The materials and methods are described in detail in [
1], so here we summarize only the basic facts. The in vivo experiment consisted in the administration of sulfathiazole suspensions with and without tenside (MLS) to rats. The drug without tenside was administered first and the corresponding series of concentration samples was recorded. Subsequently, the same process was repeated, but with added tenside. Based on the obtained concentrations, a parsimonious structure of the two-compartment pharmacokinetic model was designed. Subsequently, this model was analyzed for its parametric identifiability and then, finally, identified. The methodology used in [
1] can be applied equally well to all oral dosage forms, including those with controlled release and even sophisticated drug delivery systems (DDS).
2.1. Need for a State-Bounding Observer
In the treatment of various diseases, physicians need to know the drug concentrations in the relevant organs (compartments) of the body. However, since some of these cannot be measured, they have to be estimated by a special system: the observer.
The observer is a numeric algorithm that “observes” the measured drug amount in a chosen compartment and generates an estimate of the drug amount in the remaining compartments. From the system’s point of view, the state-bounding observer is an additional tool processing the output of the pharmacokinetic model.
For all the compartments, two independent observers, each producing a limiting curve, are designed. The limiting curves delineate the area from which the drug concentration cannot “escape”, even if the values of the pharmacokinetic parameters are estimated with limited accuracy. Resolving the state estimation problem is especially important for compartments that are not available (for technical, medical, or ethical reasons) through the collection of blood samples. These involve, for instance, the gastrointestinal tract (GIT) and the veins.
Each observer is driven by the instantaneous drug concentration in the output compartment of the pharmacokinetic model, i.e., the compartment from which the blood samples are collected. This output compartment is commonly chosen as the most suitable for sample collection.
The derivation of state-bounding observers for typical bio-systems is hampered by the fact that characteristic quantities must take exclusively non-negative values. These, involve, for example, the drug concentration, drug amount, blood pressure, body temperature, etc. These types of system belong to the category of “positive systems”, of which compartmental pharmacokinetic models are a subset [
6].
The requirement for system positivity makes the derivation of equations describing “positive” state-bounding observers rather a demanding problem. For this reason, any mathematical rigor is omitted, so only the equations that are essential to understanding the main ideas are presented. For interested readers, we reveal that the derivations are commonly based on the theory of linear programming (LP) or linear matrix inequalities (LMI) [
7,
8]. The approach adopted in this paper follows the theory presented in [
9,
10,
11,
12,
13,
14,
15].
2.2. Uncertain Compartmental Model
Perhaps the most difficult problem one may encounter while trying to predict the drug concentrations in body compartments is the lack of an exact and reliable pharmacokinetic model. Pharmacists know that the model structure is typically chosen on the basis of the properties of the drug at hand, the method of administration, the required level of model comprehensiveness, and last, but not least, the experience of the researcher. Clearly, the structure must be as parsimonious as possible, while its in silico behavior should reliably reflect the observations of real processes.
This paper presents two uncertain compartmental pharmacokinetic models. The first is intended to explain how an uncertain model can be derived. The second is proposed to demonstrate a more practical application of the observer’s prediction of whether the drug concentration can potentially extend beyond the therapeutic range if there is a mismatch between the living body and its model. It should be mentioned that the first model was already identified in our recent work [
1], whereas the second proposed model is identified in this paper.
There are numerous reasons why the parameters of pharmacokinetic models cannot be determined exactly and with high confidence. First, there is a relatively small number of blood samples that can be collected compared to the number of parameters that can be identified, which makes the identification problem underdetermined and poorly conditioned. In addition, the sensors feature limited accuracy, the numerical computations feature rounding errors, the conditions in which the collection of samples is carried out are variable, etc. These factors cannot be simply neglected, so parametric uncertainties should be included in both the pharmacokinetic model and the state-observer algorithm.
The blood volume of rats can be easily calculated, as reported in [
16]. The administered dose of suspension in [
1] was expressed in milligrams, so the concentrations were converted to amounts. Accordingly, all further variables will be presented in terms of drug amounts rather than using their concentrations.
2.3. Nominal and Uncertain Pharmacokinetic Models
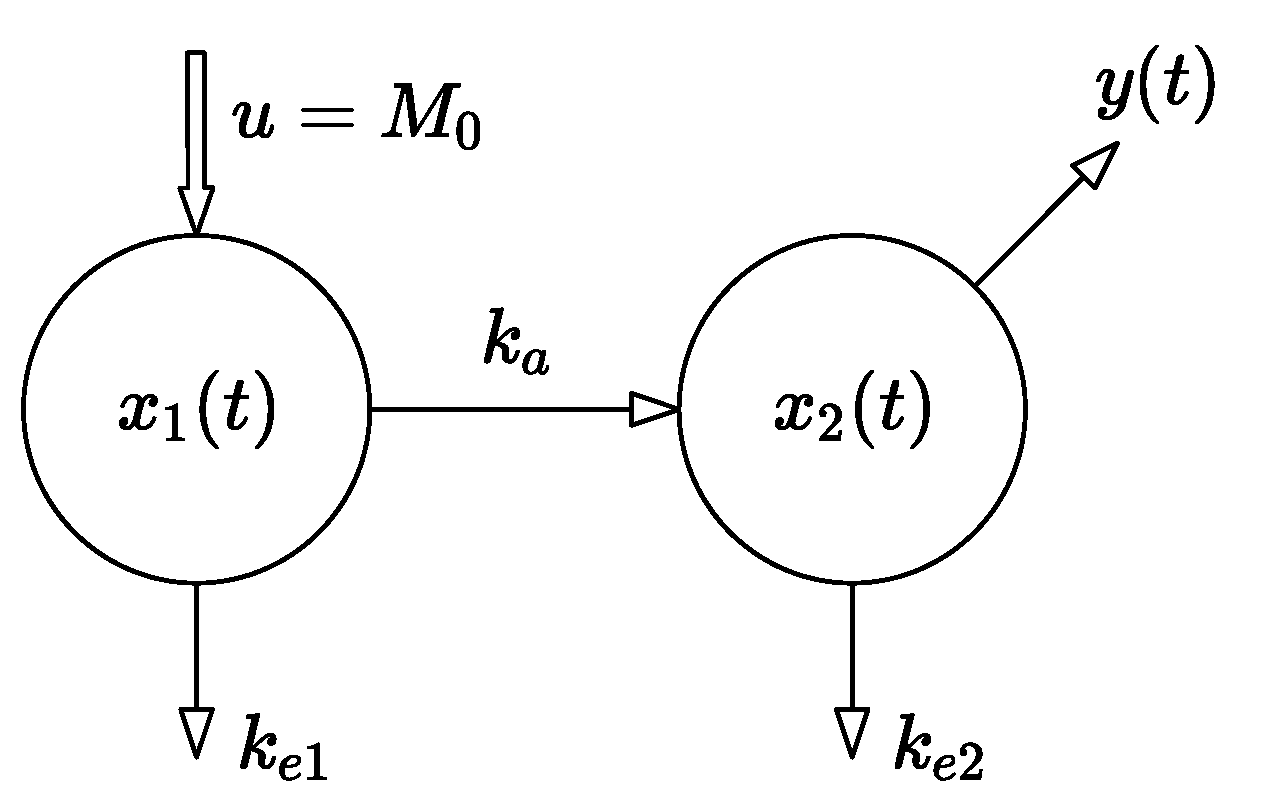
These two notions are represented by the two-compartment model with a single input and a single output (SISO) depicted in
Figure 1. The dynamics of the model are described by the set of linear differential equations (1). The state of the system represents the set of instantaneous drug amounts in the individual compartments, so the state vector
comprises
components as the number of compartments.
is the system input—an instantaneously administered dose ;
, are the rate constants of the drug elimination;
is the absorption-rate constant;
is the time-dependent drug amount in the GIT;
is the time-dependent drug amount in the central compartment—the blood;
is the system output—the observed (measurable) drug amount equal to .
The equations of the nominal compartmental model are defined as follows:
where the symbol
denotes the system matrix, i.e., the relation between the state vector
and its time derivatives, and
and
are column vectors, known as the control and the observation vectors, respectively [
5,
7]. The symbol
means the transposition operation of the column vector
; hence,
is a row vector. As mentioned above, the parameters of model (1) were identified from the results of the in vivo experiments performed on the rats. The aim of our earlier research was to analyze the effect of the tenside on the absorption-rate constant. The methods and materials used in the experiment are discussed in detail in [
1], while the crucial results of the in vivo measurements are summarized in
Table 1.
Based on these results, the following values of the model parameters were identified [
1]. Before the addition of the tenside:
After addition of the tenside:
It is obvious that the administered dose 50 mg is always known exactly, but we do not know the drug amount that enters the GIT just after the oral administration. Therefore, this value should be considered as an uncertain quantity, namely
. The model parameters are also uncertain because they are identified with finite accuracy and limited confidence. The uncertainties of the parameters are represented by the uncertain matrix
and the vector
. A general form of the uncertain pharmacokinetic model is given as follows:
where
is a fraction by which the corresponding quantity can change.
In particular, the uncertain model corresponding to (1) takes the following form:
Substituting the identified parameter values (2) into (1) yields Equation (6), which describes the nominal model:
Assuming that the parametric uncertainties do not exceed 10% of their nominal values (
, according to (4), the upper and lower matrices
,
and the vectors
,
corresponding to nominal
and
are equal to:
Let
and
represent the upper and lower limits that the drug amounts
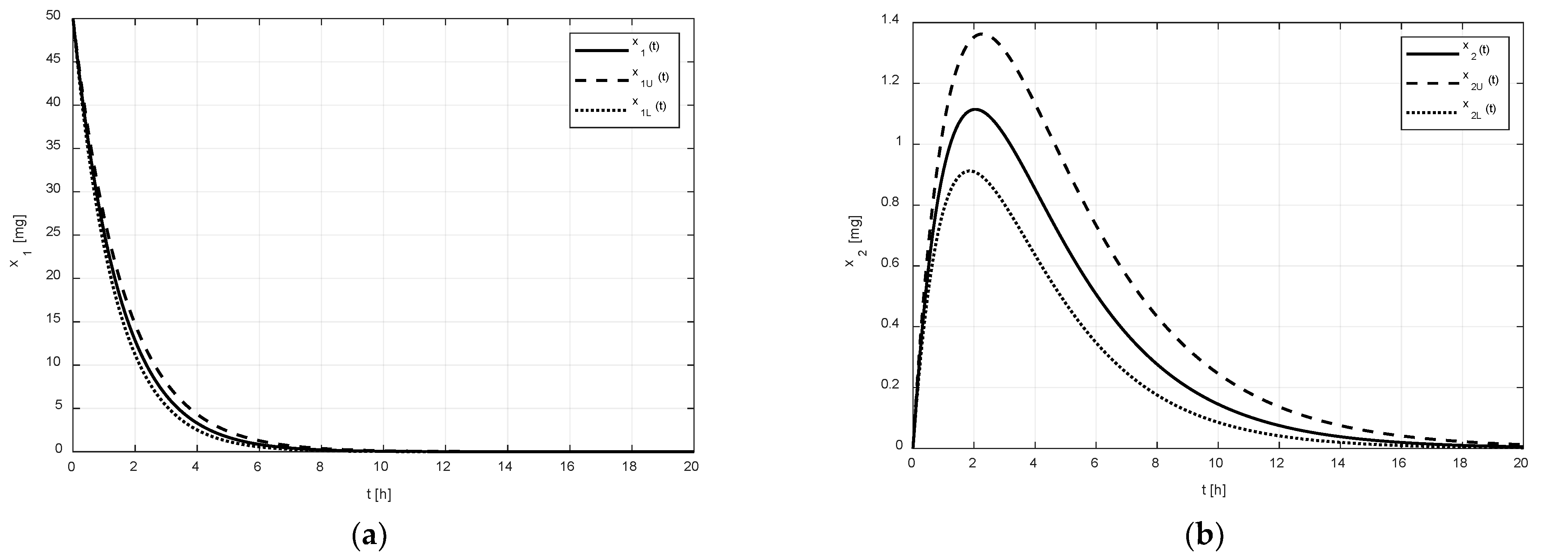
may reach due to the uncertain pharmacokinetics. The numeric solutions of Equations (1) and (5) for the nominal amount
, for the upper amount
, and for the lower amount
, corresponding to the drug without the tenside and the parametric uncertainty
, are shown in
Figure 2. The regions between the couples of curves
,
, and
,
represent possible variations in the drug amounts
and
in the corresponding compartments. As can be observed, the uncertain values of parameters (2), (3) virtually do not affect the amount
in the GIT, but the amount
in the central compartment is significantly affected.
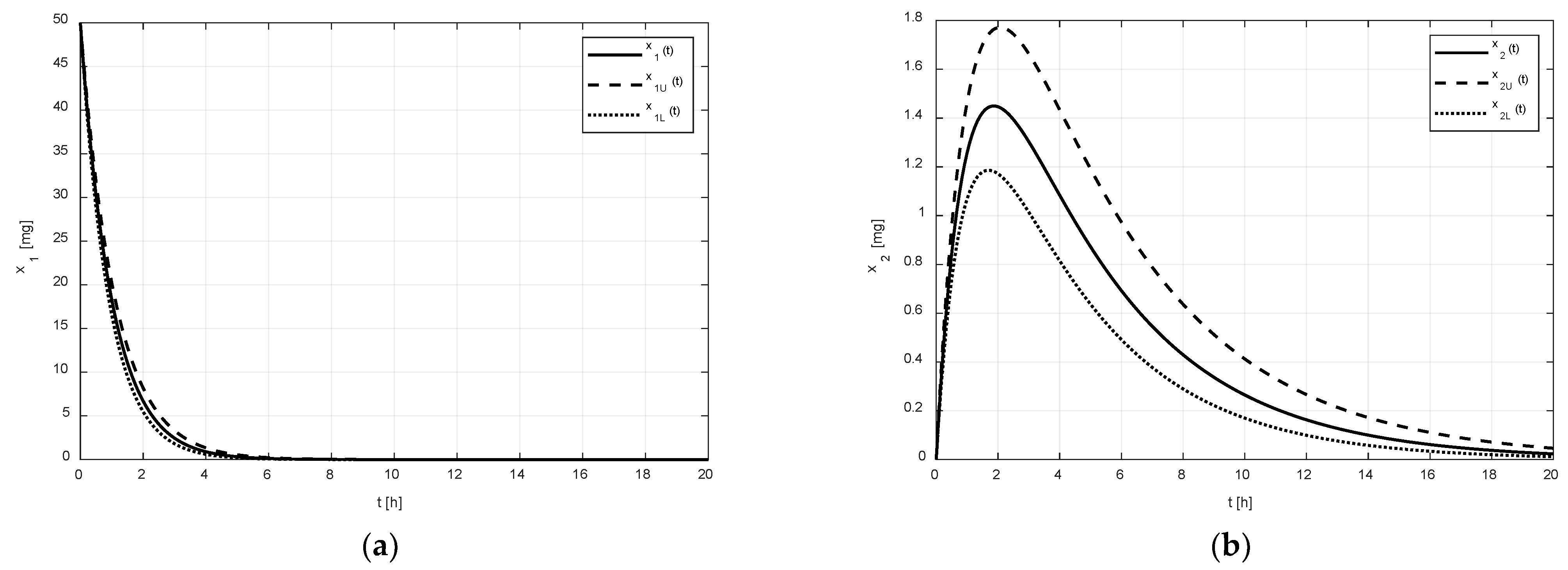
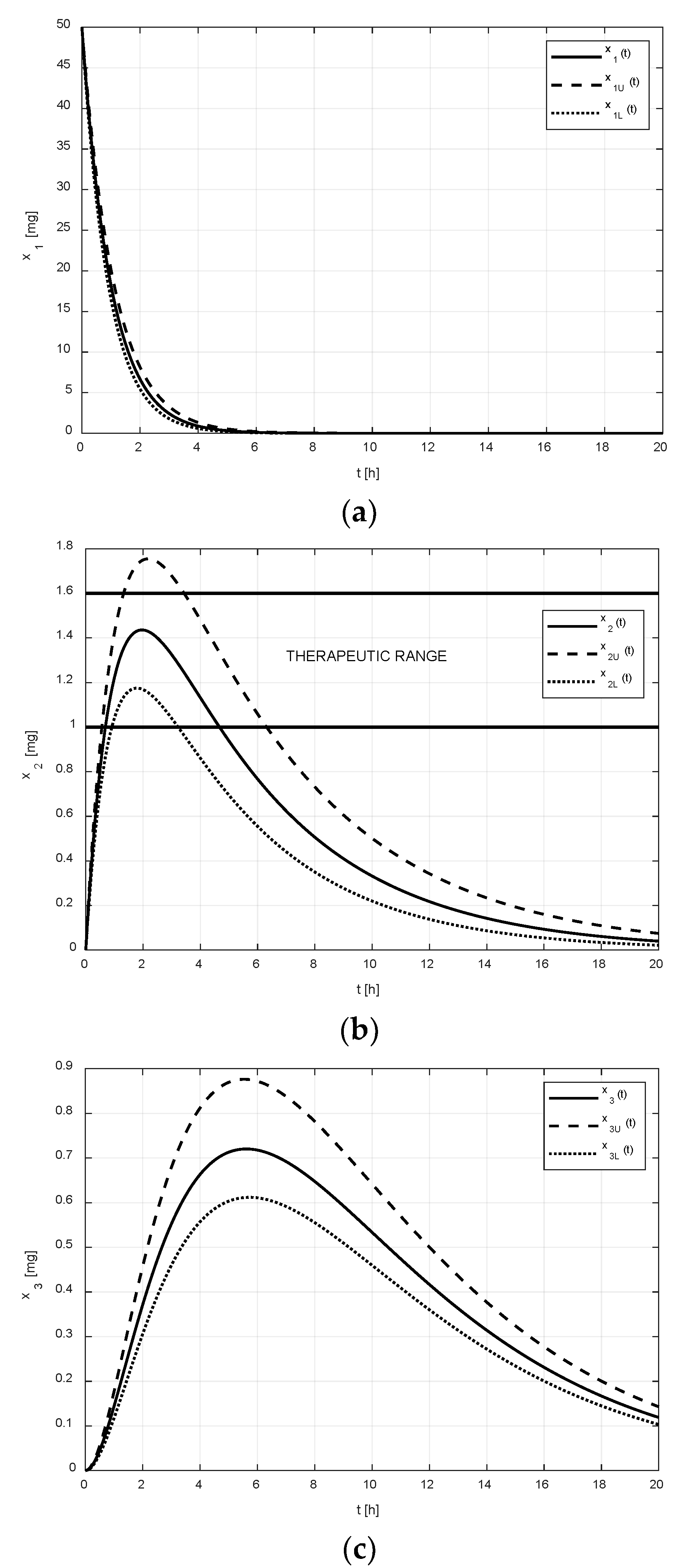
After the addition of the tenside, the second set of pharmacokinetic parameters was identified (see Equation (3)). The corresponding numeric solutions for the states in
Figure 3 are quite different.
As shown in
Figure 2a and
Figure 3a, it is evident that the difference between the limiting amounts
and
within which the amount
could vary was not significant before or after the addition of tenside. The only observable difference is that the settling time of
was shortened due to the presence of tenside from cca 8 h to cca 5 h. This indicates that the tenside sped up the drug release and, in turn, the drug absorption as well.
On the other hand, as shown in
Figure 2b and
Figure 3b, the effect of the tenside on the amount
in the central compartment was much more strongly manifested. The amount
without tenside reached its peak value (cca 1.1 mg) 2 h after the administration, and this peak value may have ranged from cca 0.9 to 1.38 mg. Thus, the absolute range of the variation was 0.48 mg.
Contrary to this, the peak value of with tenside was much greater (cca 1.43 mg) and the possible range of its variations was approximately from 1.2 mg to 1.8 mg, that is, 0.6 mg in absolute terms; hence, the addition of tenside significantly increased not only the peak value, but also the range of its variations.
Within the experiments described above, only the effects of tenside on the dynamics of the drug amounts under the assumption of uncertain pharmacokinetics were monitored. This information is undoubtedly very useful for the drug designer on its own, but the upper and lower matrices , and vectors , can be much more effectively utilized after the augmentation of the uncertain model (5) by the state-bounding observer. In the next section, it is shown that such a combination may effectively work as a predictor of drug amounts in all compartments, while the performance of measurements is required in only one compartment.
2.4. State-Bounding Observer as a Predictor of Dangerous Therapy
In this section, it is proposed that the limiting amounts and are not generated according to Equation (5) for a certain , but that these are predicted by the state-bounding observers. It is also suggested that this arrangement may significantly improve the safety of drug therapy.
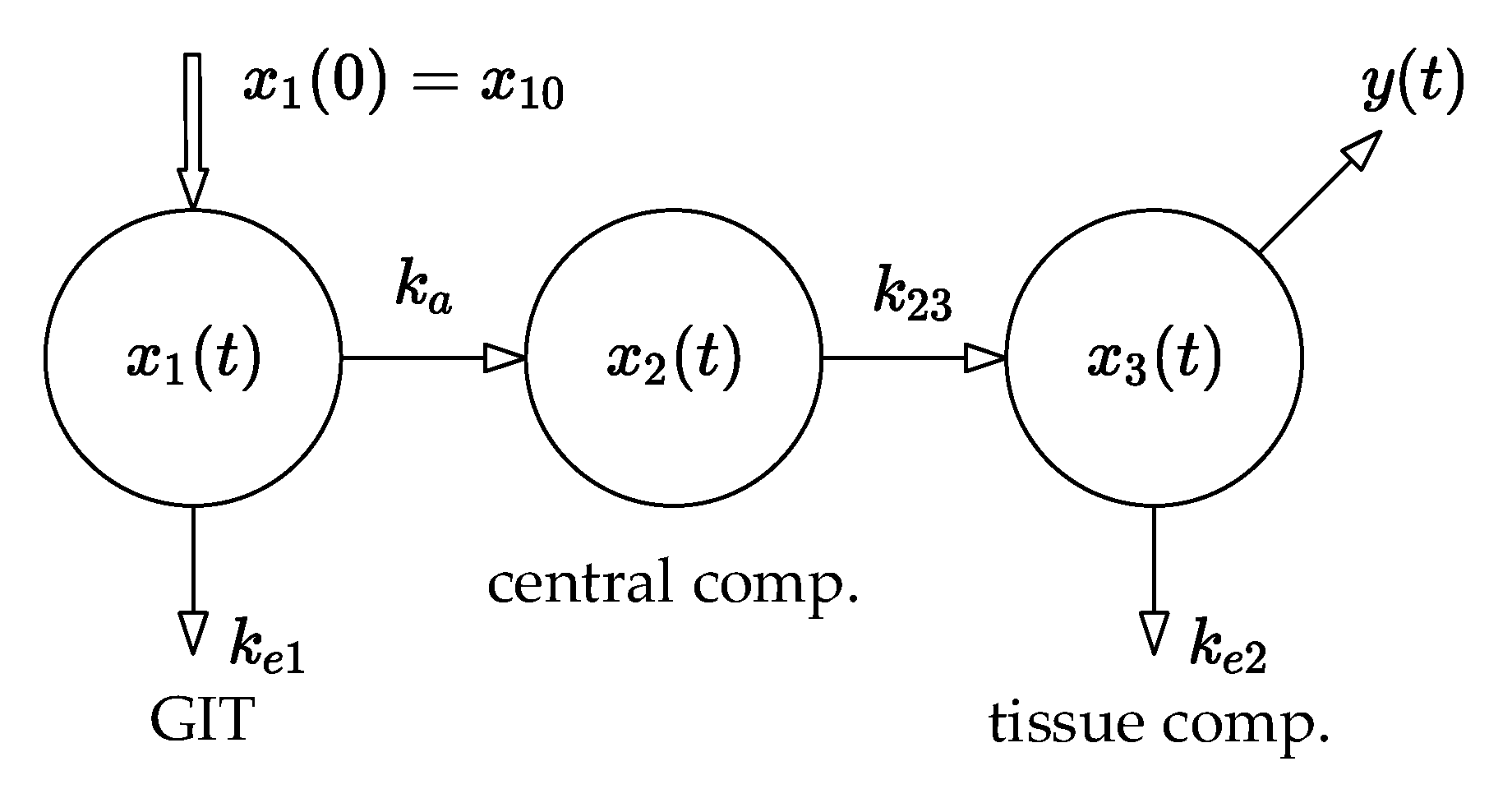
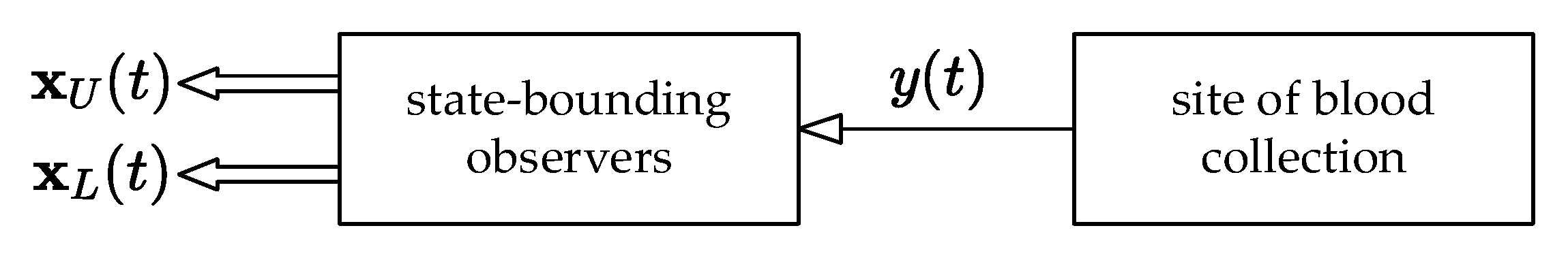
This idea is demonstrated by the compartmental model, shown in
Figure 4, which has three compartments: the GIT into which the drug is administered, the central compartment (the systemic blood circulation), and the tissue compartment from which the blood samples are typically collected.
Now, assume that blood samples are about to be collected repeatedly, but considering that the actions of various exogenous and endogenous effects cause the pharmacokinetic parameters to continually change over time. Therefore, it might be virtually impossible to receive unbiased information about the amount of drug in various body compartments. Obviously, a feasible approach is to augment the nominal pharmacokinetic model with the state-bounding observer, which is able to estimate and predict the drug amounts in all the compartments.
Assume that the same in vivo experiment as described above is performed with the same drug amounts measured, and consider again that there is a need to examine the effect of tenside on the drug amounts in the compartments. For a good illustration, the compartmental model shown in
Figure 4 is used, while the blood samples are collected from the tissue compartment, i.e., from the system output
.
The model is described by the following set of differential Equations (8):
The upper
and lower
limits of the drug amounts are estimated by two state-bounding observers in the form [
5,
12,
13,
14,
15]:
where
is the output of the system (8) and
is the observer-gain vector.
Note that the dynamics of the compartmental model (8) are directly embedded into the observers (9) via the matrices
,
and the vectors
,
. Both observers are driven by the output
of the nominal model (8). The facts mentioned above are essential for further explanation. The arrangement of the whole system, i.e., the connection between the nominal model (8) and the state-bounding observer (9), is illustrated in
Figure 5.
It should be remarked that
Figure 5 does not illustrate any form of physical back-transport of the drug beyond the information-flow diagram. The observer only mathematically estimates and predicts the amount of the drug in the remaining compartments based on the measurements from the site of blood collection. We have to mention that the theory of state-bounding observers is beyond the scope of this paper; however, we stress that the drug amounts in every compartment must be non-negative and that the upper and lower limits
,
, must be non-negative as well. Accordingly, the matrices
and
must result in observers (9) that are positive systems as well [
6].










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}