
Navitoclax Enhances the Therapeutic Effects of PLK1 Targeting on Lung Cancer Cells in 2D and 3D Culture Systems

 ,
,  ,
, 
 and
and 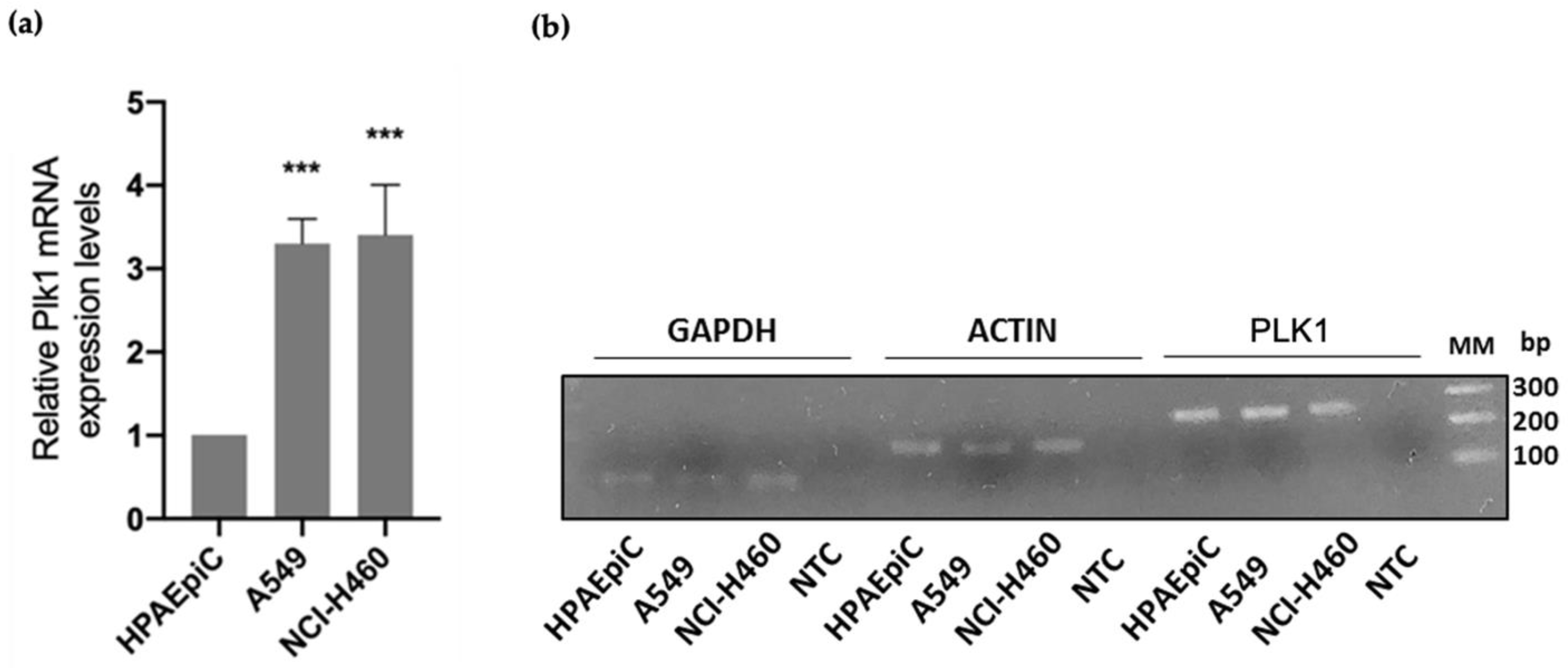{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Lines and Culture Conditions
2.3. RNA Isolation and Real-Time PCR Analysis
2.4. MTT Assay
2.5. Mitotic Index Determination
2.6. Flow Cytometry Analysis for Apoptosis Detection
2.7. Death Fluorometric TUNEL Assay
2.8. Live-Cell Imaging
2.9. 3D Spheroid Formation and Drug Treatment
2.10. CellTiter-Glo Viability Assay
2.11. Statistical Analysis
3. Results
3.1. PLK1 Is Upregulated in Lung Cancer Cells
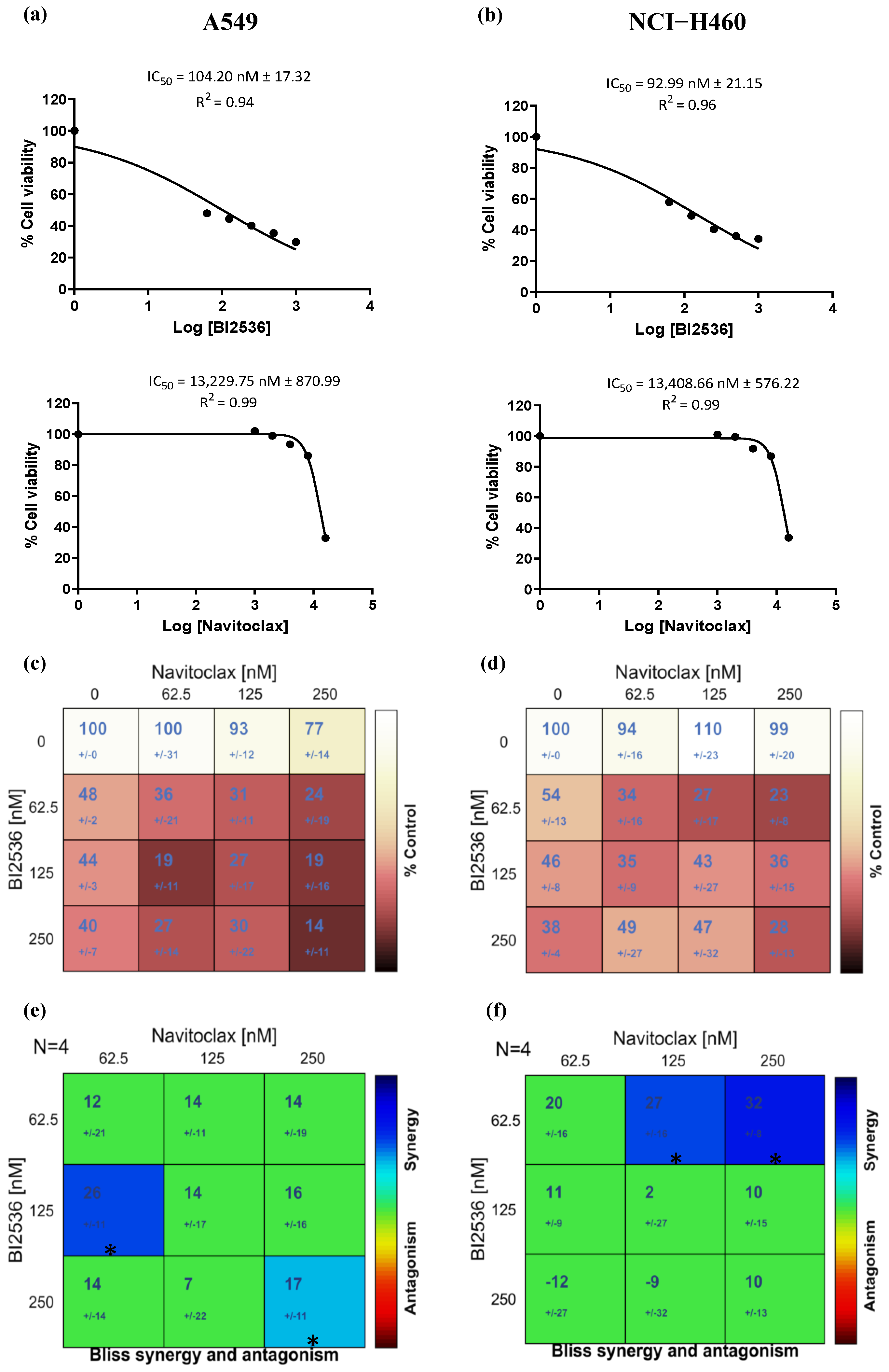
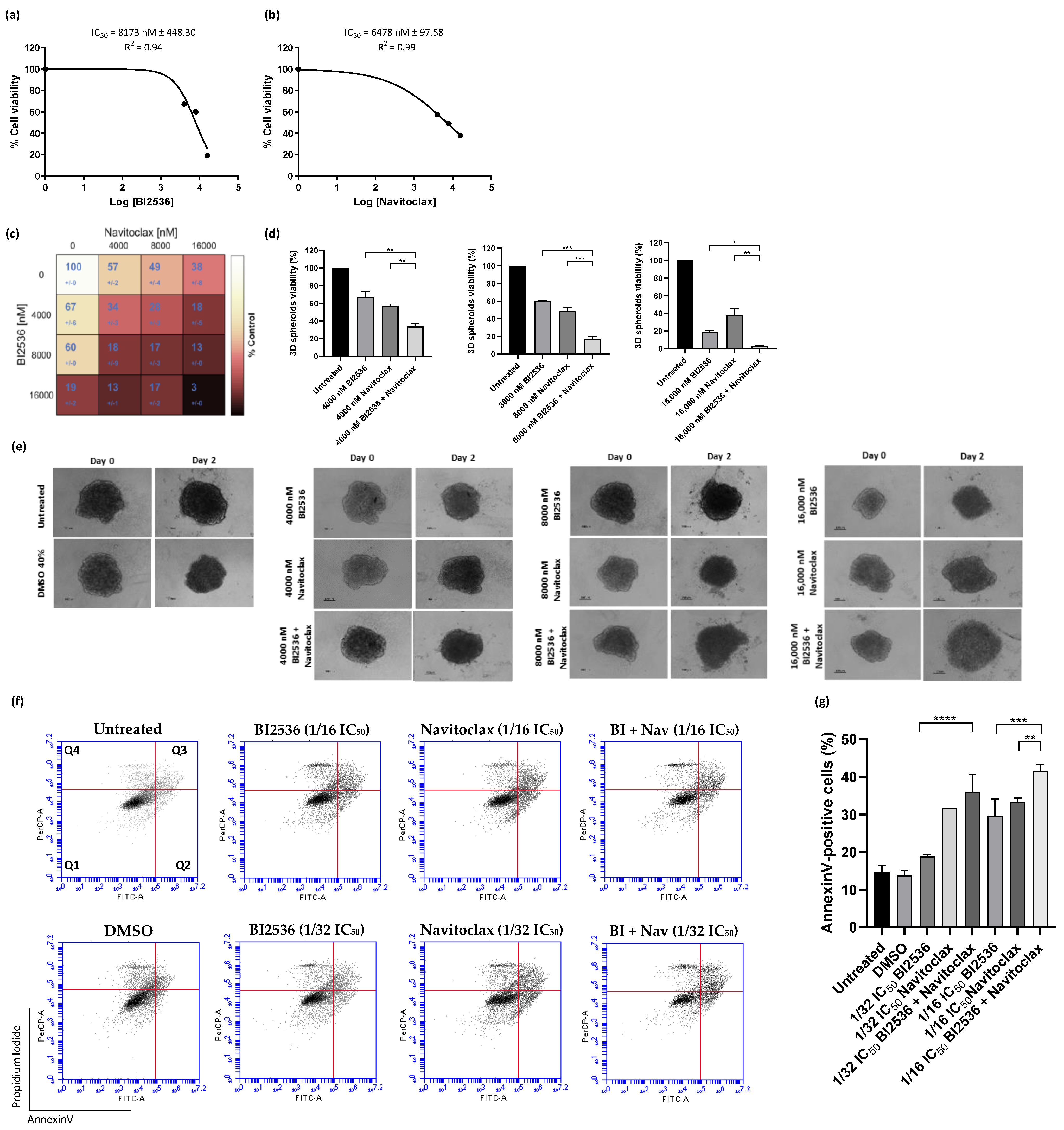
3.2. Navitoclax Enhances the Antiproliferative Effect of BI2536-Mediated PLK1 Inhibition in Lung Cancer Cells
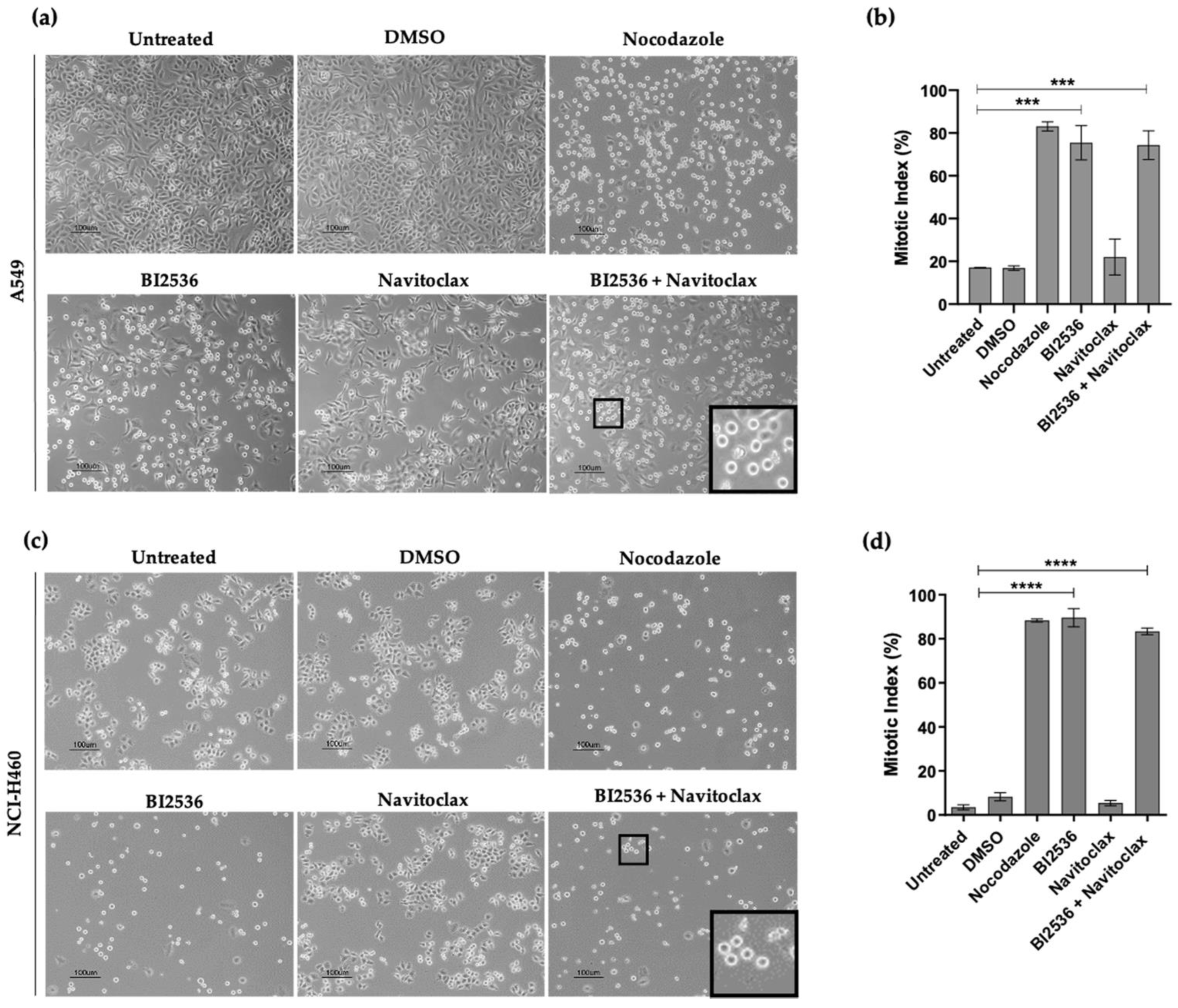
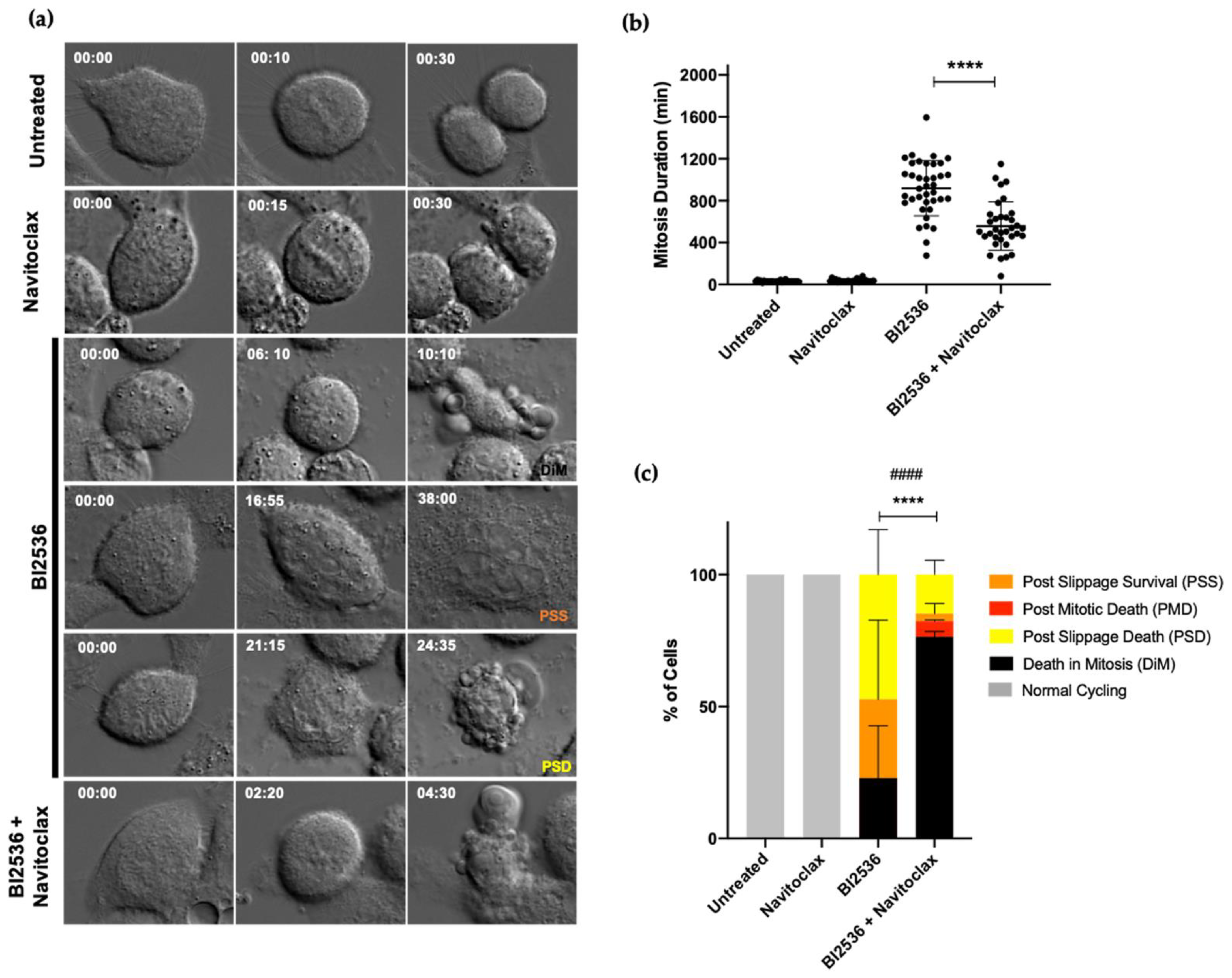
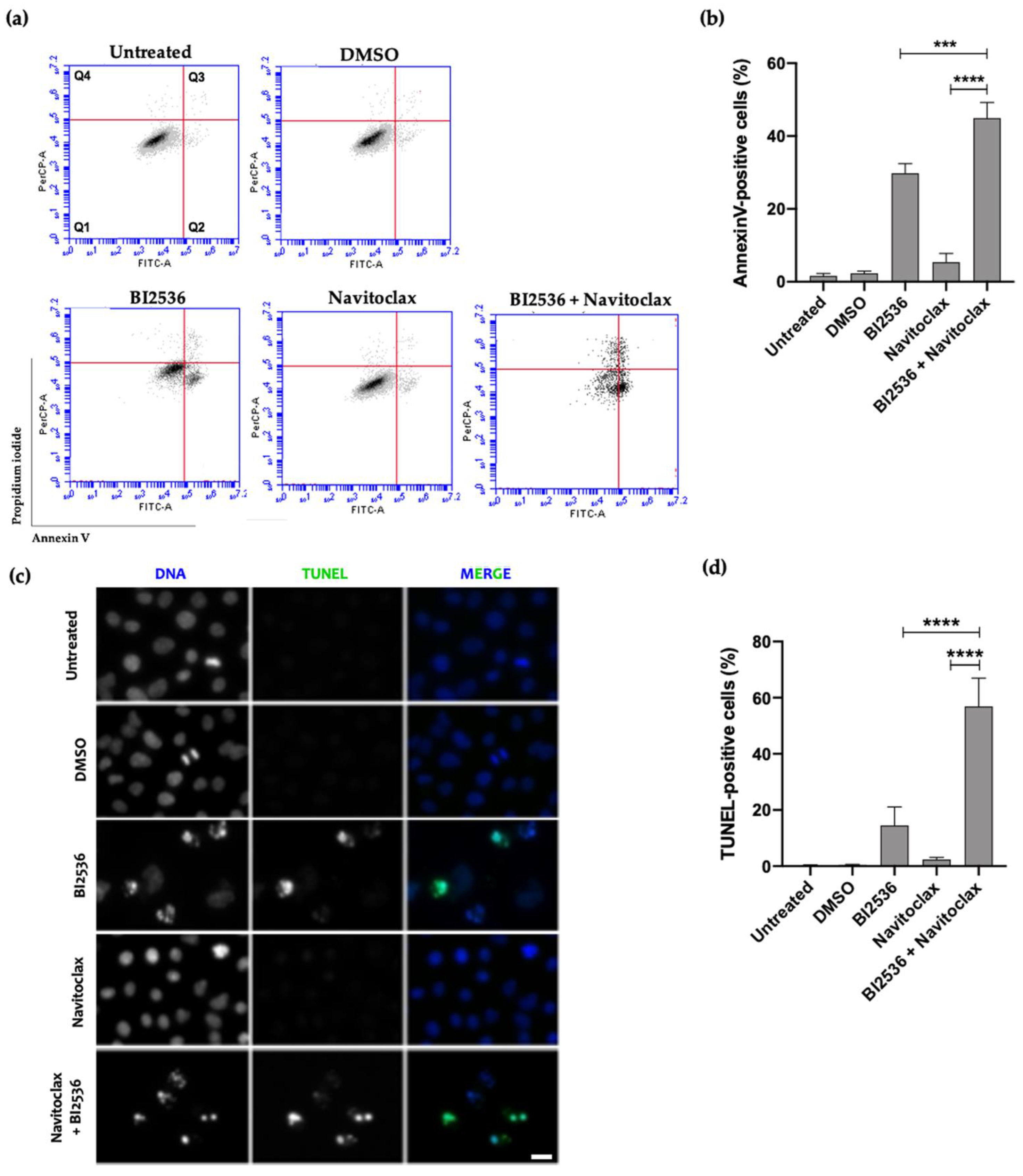
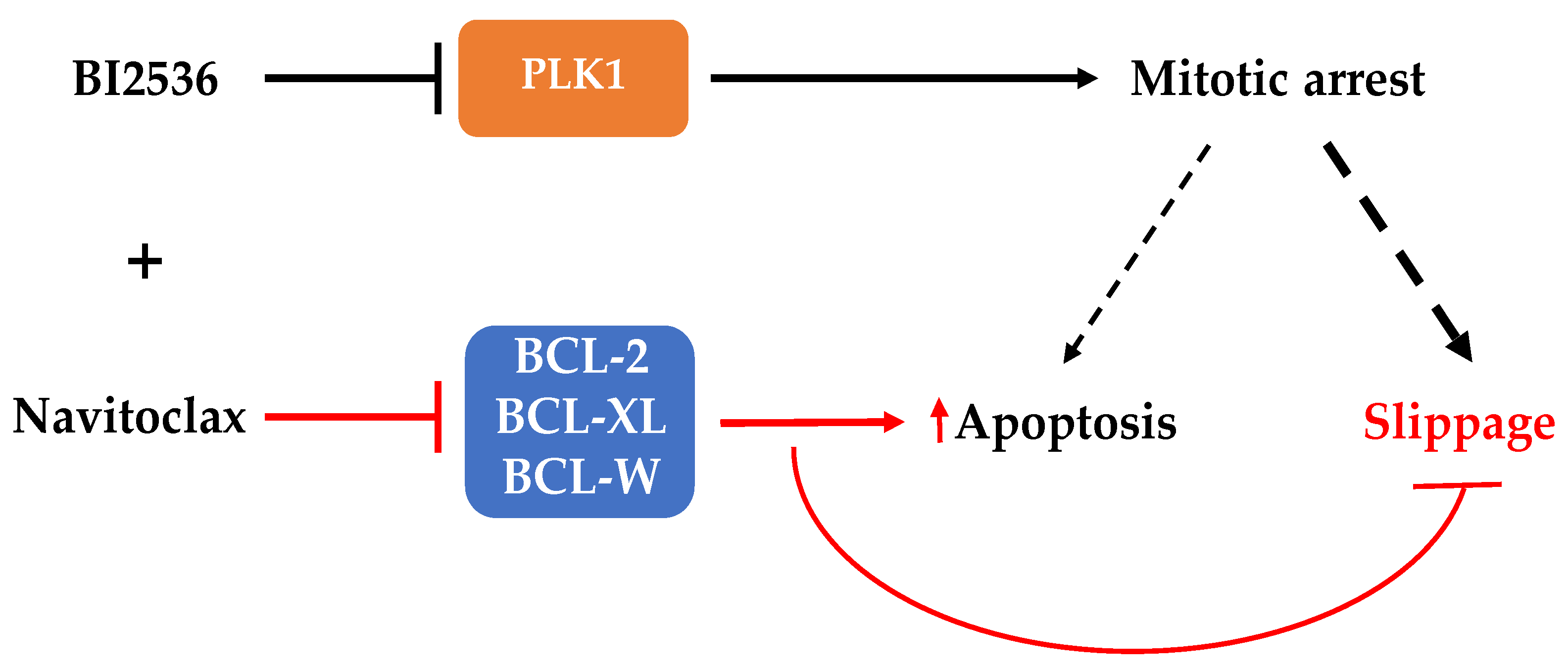
3.3. Combining BI2536-Mediated PLK1 Inhibition with Navitoclax Overcomes Slippage and Shifts the Cancer Cell Fate to Accelerated Death in Mitosis
3.4. Co-Treatment with BI2536 and Navitoclax Decrease 3D Cancer Spheroid Viability
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2020. Available online: https://seer.cancer.gov/csr/1975_2016/ (accessed on 15 November 2021).
- Ashrafizadeh, M.; Mirzaei, S.; Hushmandi, K.; Rahmanian, V.; Zabolian, A.; Raei, M.; Farahani, M.V.; Goharrizi, M.A.S.B.; Khan, H.; Zarrabi, A.; et al. Therapeutic potential of AMPK signaling targeting in lung cancer: Advances, challenges and future prospects. Life Sci. 2021, 278, 119649. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Najafi, M.; Makvandi, P.; Zarrabi, A.; Farkhondeh, T.; Samarghandian, S. Versatile role of curcumin and its derivatives in lung cancer therapy. J. Cell. Physiol. 2020, 235, 9241–9268. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H. Comparison of Four Chemotherapy Regimens for Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2002, 346, 92–98. [Google Scholar] [CrossRef]
- Peters, S.; Adjei, A.A.; Gridelli, C.; Reck, M.; Kerr, K.; Felip, E. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, vii56–vii64. [Google Scholar] [CrossRef]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, A.C.; Ribeiro, D.; Pedrosa, J.; Sarmento, B.; Silva, P.M.A.; Bousbaa, H. Mitosis inhibitors in anticancer therapy: When blocking the exit becomes a solution. Cancer Lett. 2019, 440–441, 64–81. [Google Scholar] [CrossRef]
- Lara-Gonzalez, P.; Pines, J.; Desai, A. Spindle assembly checkpoint activation and silencing at kinetochores. Semin. Cell Dev. Biol. 2021, 117, 86–98. [Google Scholar] [CrossRef]
- Silva, P.; Barbosa, J.; Nascimento, A.V.; Faria, J.; Reis, R.; Bousbaa, H. Monitoring the fidelity of mitotic chromosome segregation by the spindle assembly checkpoint. Cell Prolif. 2011, 44, 391–400. [Google Scholar] [CrossRef]
- Marques, S.; Fonseca, J.; Silva, P.; Bousbaa, H. Targeting the Spindle Assembly Checkpoint for Breast Cancer Treatment. Curr. Cancer Drug Targets 2015, 15, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Čermák, V.; Dostál, V.; Jelínek, M.; Libusová, L.; Kovář, J.; Rösel, D.; Brábek, J. Microtubule-targeting agents and their impact on cancer treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Novais, P.; Silva, P.M.A.; Amorim, I.; Bousbaa, H. Second-Generation Antimitotics in Cancer Clinical Trials. Pharmaceutics 2021, 13, 1011. [Google Scholar] [CrossRef]
- Colicino, E.G.; Hehnly, H. Regulating a key mitotic regulator, polo-like kinase 1 (PLK1). Cytoskeleton 2018, 75, 481–494. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Wang, Y.; Yang, D.; Gong, Y.; Rao, F.; Liu, R.; Danna, Y.; Li, J.; Fan, J.; Chen, J.; et al. A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. EBioMedicine 2019, 41, 244–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajtler, K.W.; Sadowski, N.; Ackermann, S.; Althoff, K.; Schönbeck, K.; Batzke, K.; Schäfers, S.; Odersky, A.; Heukamp, L.; Astrahantseff, K.; et al. The GSK461364 PLK1 inhibitor exhibits strong antitumoral activity in preclinical neuroblastoma models. Oncotarget 2017, 8, 6730–6741. [Google Scholar] [CrossRef]
- Raab, M.; Krämer, A.; Hehlgans, S.; Sanhaji, M.; Kurunci-Csacsko, E.; Dötsch, C.; Bug, G.; Ottmann, O.; Becker, S.; Pachl, F.; et al. Mitotic arrest and slippage induced by pharmacological inhibition of Polo-like kinase 1. Mol. Oncol. 2015, 9, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. Cancer Cells Display Profound Intra- and Interline Variation following Prolonged Exposure to Antimitotic Drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, D.; Duijf, P.H.G.; Khanna, K.K. Mitotic slippage: An old tale with a new twist. Cell Cycle 2019, 18, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Silva, P.M.A.; Ribeiro, N.; Lima, R.T.; Andrade, C.; Diogo, V.; Teixeira, J.; Florindo, C.; Tavares, Á.; Vasconcelos, M.H.; Bousbaa, H. Suppression of spindly delays mitotic exit and exacerbates cell death response of cancer cells treated with low doses of paclitaxel. Cancer Lett. 2017, 394, 33–42. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 129. [Google Scholar] [CrossRef]
- Lénárt, P.; Petronczki, M.; Steegmaier, M.; Di Fiore, B.; Lipp, J.J.; Hoffmann, M.; Rettig, W.J.; Kraut, N.; Peters, J.-M. The Small-Molecule Inhibitor BI 2536 Reveals Novel Insights into Mitotic Roles of Polo-like Kinase 1. Curr. Biol. 2007, 17, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Mross, K.; Frost, A.; Steinbild, S.; Hedbom, S.; Rentschler, J.; Kaiser, R.; Rouyrre, N.; Trommeshauser, D.; Hoesl, C.E.; Munzert, G. Phase I Dose Escalation and Pharmacokinetic Study of BI 2536, a Novel Polo-Like Kinase 1 Inhibitor, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2008, 26, 5511–5517. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Dinsdale, D.; Dyer, M.J.S.; Cohen, G.M. Bcl-2 inhibitors: Small molecules with a big impact on cancer therapy. Cell Death Differ. 2009, 16, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G.; Elez, R.; Doermer, A.; Holtrich, U.; Ackermann, H.; Stutte, H.J.; Altmannsberger, H.M.; Rübsamen-Waigmann, H.; Strebhardt, K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 1997, 14, 543–549. [Google Scholar] [CrossRef]
- Zeng, Y.; Li, N.; Liu, W.; Zeng, M.; Cheng, J.; Huang, J. Analyses of expressions and prognostic values of Polo-like kinases in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2447–2460. [Google Scholar] [CrossRef] [PubMed]
- Mohamad Anuar, N.N.; Nor Hisam, N.S.; Liew, S.L.; Ugusman, A. Clinical Review: Navitoclax as a Pro-Apoptotic and Anti-Fibrotic Agent. Front. Pharmacol. 2020, 11, 1817. [Google Scholar] [CrossRef]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [Green Version]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef]
- Liu, N.; Hu, G.; Wang, H.; Li, Z.; Guo, Z. PLK1 inhibitor facilitates the suppressing effect of temozolomide on human brain glioma stem cells. J. Cell. Mol. Med. 2018, 22, 5300–5310. [Google Scholar] [CrossRef]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lénárt, P.; Petronczki, M.; Krššák, M.; Gürtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a Potent and Selective Inhibitor of Polo-like Kinase 1, Inhibits Tumor Growth In Vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.; Chen, G.; Liu, S.; Li, Y.; Zhong, J.; Zhang, B.; Li, L.; Huang, H.; Wang, Z.; Xu, Q.; et al. Discovery of methyl 3-((2-((1-(dimethylglycyl)-5-methoxyindolin-6-yl)amino)-5-(trifluoro-methyl) pyrimidin-4-yl)amino)thiophene-2-carboxylate as a potent and selective polo-like kinase 1 (PLK1) inhibitor for combating hepatocellular carcinoma. Eur. J. Med. Chem. 2020, 206, 112697. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Li, J.; Luo, Q.; Wang, R.; Kong, Y.; Carlock, C.; Liu, Z.; Elzey, B.D.; Liu, X. Plk1 inhibition enhances the efficacy of BET epigenetic reader blockade in castration-resistant prostate cancer. Mol. Cancer Ther. 2018, 17, 1554–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Martino, D.; Yilmaz, E.; Orlacchio, A.; Ranieri, M.; Zhao, K.; Di Cristofano, A. PI3K blockage synergizes with PLK1 inhibition preventing endoreduplication and enhancing apoptosis in anaplastic thyroid cancer. Cancer Lett. 2018, 439, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhou, Y.; Huang, H.C.; Mitchison, T.J. Navitoclax (ABT-263) accelerates apoptosis during drug-induced mitotic arrest by antagonizing Bcl-xL. Cancer Res. 2011, 71, 4518–4526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, N.; Malek, M.; Zha, J.; Yue, P.; Kassees, R.; Berry, L.; Fairbrother, W.J.; Sampath, D.; Belmont, L.D. Navitoclax enhances the efficacy of taxanes in non-small cell lung cancer models. Clin. Cancer Res. 2011, 17, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- Henriques, A.C.; Silva, P.M.A.; Sarmento, B.; Bousbaa, H. Antagonizing the spindle assembly checkpoint silencing enhances paclitaxel and Navitoclax-mediated apoptosis with distinct mechanistic. Sci. Rep. 2021, 11, 4139. [Google Scholar] [CrossRef]
- Henriques, A.C.; Silva, P.M.A.; Sarmento, B.; Bousbaa, H. The Mad2-Binding Protein p31comet as a Potential Target for Human Cancer Therapy. Curr. Cancer Drug Targets 2021, 21, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, J.; Nascimento, A.V.; Faria, J.; Silva, P.; Bousbaa, H. The spindle assembly checkpoint: Perspectives in tumorigenesis and cancer therapy. Front. Biol. 2011, 6, 147–155. [Google Scholar] [CrossRef]
- Wong, M.; Tan, N.; Zha, J.; Peale, F.V.; Yue, P.; Fairbrother, W.J.; Belmont, L.D. Navitoclax (ABT-263) Reduces Bcl-x L –Mediated Chemoresistance in Ovarian Cancer Models. Mol. Cancer Ther. 2012, 11, 1026–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgovan, T.; Bellistri, J.P.S.; Slack, K.N.; Kopelovich, L.; Desai, M.; Joe, A.K. Inhibition of BCL2 expression and activity increases H460 sensitivity to the growth inhibitory effects of polyphenon E. J. Exp. Ther. Oncol. 2009, 8, 129–144. [Google Scholar]
- Bojes, H. Bcl-2 and Bcl-xLin Peroxide-Resistant A549 and U87MG Cells. Toxicol. Sci. 1998, 42, 109–116. [Google Scholar] [CrossRef]
- Amundson, S.A.; Myers, T.G.; Scudiero, D.; Kitada, S.; Reed, J.C.; Fornace, A.J. An Informatics Approach Identifying Markers of Chemosensitivity in Human Cancer Cell Lines. Cancer Res. 2000, 60, 6101–6110. [Google Scholar] [PubMed]
- Nor Hisam, N.S.; Ugusman, A.; Rajab, N.F.; Ahmad, M.F.; Fenech, M.; Liew, S.L.; Mohamad Anuar, N.N. Combination Therapy of Navitoclax with Chemotherapeutic Agents in Solid Tumors and Blood Cancer: A Review of Current Evidence. Pharmaceutics 2021, 13, 1353. [Google Scholar] [CrossRef]
- de Vos, S.; Leonard, J.P.; Friedberg, J.W.; Zain, J.; Dunleavy, K.; Humerickhouse, R.; Hayslip, J.; Pesko, J.; Wilson, W.H. Safety and efficacy of navitoclax, a BCL-2 and BCL-X L inhibitor, in patients with relapsed or refractory lymphoid malignancies: Results from a phase 2a study. Leuk. Lymphoma 2021, 62, 810–818. [Google Scholar] [CrossRef]
- Mross, K.; Dittrich, C.; Aulitzky, W.E.; Strumberg, D.; Schutte, J.; Schmid, R.M.; Hollerbach, S.; Merger, M.; Munzert, G.; Fleischer, F.; et al. A randomised phase II trial of the Polo-like kinase inhibitor BI 2536 in chemo-naïve patients with unresectable exocrine adenocarcinoma of the pancreas—A study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br. J. Cancer 2012, 107, 280–286. [Google Scholar] [CrossRef] [Green Version]
- Yim, H. Current clinical trials with polo-like kinase 1 inhibitors in solid tumors. Anticancer Drugs 2013, 24, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Bousbaa, H. Novel Anticancer Strategies. Pharmaceutics 2021, 13, 275. [Google Scholar] [CrossRef] [PubMed]
- Mathews Griner, L.A.; Zhang, X.; Guha, R.; McKnight, C.; Goldlust, I.S.; Lal-Nag, M.; Wilson, K.; Michael, S.; Titus, S.; Shinn, P.; et al. Large-scale pharmacological profiling of 3D tumor models of cancer cells. Cell Death Dis. 2016, 7, e2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2016, 10, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, B.; Novais, P.; Henriques, A.C.; Carvalho-Tavares, J.; Silva, P.M.A.; Bousbaa, H. Navitoclax Enhances the Therapeutic Effects of PLK1 Targeting on Lung Cancer Cells in 2D and 3D Culture Systems. Pharmaceutics 2022, 14, 1209. https://doi.org/10.3390/pharmaceutics14061209
Pinto B, Novais P, Henriques AC, Carvalho-Tavares J, Silva PMA, Bousbaa H. Navitoclax Enhances the Therapeutic Effects of PLK1 Targeting on Lung Cancer Cells in 2D and 3D Culture Systems. Pharmaceutics. 2022; 14(6):1209. https://doi.org/10.3390/pharmaceutics14061209
Chicago/Turabian StylePinto, Bárbara, Pedro Novais, Ana C. Henriques, Juliana Carvalho-Tavares, Patrícia M. A. Silva, and Hassan Bousbaa. 2022. "Navitoclax Enhances the Therapeutic Effects of PLK1 Targeting on Lung Cancer Cells in 2D and 3D Culture Systems" Pharmaceutics 14, no. 6: 1209. https://doi.org/10.3390/pharmaceutics14061209