
Fast In Vitro Release and In Vivo Absorption of an Anti-Schizophrenic Drug Paliperidone from Its Soluplus®/TPGS Mixed Micelles

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Preparation of the Mixed Micelles
2.4. Characterization of the Mixed Micelles
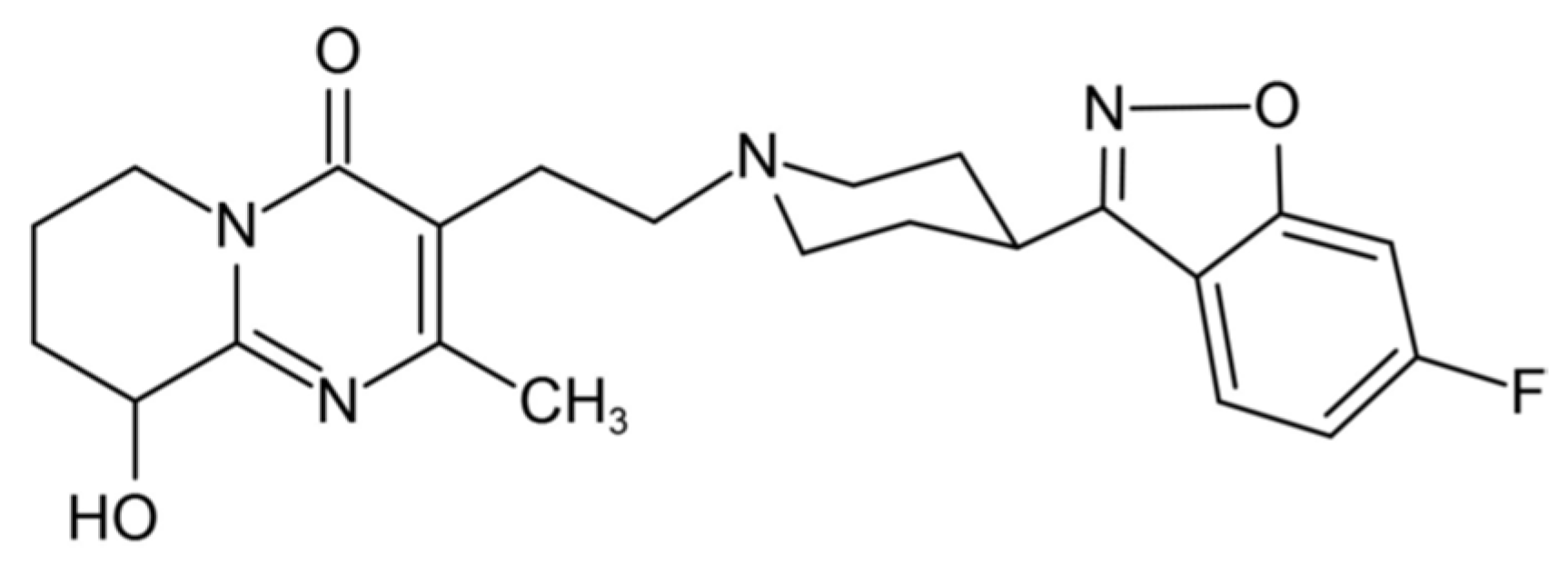
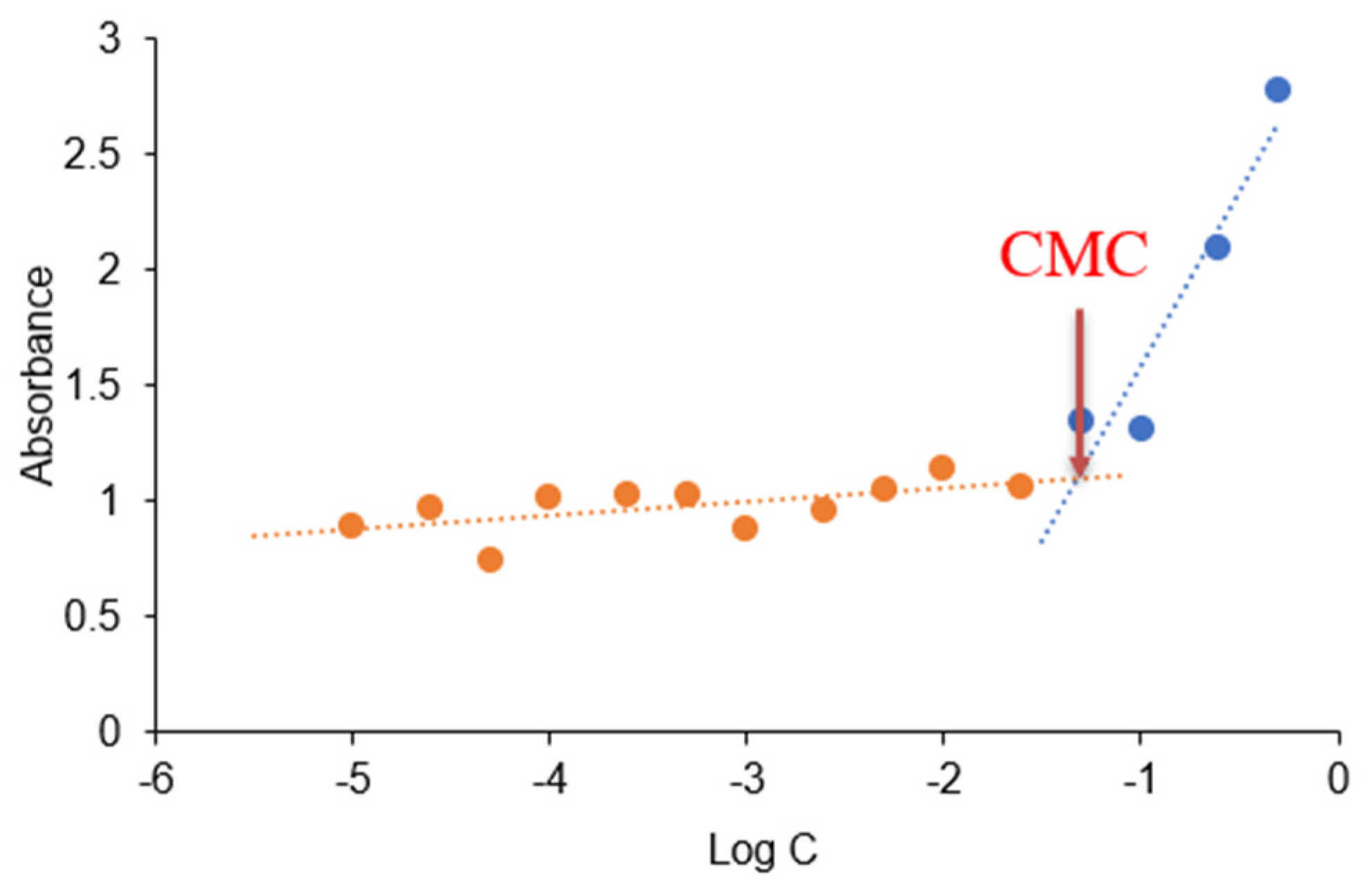
2.4.1. Critical Micelle Concentration (CMC)
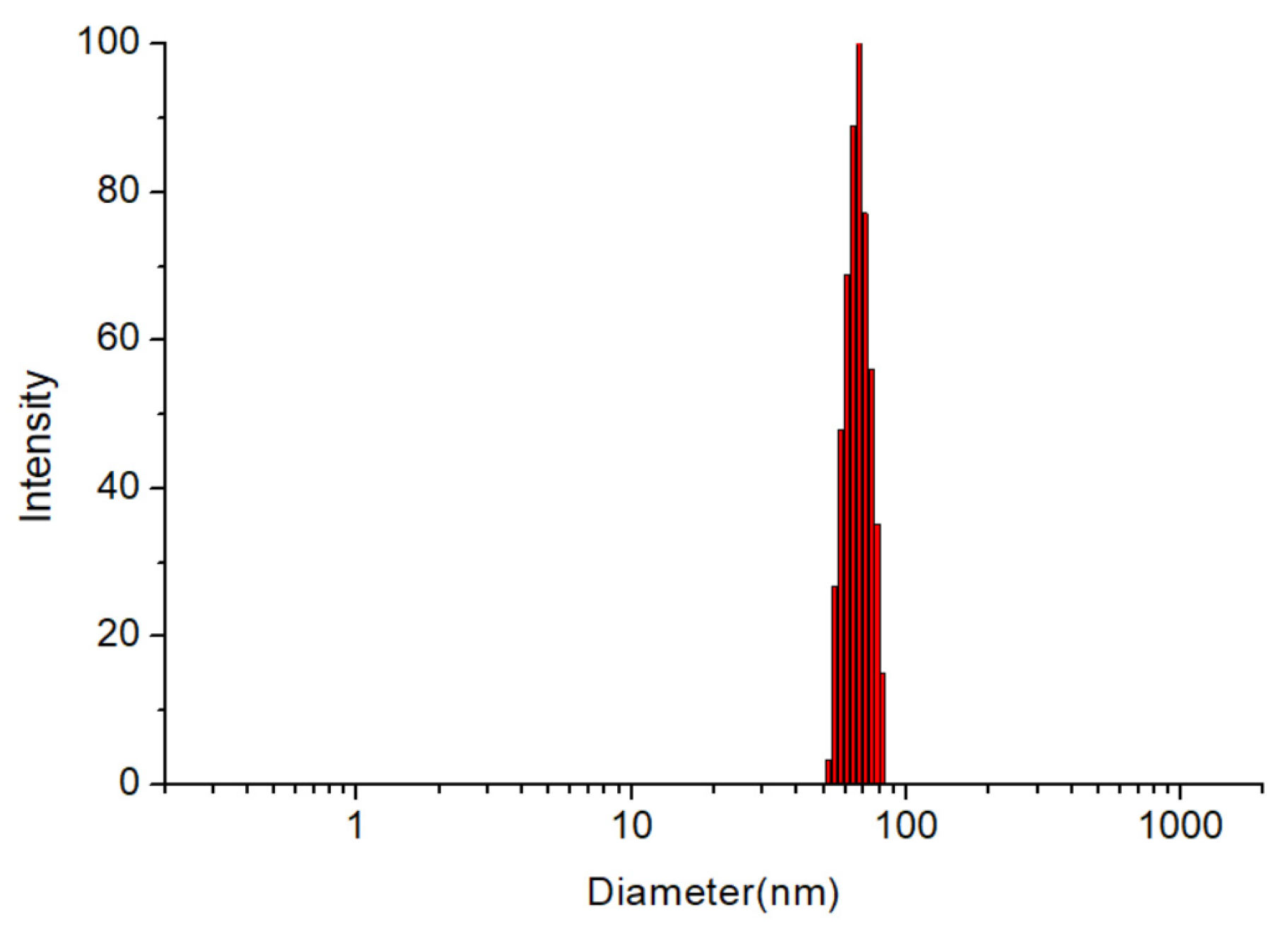
2.4.2. Particle Size, Polydispersity Index (PDI) and Zeta Potential Analysis
2.4.3. Encapsulation Efficiency and Drug Loading Efficiency
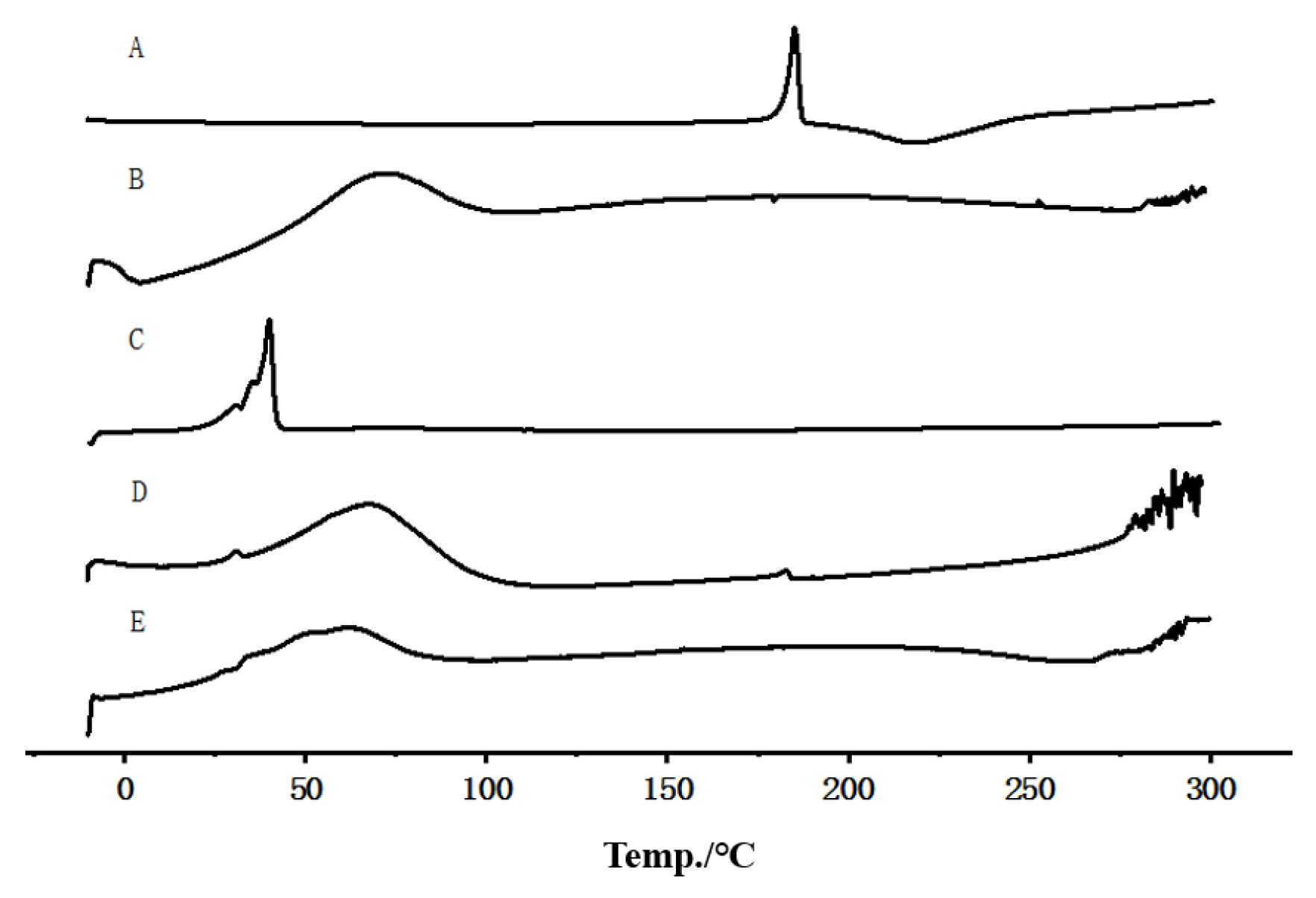
2.4.4. Differential Scanning Calorimetry (DSC)
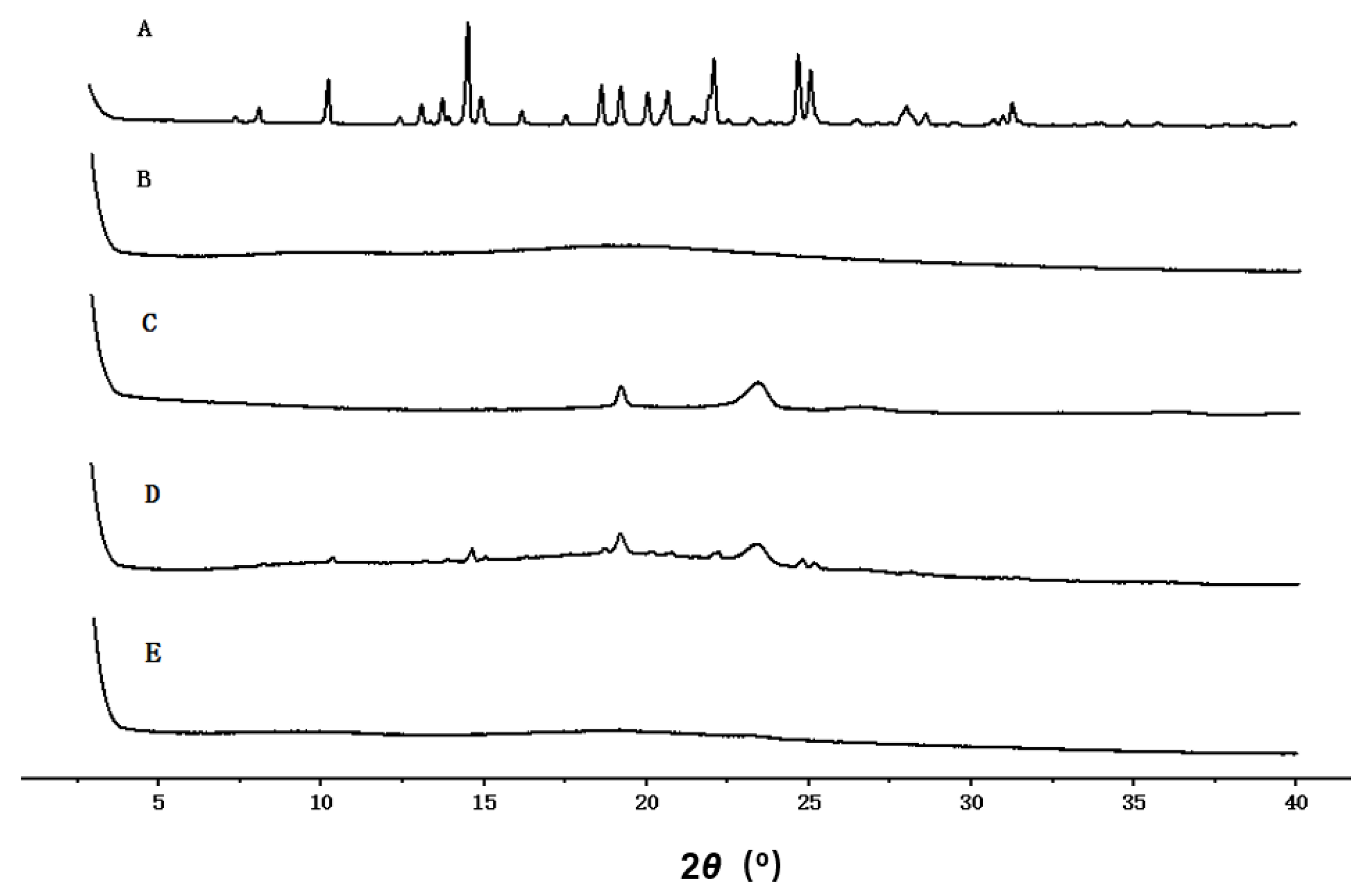
2.4.5. X-ray Diffraction (XRD)
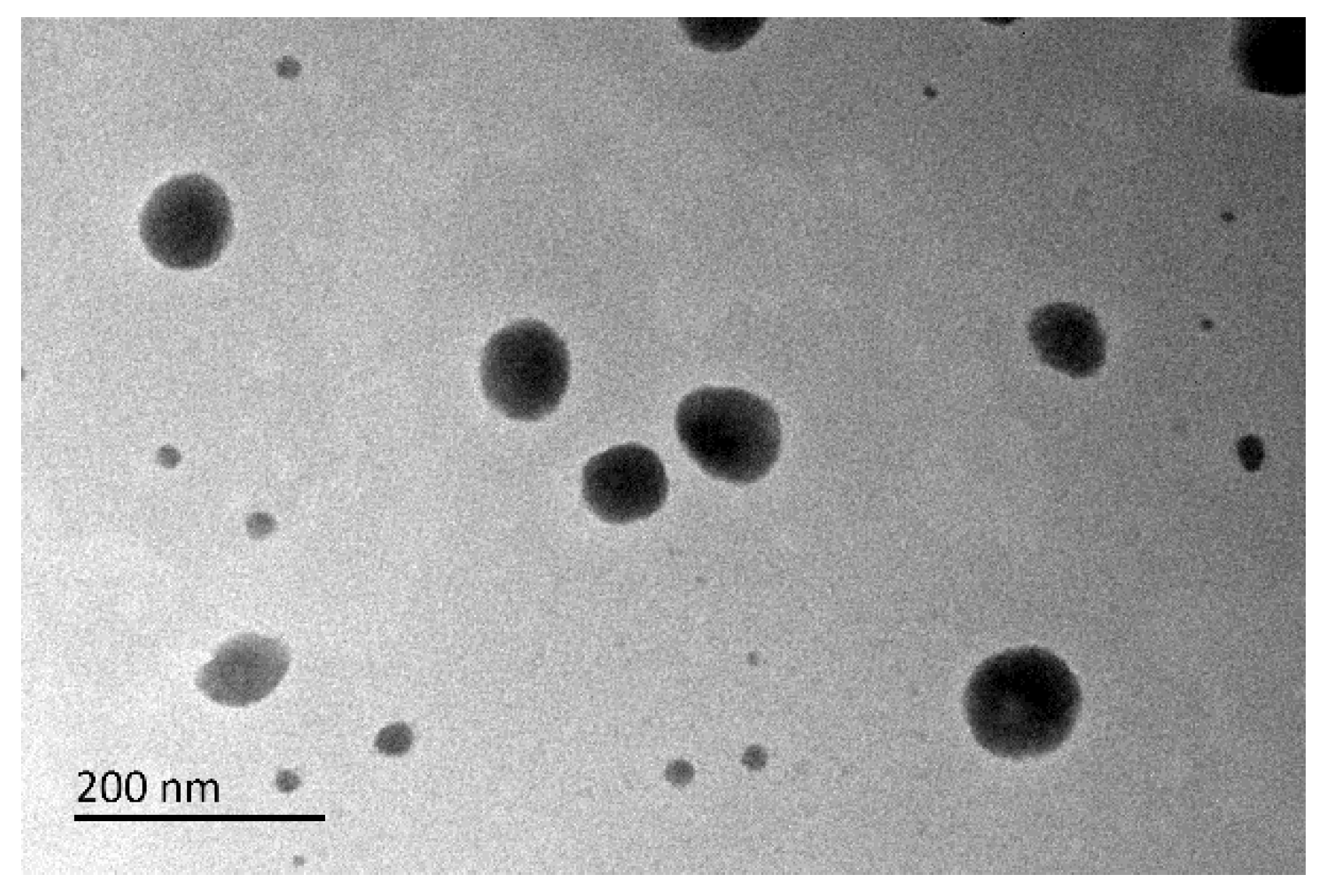
2.4.6. Transmission Electron Microscopy (TEM)
2.5. Stability Studies
2.5.1. Dilution Stability
2.5.2. pH Stability
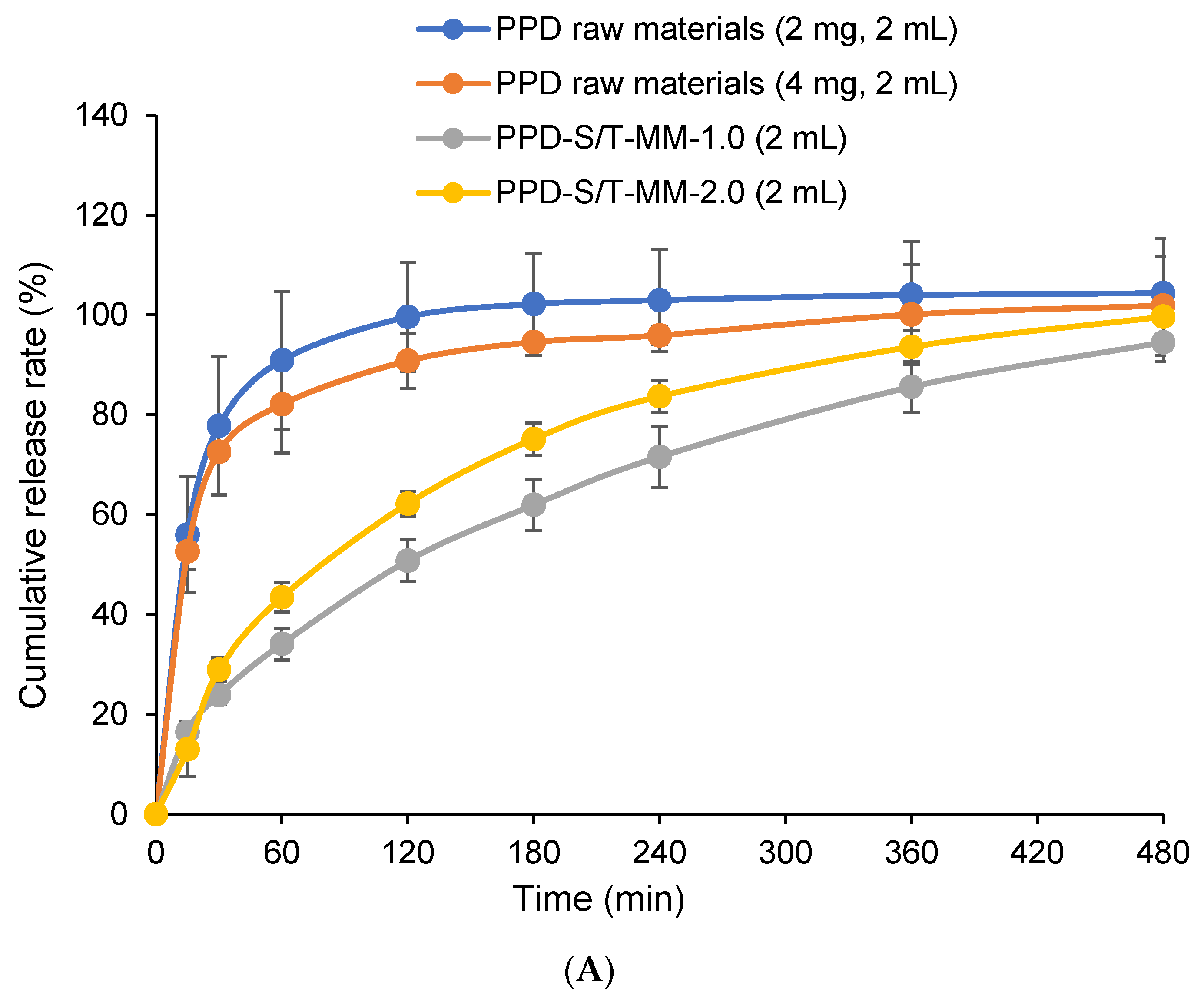
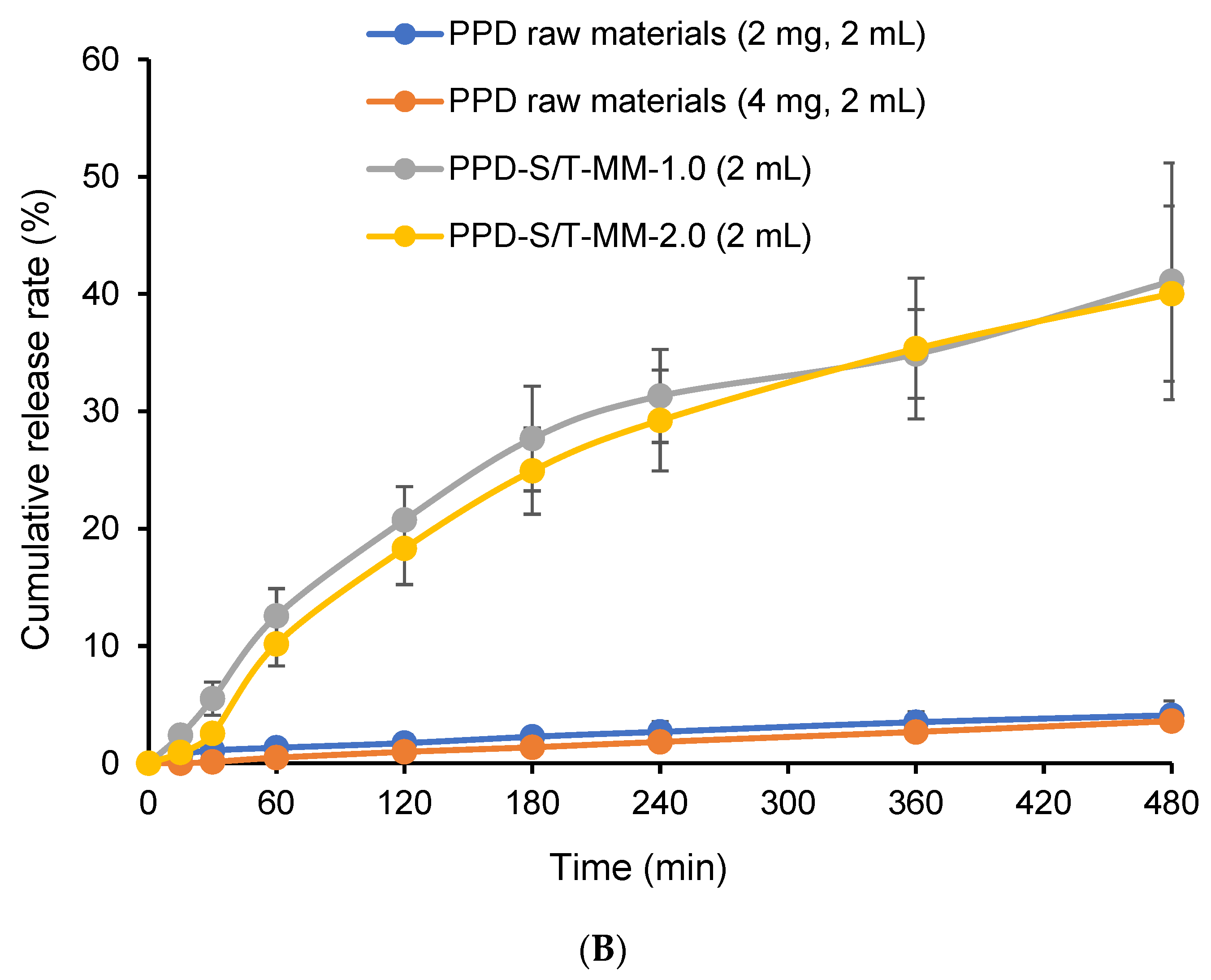
2.6. In Vitro Drug Release
2.7. Pharmacokinetic Study
2.8. Quantification of PPD
2.9. Statistical Analysis
3. Results and Discussion
3.1. CMC Determination
3.2. Particle Size, PDI and Zeta Potential Analysis
3.3. Encapsulation Efficiency and Drug Loading Capacity
3.4. DSC
3.5. XRD
3.6. TEM
3.7. Stability Studies
3.7.1. Dilution Stability
3.7.2. pH Stability
3.8. In Vitro Drug Release
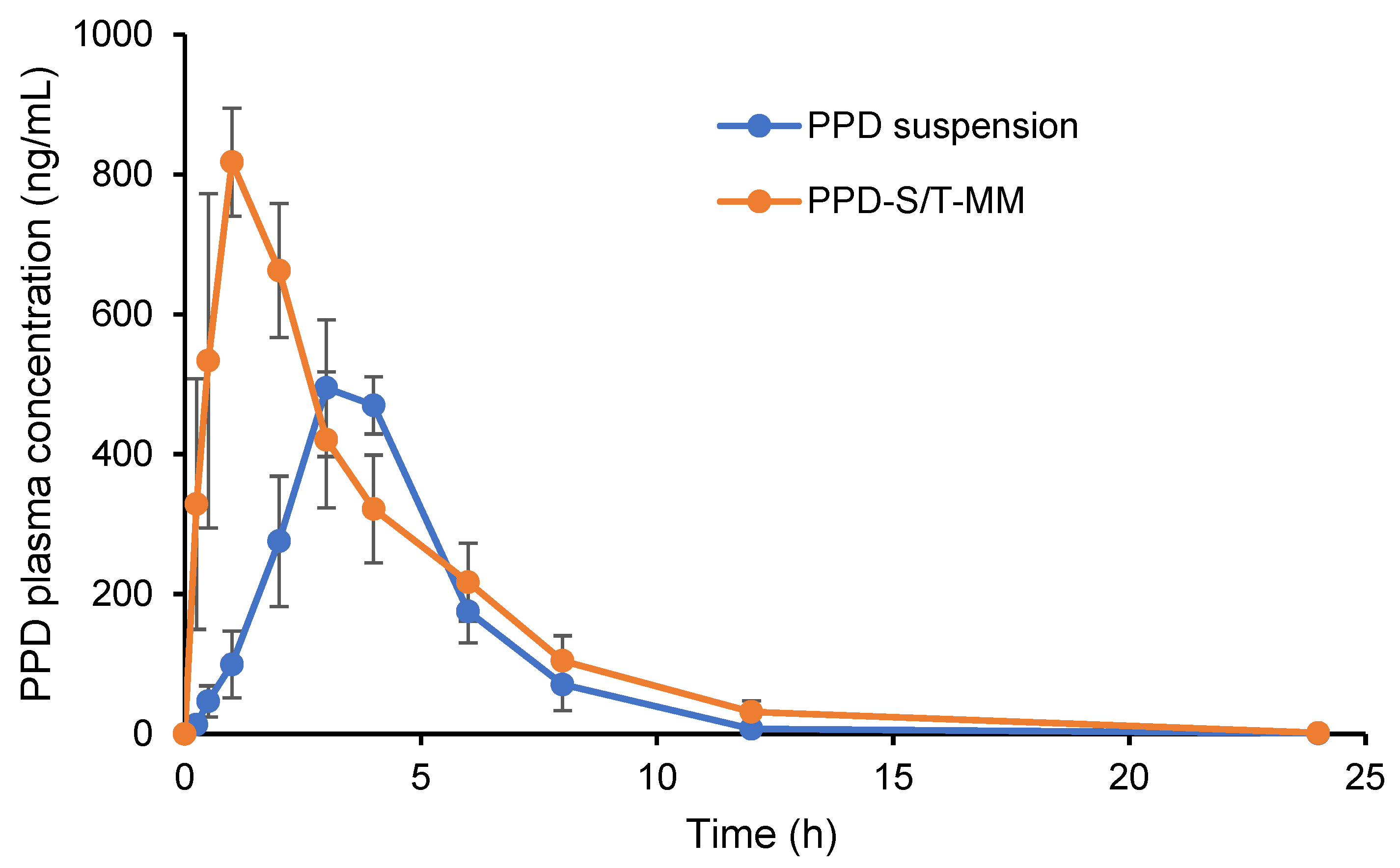
3.9. Pharmacokinetic Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Schizophrenia. Available online: https://www.who.int/news-room/fact-sheets/detail/schizophrenia (accessed on 31 March 2022).
- Xiang, Y.T.; Weng, Y.Z.; Leung, C.M.; Tang, W.K.; Ungvari, G.S. Subjective quality of life in outpatients with schizophrenia in Hong Kong and Beijing: Relationship to socio-demographic and clinical factors. Qual. Life Res. 2008, 17, 27–36. [Google Scholar] [CrossRef]
- Hatta, K. Practical pharmacotherapy for acute schizophrenia patients. Psychiatry Clin. Neurosci. 2015, 69, 674–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Välimäki, M.; Lantta, T.; Hätönen, H.M.; Kontio, R.; Zhang, S.Y. Risk assessment for aggressive behaviour in schizophrenia. Cochrane Database Syst. Rev. 2016, 2016, CD012397. [Google Scholar] [CrossRef]
- Falkai, P.; Vogeley, K. The chances of new atypical substances. Fortschr. Neurol. Psychiatr. 2000, 68, S32–S37. [Google Scholar]
- Salarvand, M.; Ramezani, V.; Salarvand, F.; Darabi, Z.A.; Akrami, M. Improvement of drug delivery properties of risperidone via preparation of fast dissolution tablet containing nanostructured microparticles. Iran. J. Pharm. Res. 2021, 20, 183–196. [Google Scholar]
- Al-Dhubiab, B.E. Aripiprazole nanocrystal impregnated buccoadhesive films for schizophrenia. J. Nanosci. Nanotechnol. 2017, 17, 2345–2352. [Google Scholar] [CrossRef]
- Pidaparthi, K.; Suares, D. Comparison of nanoemulsion and aqueous micelle systems of paliperidone for intranasal delivery. AAPS Pharm. Sci. Tech. 2017, 18, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Sobczyński, J.; Chudzik-Rząd, B. Mixed micelles as drug delivery nanocarriers. In Design and Development of New Nanocarriers, 1st ed.; Grumezescu, A.M., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 331–364. [Google Scholar]
- Lasic, D.D. Mixed micelles in drug delivery. Nature 1992, 355, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ling, P.X.; Zhang, T.M. Polymeric micelles, a promising drug delivery system to enhance bioavailability of poorly water-soluble drugs. J. Drug Deliv. 2013, 2013, 340315. [Google Scholar] [CrossRef]
- USA Food and Drug Administration (FDA). Approval Package for NDA application of Invega Extended Release. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/021999Orig1s004.pdf (accessed on 31 March 2022).
- Janicak, P.G.; Winans, E.A. Paliperidone ER: A review of the clinical trial data. Neuropsychiatr. Dis. Treat. 2007, 3, 869–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.Y.; Teng, H.; Shi, Y.N.; He, H.B.; Zhang, Y.; Yin, T.; Cai, C.F.; Tang, X. Tablets of paliperidone using compression-coated technology for controlled ascending release. Asian J. Pharm. Sci. 2018, 13, 143–154. [Google Scholar] [CrossRef]
- Piazzini, V.; Landucci, E.; Urru, M.; Chiarugi, A.; Pellegrini Giampietro, D.E.; Bilia, A.R.; Bergonzi, M.C. Enhanced dissolution, permeation and oral bioavailability of aripiprazole mixed micelles: In vitro and in vivo evaluation. Int. J. Pharm. 2020, 583, 119361. [Google Scholar] [CrossRef]
- Ji, S.P.; Lin, X.; Yu, E.J.; Dian, C.Y.; Yan, X.; Li, L.Y.; Zhang, M.M.; Zhao, W.C.; Dian, L.H. Curcumin-loaded mixed micelles: Preparation, characterization, and in vitro antitumor activity. J. Nanotechnol. 2018, 2018, 9103120. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Zhang, J.J.; Ding, R.; Fu, Y.; Gong, T.; Zhang, Z.R. Improved oral bioavailability and therapeutic efficacy of dabigatran etexilate via Soluplus-TPGS binary mixed micelles system. Drug Dev. Ind. Pharm. 2017, 43, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Sherje, A.; Londhe, V. Inclusion complexes of hydroxy propyl-β-cyclodextrin and paliperidone: Preparation and characterization. Curr. Drug Discov. Technol. 2014, 11, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Ding, Y.F.; Xu, Y.W.; Wang, C.Y.; Ding, Y.Y.; Gao, M.; Ma, C.G.; Ma, X.D.; Li, L. Mixed micelles of TPGS and Soluplus® for co-delivery of paclitaxel and fenretinide: In vitro and in vivo anticancer study. Pharm. Dev. Technol. 2020, 25, 865–873. [Google Scholar] [CrossRef] [PubMed]
- USA Food and Drug Administration (FDA). Dissolution Methods. Paliperidone. Available online: https://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_SearchResults.cfm (accessed on 31 March 2022).
- Giri, K.; Lau, M.; Kuschnerus, I.; Moroni, I.; Garcia-Bennett, A.E. A lysozyme corona complex for the controlled pharmacokinetic release of probucol from mesoporous silica particles. Biomater. Sci. 2020, 8, 3800–3803. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Cui, Y.N.; Li, Y.M.; Li, L.B. Stable phosphatidylcholine-bile salt mixed micelles enhance oral absorption of paclitaxel: Preparation and mechanism in rats. J. Drug Target. 2014, 22, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.X.; He, D.D.; Wang, S.X.; Ding, P.G.; Wang, J.N.; Ju, J.M. Preparation, characterization, and pharmacokinetics study of a novel genistein-loaded mixed micelles system. Drug Dev. Ind. Pharm. 2018, 44, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Aravagiri, M.; Marder, S.R. Brain, plasma and tissue pharmacokinetics of risperidone and 9-hydroxyrisperidone after separate oral administration to rats. Psychopharmacology 2002, 159, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Bindu, K.H.; Reddy, I.U.; Anjaneyulu, Y.; Suryanarayana, M.V. A stability-indicating ultra-performance liquid chromatographic method for estimation of related substances and degradants in paliperidone active pharmaceutical ingredient and its pharmaceutical dosage forms. J. Chromatogr. Sci. 2012, 50, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Stopková, L.; Gališinová, J.; Šuchtová, Z.; Čižmárik, J.; Andriamainty, F. Determination of critical micellar concentration of homologous 2-alkoxyphenylcarbamoyloxyethyl-morpholinium chlorides. Molecules 2018, 23, 1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BASF Corporation. Technical Information—Soluplus®. Available online: https://pharma.basf.com/technicalinformation/30446233/soluplus (accessed on 31 March 2022).
- Bernabeu, E.; Gonzalez, L.; Cagel, M.; Gergic, E.P.; Moretton, M.A.; Chiappetta, D.A. Novel Soluplus®—TPGS mixed micelles for encapsulation of paclitaxel with enhanced in vitro cytotoxicity on breast and ovarian cancer cell lines. Colloids Surf. B Biointerfaces 2016, 140, 403–411. [Google Scholar] [CrossRef]
- Alopaeus, J.F.; Hagesæther, E.; Tho, I. Micellisation mechanism and behaviour of Soluplus®–furosemide micelles: Preformulation studies of an oral nanocarrier-based system. Psychopharmacology 2019, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Gaucher, G.; Satturwar, P.; Jones, M.C.; Furtos, A.; Leroux, J.C. Polymeric micelles for oral drug delivery. Eur. J. Pharm. Biopharm. 2010, 76, 147–158. [Google Scholar] [CrossRef]
- Jones, M.C.; Ranger, M.; Leroux, J.C. pH-sensitive unimolecular polymeric micelles: Synthesis of a novel drug carrier. Bioconjug. Chem. 2003, 14, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, Y.; Li, L.; Zhang, Y.; Zhang, L.; Zhang, Z. Preparation, evaluation and metabolites study in rats of novel amentoflavone-loaded TPGS/soluplus mixed nanomicelles. Drug Deliv. 2020, 27, 137–150. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, Y.W.; Wang, C.Y.; Ding, Y.F.; Chen, M.Y.; Wang, Y.F.; Peng, J.Y.; Li, L.; Lv, L. Soluplus/TPGS mixed micelles for dioscin delivery in cancer therapy. Drug Dev. Ind. Pharm. 2017, 43, 1197–1204. [Google Scholar] [CrossRef]
- Ding, Y.Y.; Wang, C.Y.; Wang, Y.T.; Xu, Y.W.; Zhao, J.; Gao, M.; Ding, Y.F.; Peng, J.Y.; Li, L. Development and evaluation of a novel drug delivery: Soluplus®/TPGS mixed micelles loaded with piperine in vitro and in vivo. Drug Dev. Ind. Pharm. 2018, 44, 1409–1416. [Google Scholar] [CrossRef]
- USA Food and Drug Administration (FDA). Prescribing Information for INVEGA® (Paliperidone) Extended-Release Tablets. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021999s018lbl.pdf (accessed on 31 March 2022).
- Kumar, S.; Randhawa, J.K. Preparation and characterization of paliperidone loaded solid lipid nanoparticles. Colloids Surf. B Biointerfaces. 2013, 102, 562–568. [Google Scholar] [CrossRef]
- Altamimi, M.A.; Neau, S.H. Investigation of the in vitro performance difference of drug-Soluplus® and drug-PEG 6000 dispersions when prepared using spray drying or lyophilization. Saudi Pharm. J. 2017, 25, 419–439. [Google Scholar] [CrossRef] [Green Version]
- Wijiani, N.; Isadiartuti, D.; Rijal, M.A.S.; Yusuf, H. Characterization and dissolution study of micellar curcumin-spray dried powder for oral delivery. Int. J. Nanomed. 2020, 15, 1787–1796. [Google Scholar] [CrossRef] [Green Version]
- Hattori, Y.; Haruna, Y.; Otsuka, M. Dissolution process analysis using model-free Noyes-Whitney integral equation. Colloids Surf. B Biointerfaces 2013, 102, 227–231. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, E.; Yang, J.H.; Cao, Z.Q. Strategies to improve micelle stability for drug delivery. Nano Res. 2018, 11, 4985–4998. [Google Scholar] [CrossRef]
- Wang, J.; Ma, W.; Tu, P. The mechanism of self-assembled mixed micelles in improving curcumin oral absorption: In vitro and in vivo. Colloids Surf. B Biointerfaces. 2015, 133, 108–119. [Google Scholar] [CrossRef]
- Zhong, Y.; Jing, G.H.; Tian, B.; Huang, H.; Zhang, Y.Y.; Gou, J.X.; Tang, X.; He, H.B.; Wang, Y.J. Supersaturation induced by itraconazole/Soluplus® micelles provided high GI absorption in vivo. Asian J. Pharm. Sci. 2016, 11, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Collnot, E.M.; Baldes, C.; Wempe, M.F.; Kappl, R.; Hüttermann, J.; Hyatt, J.A.; Edgar, K.J.; Schaefer, U.F.; Lehr, C.M. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: Influence on ATPase activity and membrane fluidity. Mol. Pharm. 2007, 4, 465–474. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brand Name | Active Ingredient | Dosage Form | Route of Administration | Strength | Manufacturer |
|---|---|---|---|---|---|
| Invega® | Paliperidone | Extended-release tablets | Oral administration | 1.5 mg, 3 mg, 6 mg, 9 mg | JANSSEN PHARMS |
| Invega Sustenna® | Paliperidone palmitate | Long-acting injectables | Intramuscular injection | 156 mg/mL | JANSSEN PHARMS |
| Invega Trinza® | Paliperidone palmitate | Long-acting injectables | Intramuscular injection | 312 mg/mL | JANSSEN PHARMS |
| Types of the Mixed Micelles | Particle Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|
| Blank S/T-MM | 61.3 ± 0.3 | 0.06 ± 0.02 | −(1.65 ± 1.97) |
| PPD-S/T-MM-0.4 | 61.5 ± 0.7 | 0.05 ± 0.01 | −(1.73 ± 4.63) |
| PPD-S/T-MM-1.0 | 61.3 ± 0.8 | 0.05 ± 0.01 | −(2.89 ± 2.50) |
| PPD-S/T-MM-2.0 | 61.8 ± 0.9 | 0.04 ± 0.00 | −(4.57 ± 1.64) |
| Types of the Mixed Micelles | |||
|---|---|---|---|
| PPD-S/T-MM-0.4 | PPD-S/T-MM-1.0 | PPD-S/T-MM-2.0 | |
| DL (%) | 0.78 ± 0.05 | 1.92 ± 0.01 | 3.69 ± 0.72 |
| EE (%) | 98.29 ± 1.67 | 97.41 ± 0.66 | 94.69 ± 2.19 |
| Types of the Mixed Micelles | Dilution | Particle Size (nm) | PDI |
|---|---|---|---|
| PPD-S/T-MM-1.0 | 5-fold | 61.34 ± 0.76 | 0.05 ± 0.01 |
| 50-fold | 62.44 ± 0.48 | 0.06 ± 0.01 | |
| 100-fold | 62.89 ± 0.22 | 0.06 ± 0.02 | |
| PPD-S/T-MM-2.0 | 5-fold | 61.77 ± 0.88 | 0.04 ± 0.00 |
| 50-fold | 61.12 ± 0.31 | 0.03 ± 0.02 | |
| 100-fold | 62.93 ± 0.39 | 0.02 ± 0.01 |
| Types of The Mixed Micelles | Duration of Exposure in SGF (h) | Duration of Exposure in SIF (h) | ||||
|---|---|---|---|---|---|---|
| 0 | 2 | 4 | 0 | 2 | 4 | |
| PPD-S/T-MM-1.0 | 61.2 ± 0.8 | 60.8 ± 1.5 | 58.3 ± 2.2 | 65.7 ± 0.8 | 64.2 ± 1.1 | 63.5 ± 0.4 |
| PPD-S/T-MM-2.0 | 64.2 ± 1.1 | 62.7 ± 2.7 | 57.4 ± 1.4 | 67.2 ± 0.7 | 64.5 ± 1.1 | 63.6 ± 2.1 |
| Parameters | PPD-S/T-MM | PPD Suspension |
|---|---|---|
| Tmax (h) | 0.83 ± 0.29 * | 3.33 ± 0.58 |
| Cmax (ng/mL) | 844.33 ± 93.73 * | 531.33 ± 77.10 |
| T1/2 (h) | 2.47 ± 0.20 | 2.08 ± 0.16 |
| AUC0→24 h (ng/mL·h) | 3467.33 ± 705.33 * | 2191.88 ± 607.99 |
| AUC0→∞ (ng/mL·h) | 3472.29 ± 709.18 * | 2193.95 ± 609.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Wang, C.; Liu, W.; Yang, M.; Xu, B.; Chen, Y. Fast In Vitro Release and In Vivo Absorption of an Anti-Schizophrenic Drug Paliperidone from Its Soluplus®/TPGS Mixed Micelles. Pharmaceutics 2022, 14, 889. https://doi.org/10.3390/pharmaceutics14050889
Zhou Y, Wang C, Liu W, Yang M, Xu B, Chen Y. Fast In Vitro Release and In Vivo Absorption of an Anti-Schizophrenic Drug Paliperidone from Its Soluplus®/TPGS Mixed Micelles. Pharmaceutics. 2022; 14(5):889. https://doi.org/10.3390/pharmaceutics14050889
Chicago/Turabian StyleZhou, Ye, Chenhui Wang, Wenqian Liu, Meiqing Yang, Bohui Xu, and Yong Chen. 2022. "Fast In Vitro Release and In Vivo Absorption of an Anti-Schizophrenic Drug Paliperidone from Its Soluplus®/TPGS Mixed Micelles" Pharmaceutics 14, no. 5: 889. https://doi.org/10.3390/pharmaceutics14050889