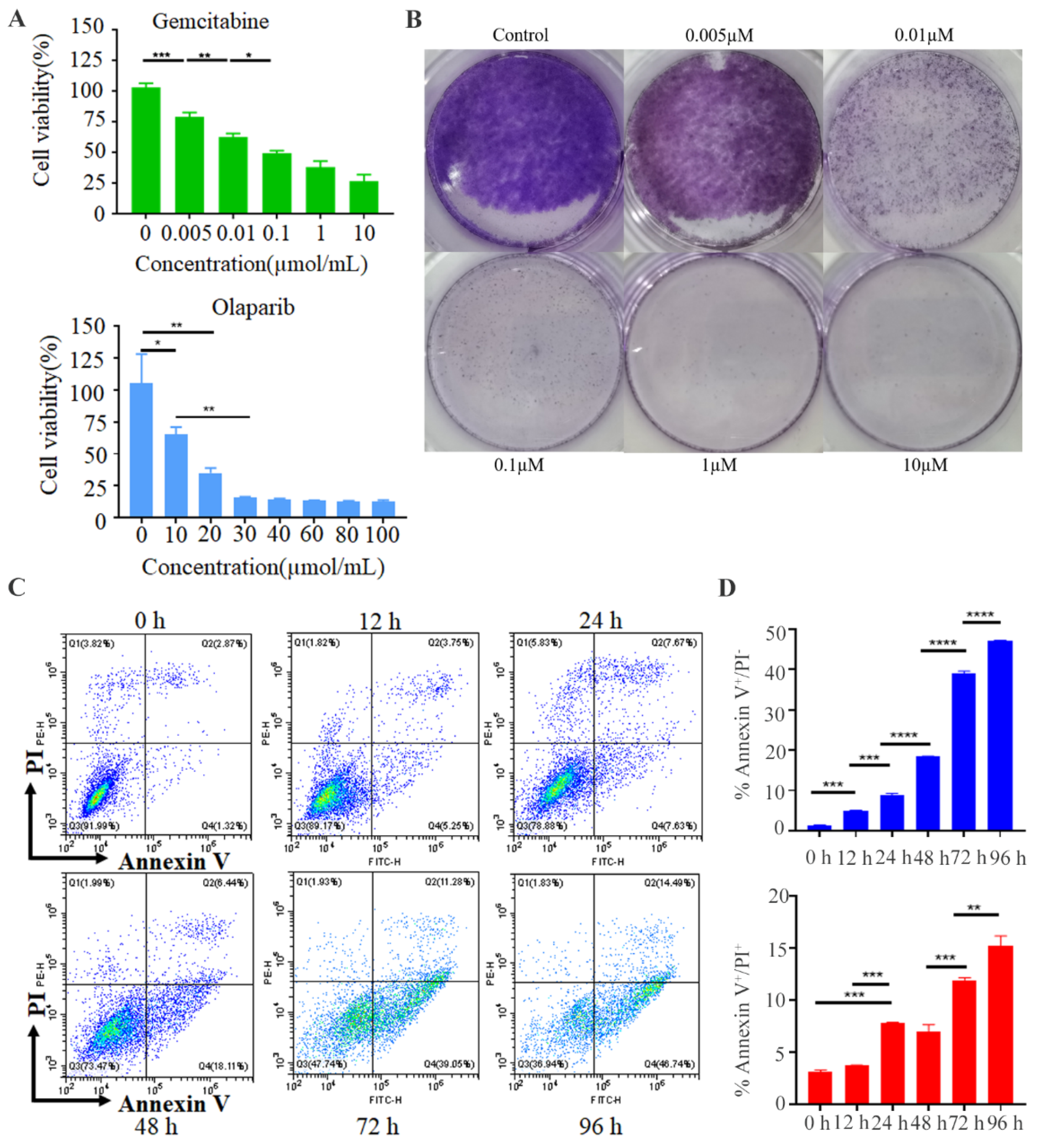
3.3. Gemcitabine Inhibited the Tumor Cell Growth and PD-1&GEM NVs Induced Cell Apoptosis of Breast Cancer In Vitro
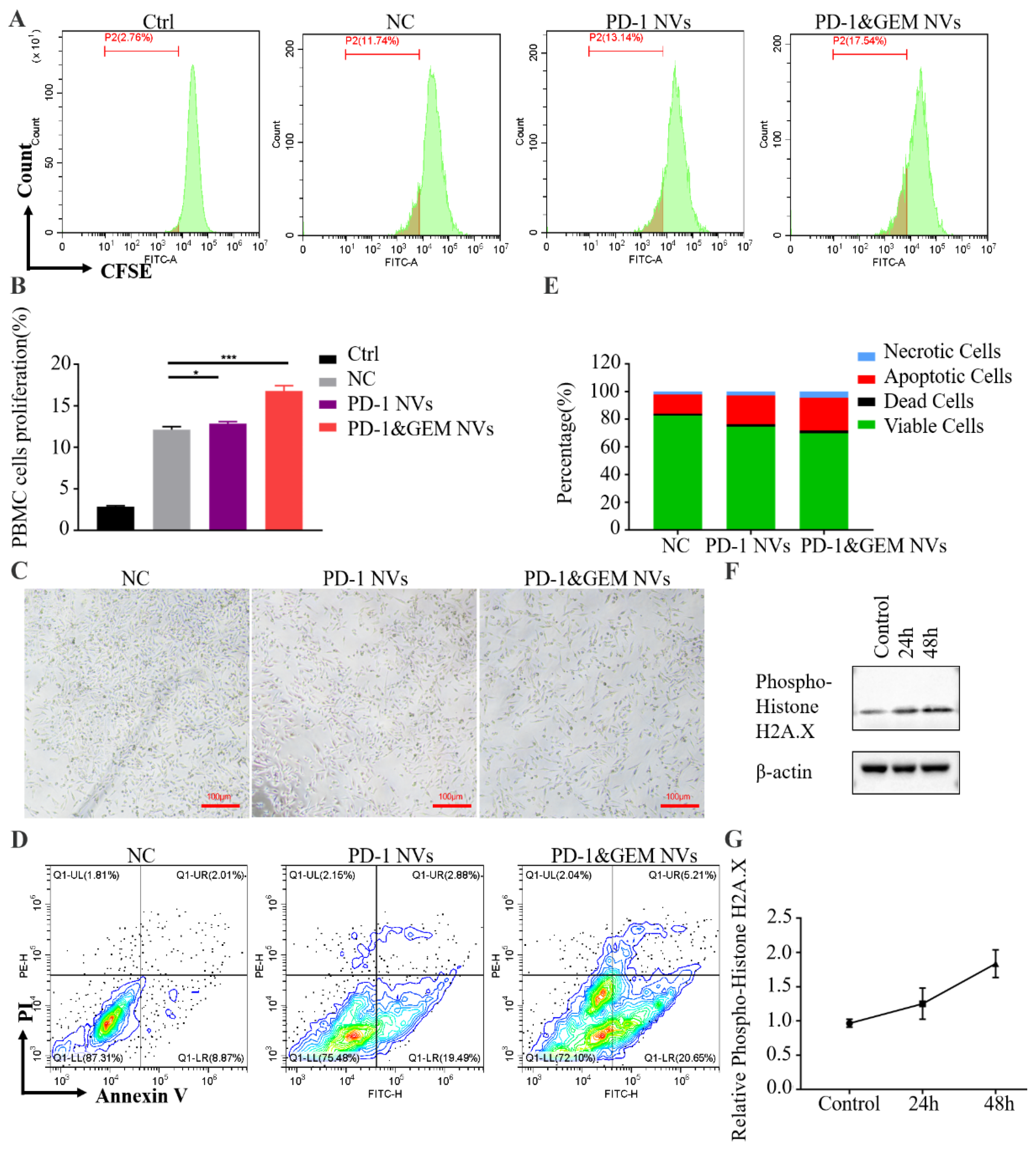
3.4. PD-1&GEM NVs Promoted PBMC Activation and MDA-MB-231 Cell Apoptosis When Co-Cultured
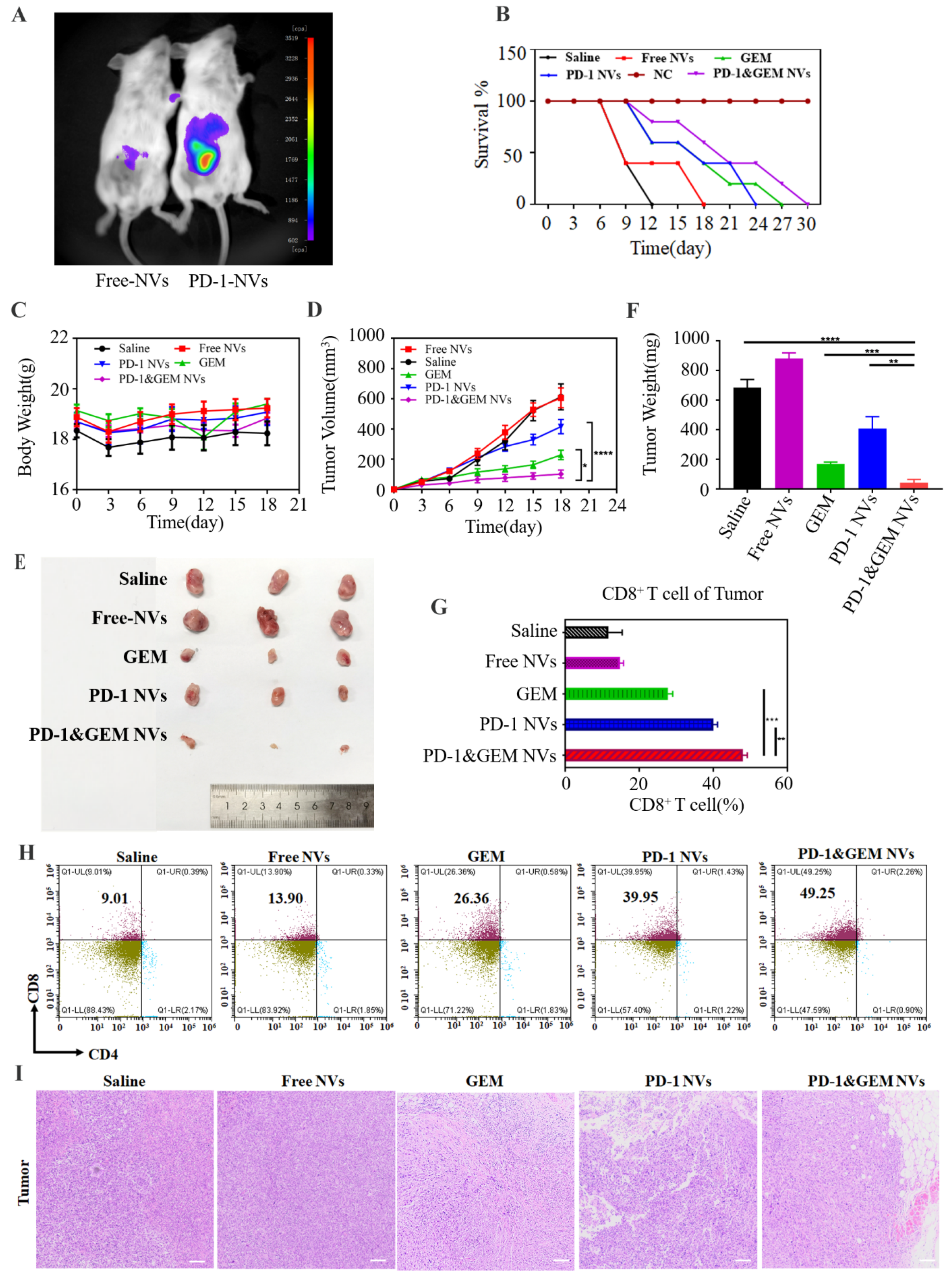
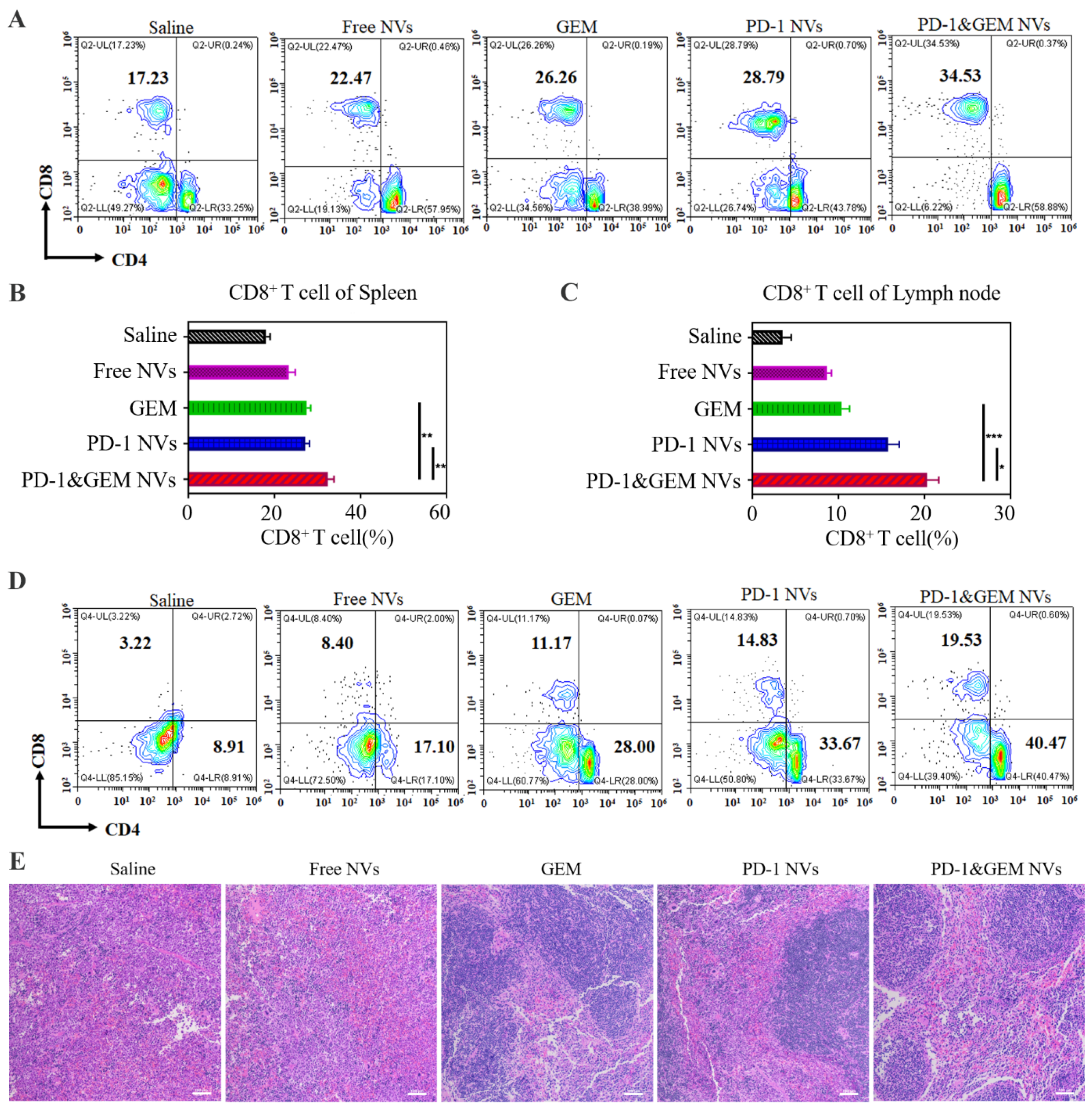
3.5. GEM&PD-1 NVs Inhibited the Growth of TNBC Breast Cancer In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Wengner, A.M.; Scholz, A.; Haendler, B. Targeting DNA Damage Response in Prostate and Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8273. [Google Scholar] [CrossRef] [PubMed]
- Paroha, S.; Verma, J.; Dubey, R.D.; Dewangan, R.P.; Molugulu, N.; Bapat, R.A.; Sahoo, P.K.; Kesharwani, P. Recent advances and prospects in gemcitabine drug delivery systems. Int. J. Pharm. 2021, 592, 120043. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Salas-Benito, D.; Pérez-Gracia, J.L.; Ponz-Sarvisé, M.; Rodriguez-Ruiz, M.E.; Martínez-Forero, I.; Castañón, E.; López-Picazo, J.M.; Sanmamed, M.F.; Melero, I. Paradigms on Immunotherapy Combinations with Chemotherapy. Cancer Discov. 2021, 11, 1353–1367. [Google Scholar] [CrossRef] [PubMed]
- Rosa, K. Roche Withdraws Application for Atezolizumab in Early or Locally Advanced TNBC in Europe. Available online: https://www.onclive.com/view/roche-withdraws-application-for-atezolizumab-in-early-or-locally-advanced-tnbc-in-europe (accessed on 27 July 2021).
- Robbins, P.D.; Dorronsoro, A.; Booker, C.N. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J. Clin. Investig. 2016, 126, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Zhang, Y.; Tang, K.; Zhang, H.; Yin, X.; Li, Y.; Xu, P.; Sun, Y.; Ma, R.; Ji, T.; et al. Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles. Cell Res. 2016, 26, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Guo, J.; Liu, Z.; Li, Y.; Jin, S.; Wang, T. The Role of Exosomes in the Female Reproductive System and Breast Cancers. OncoTargets Ther. 2020, 13, 12567–12586. [Google Scholar] [CrossRef]
- Wang, X.; Xu, C.; Hua, Y.; Sun, L.; Cheng, K.; Jia, Z.; Han, Y.; Dong, J.; Cui, Y.; Yang, Z. Exosomes play an important role in the process of psoralen reverse multidrug resistance of breast cancer. J. Exp. Clin. Cancer Res. 2016, 35, 186. [Google Scholar] [CrossRef] [Green Version]
- Lowry, M.C.; Gallagher, W.M.; O’Driscoll, L. The Role of Exosomes in Breast Cancer. Clin. Chem. 2015, 61, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, C.; Wang, J.; Hu, Q.; Langworthy, B.; Ye, Y.; Sun, W.; Lin, J.; Wang, T.; Fine, J.; et al. PD-1 Blockade Cellular Vesicles for Cancer Immunotherapy. Adv. Mater. 2018, 30, e1707112. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Chen, Z.; Hu, Q.; Wang, C.; Yan, J.; Dotti, G.; Huang, P.; Gu, Z. Engineering PD-1-Presenting Platelets for Cancer Immunotherapy. Nano Lett. 2018, 18, 5716–5725. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, X.; Chen, X.; Li, J. Multifunctional Biomedical Materials Derived from Biological Membranes. Adv. Mater. 2021, e2107406. [Google Scholar] [CrossRef]
- Tan, S.; Wu, T.; Zhang, D.; Zhang, Z. Cell or cell membrane-based drug delivery systems. Theranostics 2015, 5, 863–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Gregorio, A.; Häberle, L.; Fasching, P.A.; Müller, V.; Schrader, I.; Lorenz, R.; Forstbauer, H.; Friedl, T.W.P.; Bauer, E.; de Gregorio, N.; et al. Gemcitabine as adjuvant chemotherapy in patients with high-risk early breast cancer-results from the randomized phase III SUCCESS-A trial. Breast Cancer Res. 2020, 22, 111. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, Y.; Ji, Y.; Yu, Y.; Jin, Y.; Zhang, X.; Zhou, J. Gemcitabine induces apoptosis and autophagy via the AMPK/mTOR signaling pathway in pancreatic cancer cells. Biotechnol. Appl. Biochem. 2018, 65, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D. Overview: Gemcitabine as single-agent therapy for advanced breast cancer. Clin. Breast Cancer 2002, 3 (Suppl. S1), 8–11. [Google Scholar] [CrossRef]
- Yang, M.; Xu, Z.; Yan, H.; Tsai, H.I.; Su, D.; Yan, F.; Lu, Q.; Feng, J.; Zeng, W.; Xi, L.; et al. PD-L1 cellular nanovesicles carrying rapamycin inhibit alloimmune responses in transplantation. Biomater. Sci. 2021, 9, 1246–1255. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- Nikanjam, M.; Patel, H.; Kurzrock, R. Dosing immunotherapy combinations: Analysis of 3,526 patients for toxicity and response patterns. Oncoimmunology 2017, 6, e1338997. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Brahmer, J.R.; Juergens, R.A.; Borghaei, H.; Gettinger, S.; Chow, L.Q.; Gerber, D.E.; Laurie, S.A.; Goldman, J.W.; et al. Nivolumab in Combination With Platinum-Based Doublet Chemotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 2969–2979. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.; Gligorov, J.; André, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol. 2021, 32, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Rathore, S.; Munshi, A.; Ramesh, R. Organically derived exosomes as carriers of anticancer drugs and imaging agents for cancer treatment. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2022. [Google Scholar] [CrossRef]
- Ramesh, A.; Kumar, S.; Nandi, D.; Kulkarni, A. CSF1R- and SHP2-Inhibitor-Loaded Nanoparticles Enhance Cytotoxic Activity and Phagocytosis in Tumor-Associated Macrophages. Adv. Mater. 2019, 31, e1904364. [Google Scholar] [CrossRef] [PubMed]
- Caro, C.; Avasthi, A.; Paez-Muñoz, J.M.; Pernia Leal, M.; García-Martín, M.L. Passive targeting of high-grade gliomas via the EPR effect: A closed path for metallic nanoparticles? Biomater. Sci. 2021, 9, 7984–7995. [Google Scholar] [CrossRef] [PubMed]
- Islam, W.; Fang, J.; Imamura, T.; Etrych, T.; Subr, V.; Ulbrich, K.; Maeda, H. Augmentation of the Enhanced Permeability and Retention Effect with Nitric Oxide-Generating Agents Improves the Therapeutic Effects of Nanomedicines. Mol. Cancer Ther. 2018, 17, 2643–2653. [Google Scholar] [CrossRef] [Green Version]
- Donahue, N.D.; Acar, H.; Wilhelm, S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv. Drug Deliv. Rev. 2019, 143, 68–96. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primer Sequence 5′→3′ | Reverse Primer Sequence 5′→3′ |
|---|---|---|
| Mouse-β-Actin Mouse-Pd-l1 Human-Pd-1 Human-Pd-l1 | GGCTGTATTCCCCTCCATCG TCTGATCGTCGATTGGCAGC CCCAAGGCGCAGATCAA GGTGAGGATGGTTCTACACAG | CCAGTTGGTAACAATGCCATGT CGTTGTTCCAGGCTCCTCTC CTGGCGAGCCTTAGTTTGGAC GAGAACTGCATGAGGTTGC |
| Human-β-Actin | CCACACTGTGCCCATCTAC | AGGATCTTCATGAGGTAGTCAGTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zha, H.; Xu, Z.; Xu, X.; Lu, X.; Shi, P.; Xiao, Y.; Tsai, H.-I.; Su, D.; Cheng, F.; Cheng, X.; et al. PD-1 Cellular Nanovesicles Carrying Gemcitabine to Inhibit the Proliferation of Triple Negative Breast Cancer Cell. Pharmaceutics 2022, 14, 1263. https://doi.org/10.3390/pharmaceutics14061263
Zha H, Xu Z, Xu X, Lu X, Shi P, Xiao Y, Tsai H-I, Su D, Cheng F, Cheng X, et al. PD-1 Cellular Nanovesicles Carrying Gemcitabine to Inhibit the Proliferation of Triple Negative Breast Cancer Cell. Pharmaceutics. 2022; 14(6):1263. https://doi.org/10.3390/pharmaceutics14061263
Chicago/Turabian StyleZha, Hualian, Zhanxue Xu, Xichao Xu, Xingyu Lu, Peilin Shi, Youmei Xiao, Hsiang-I Tsai, Dandan Su, Fang Cheng, Xiaoli Cheng, and et al. 2022. "PD-1 Cellular Nanovesicles Carrying Gemcitabine to Inhibit the Proliferation of Triple Negative Breast Cancer Cell" Pharmaceutics 14, no. 6: 1263. https://doi.org/10.3390/pharmaceutics14061263