
Screening for Best Neuronal-Glial Differentiation Protocols of Neuralizing Agents Using a Multi-Sized Microfluidic Embryoid Body Array

 , ,
, ,
Abstract
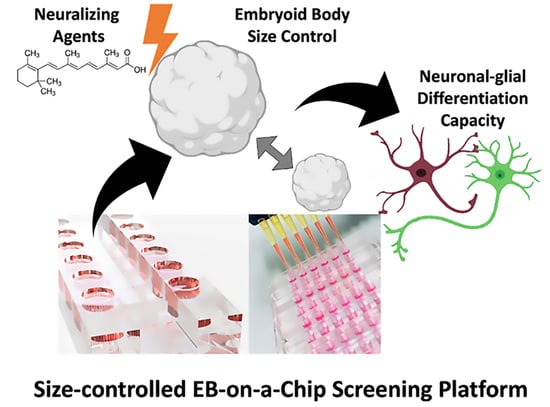
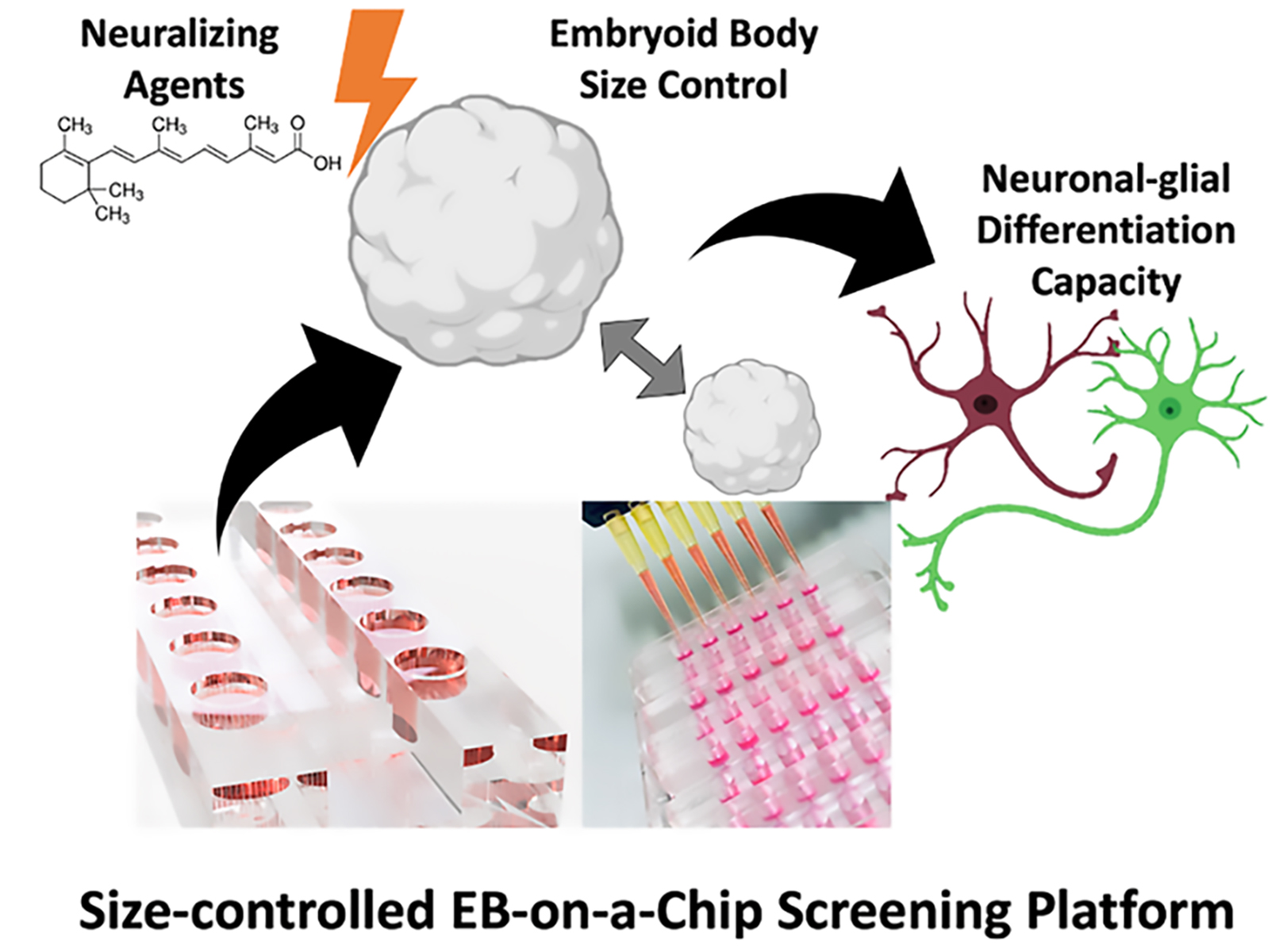
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- (1)
- optimal cultivation conditions over several days,
- (2)
- precise control of nutrient and gas exchange,
- (3)
- user-friendly cell loading capacities,
- (4)
- parallel embryoid body production and harvesting option,
- (5)
- simple operation to ensure tissue maintenance.
2. Materials and Methods
2.1. Cell Culture
2.2. Microfluidic Chip Fabrication
2.3. Microfluidic Chip Priming and Cell Seeding
2.4. Viability Analysis
2.5. P19 Induction and Embryoid Body Differentiation
2.6. Immunofluorescence Imaging
2.7. General Microscopy and Morphometric Analysis
2.8. Statistical Analysis
3. Results and Discussion
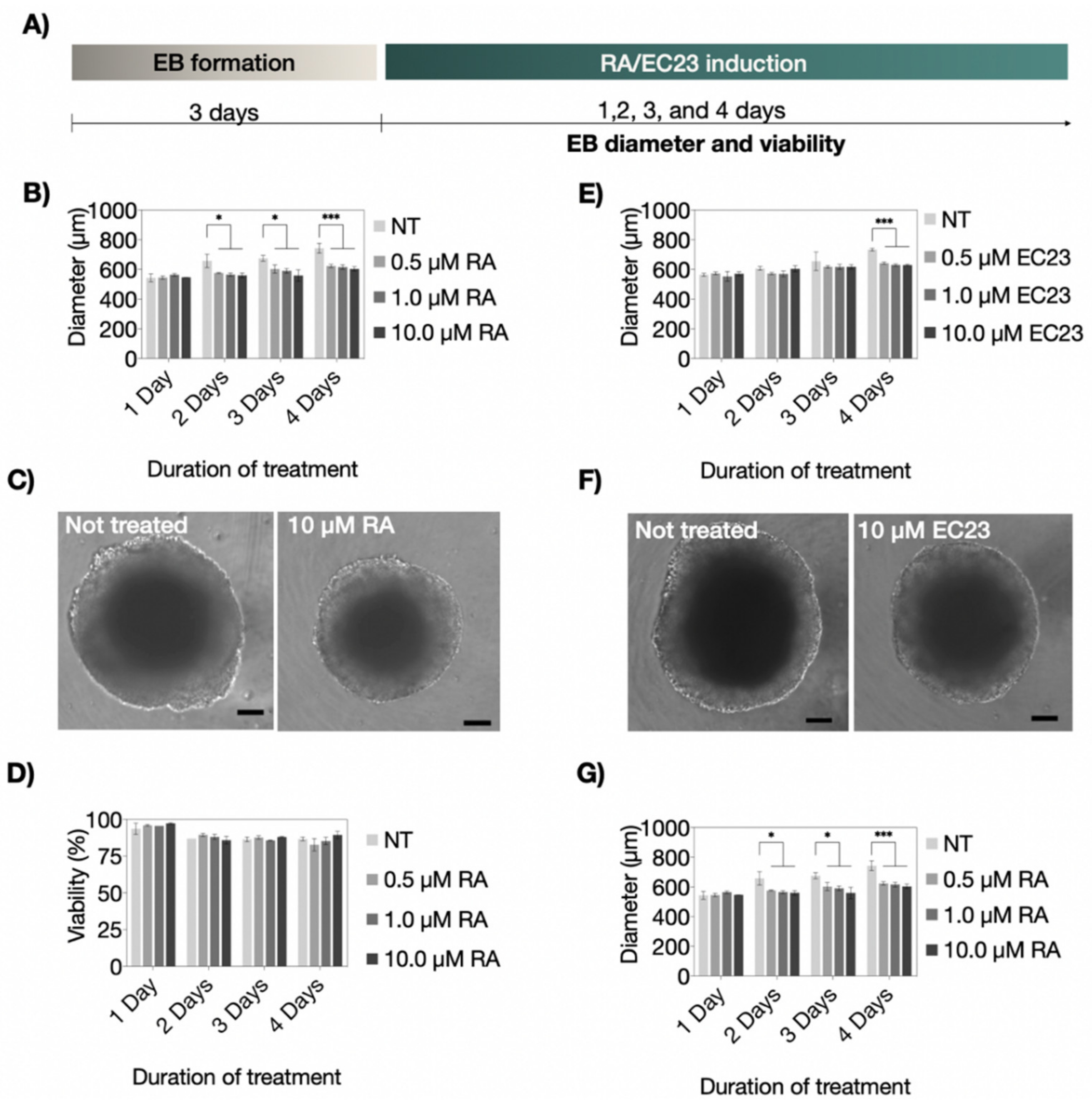
3.1. Influence of RA and EC23 Concentration on Embryoid Body Formation
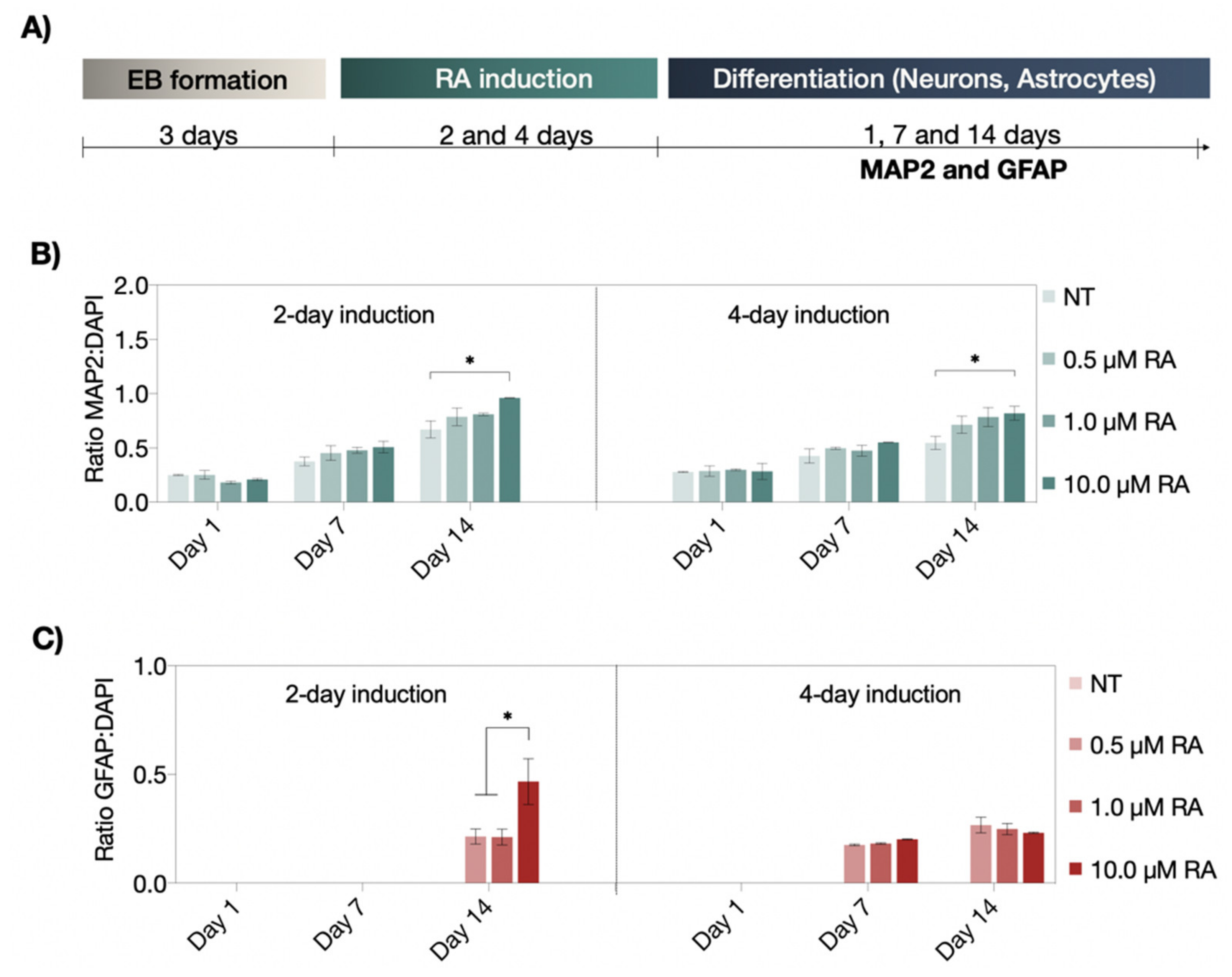
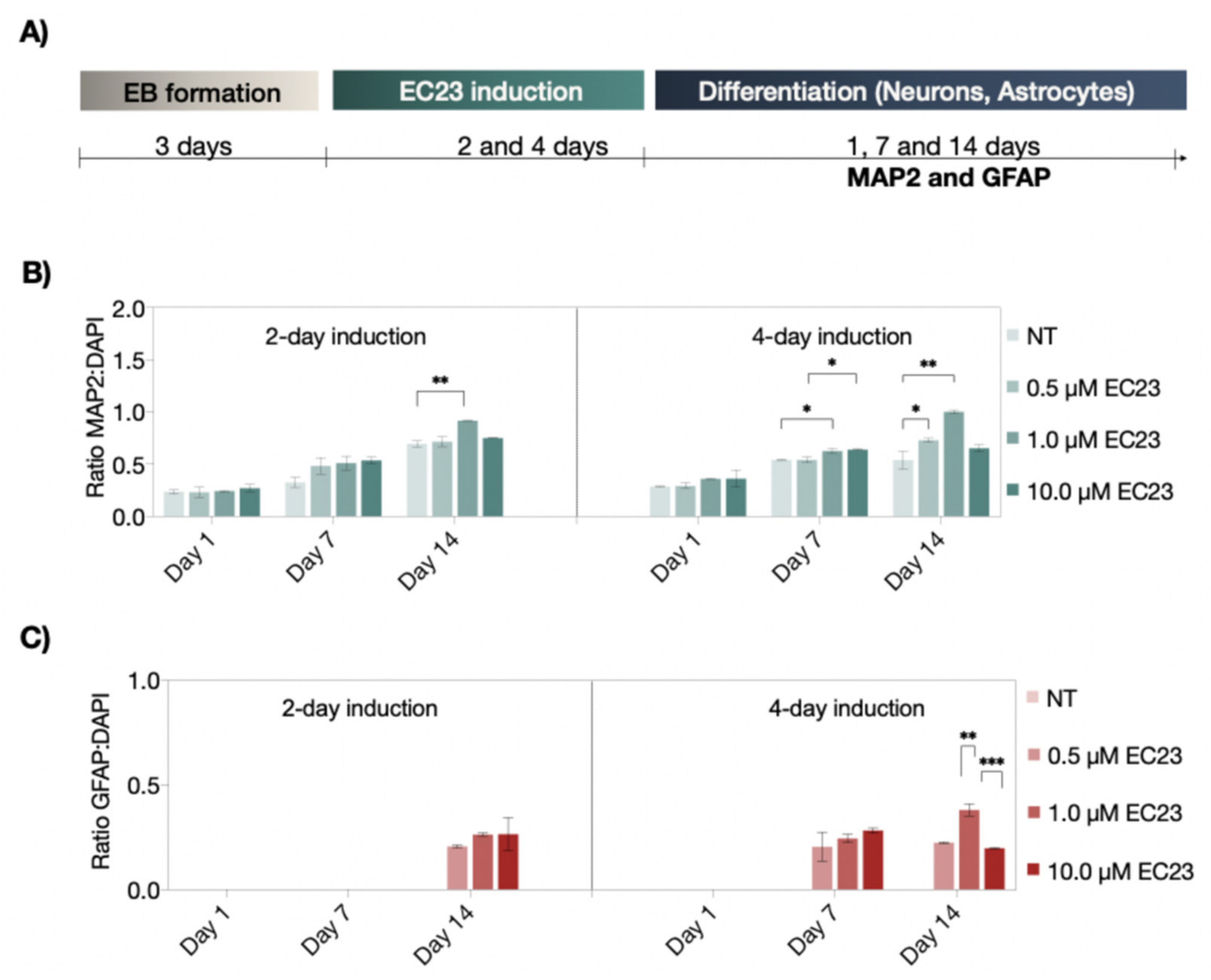
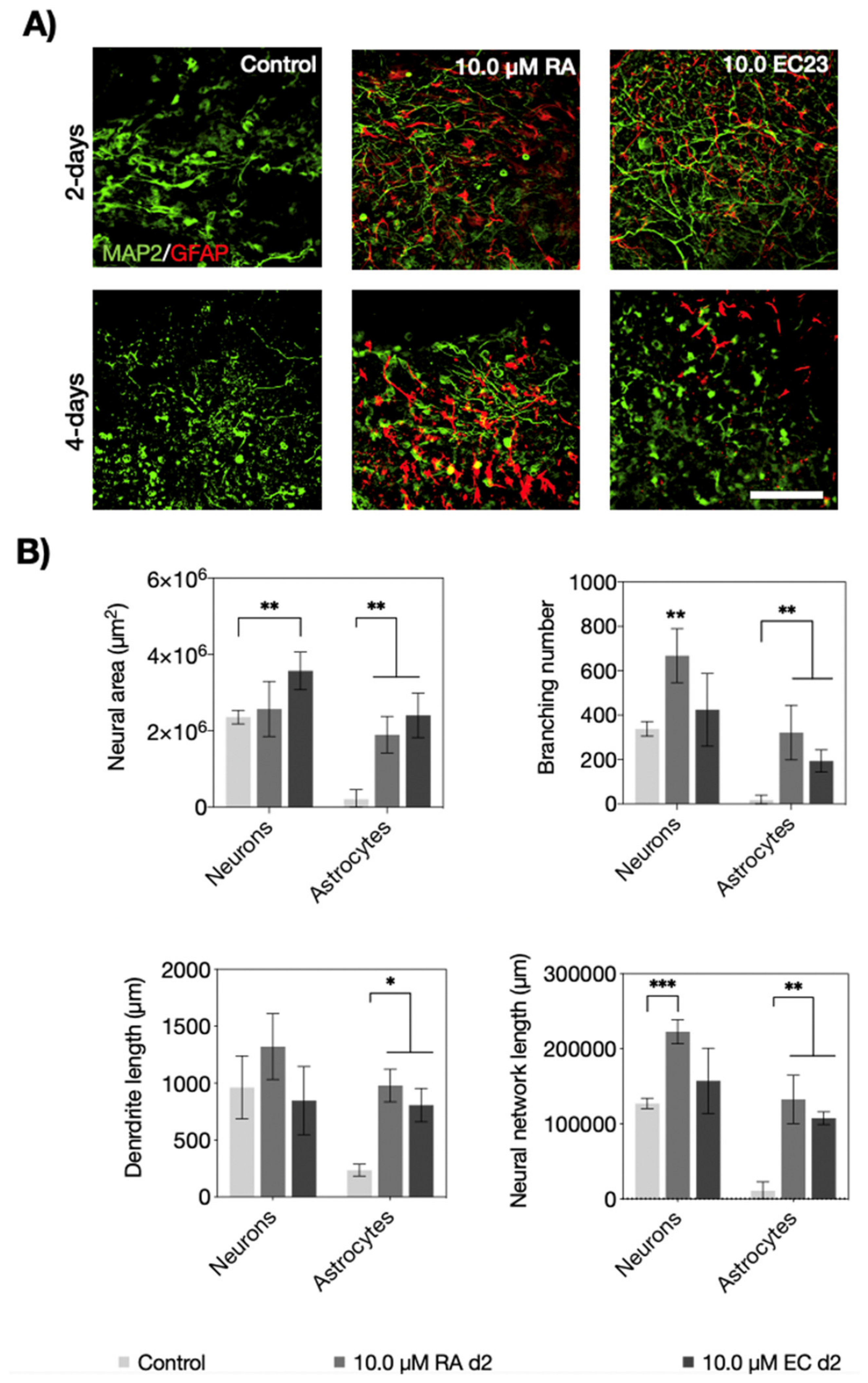
3.2. Neuronal and Astrocyte Differentiation Capacity of RA and EC23 Treated EBs
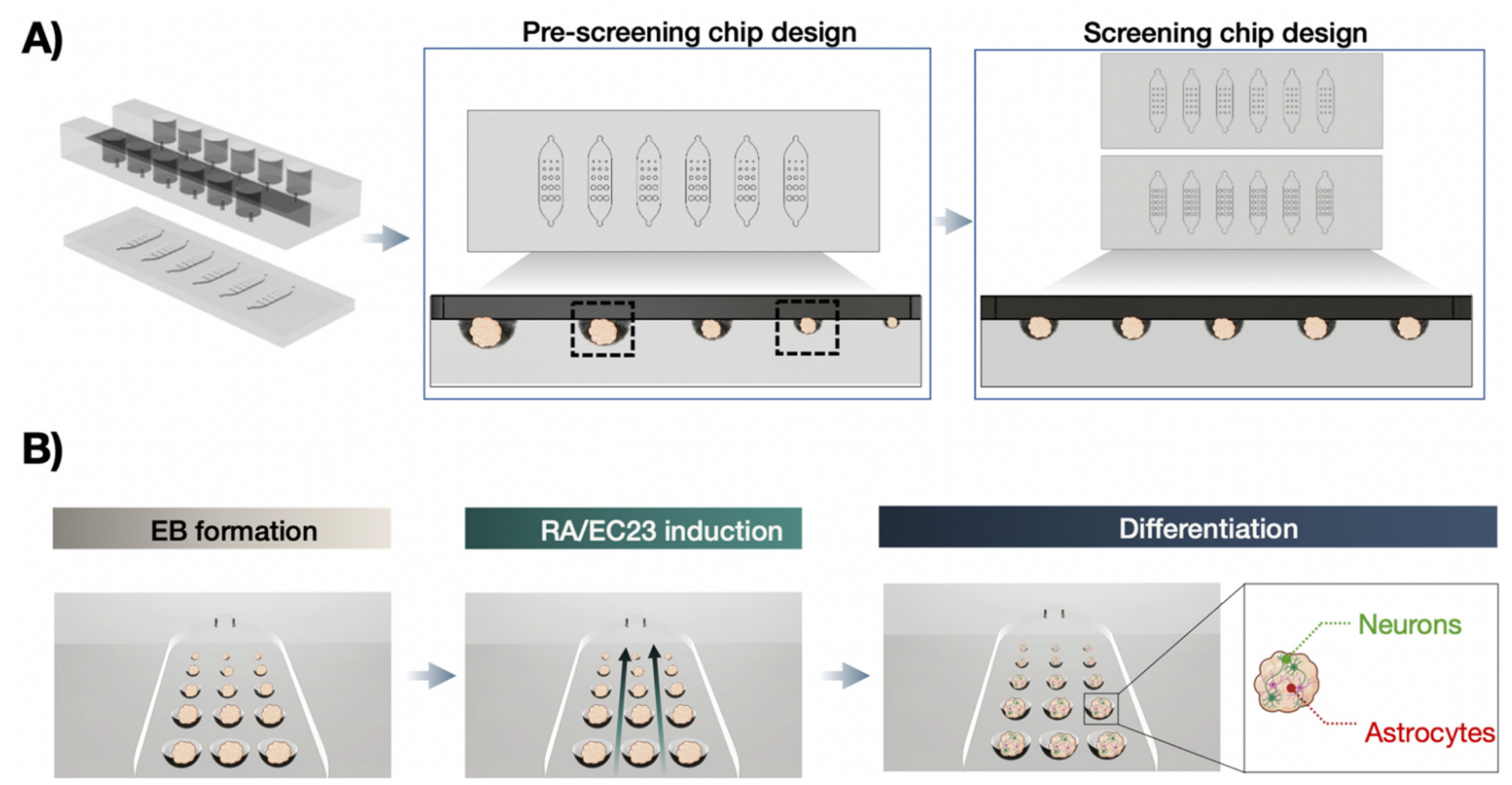
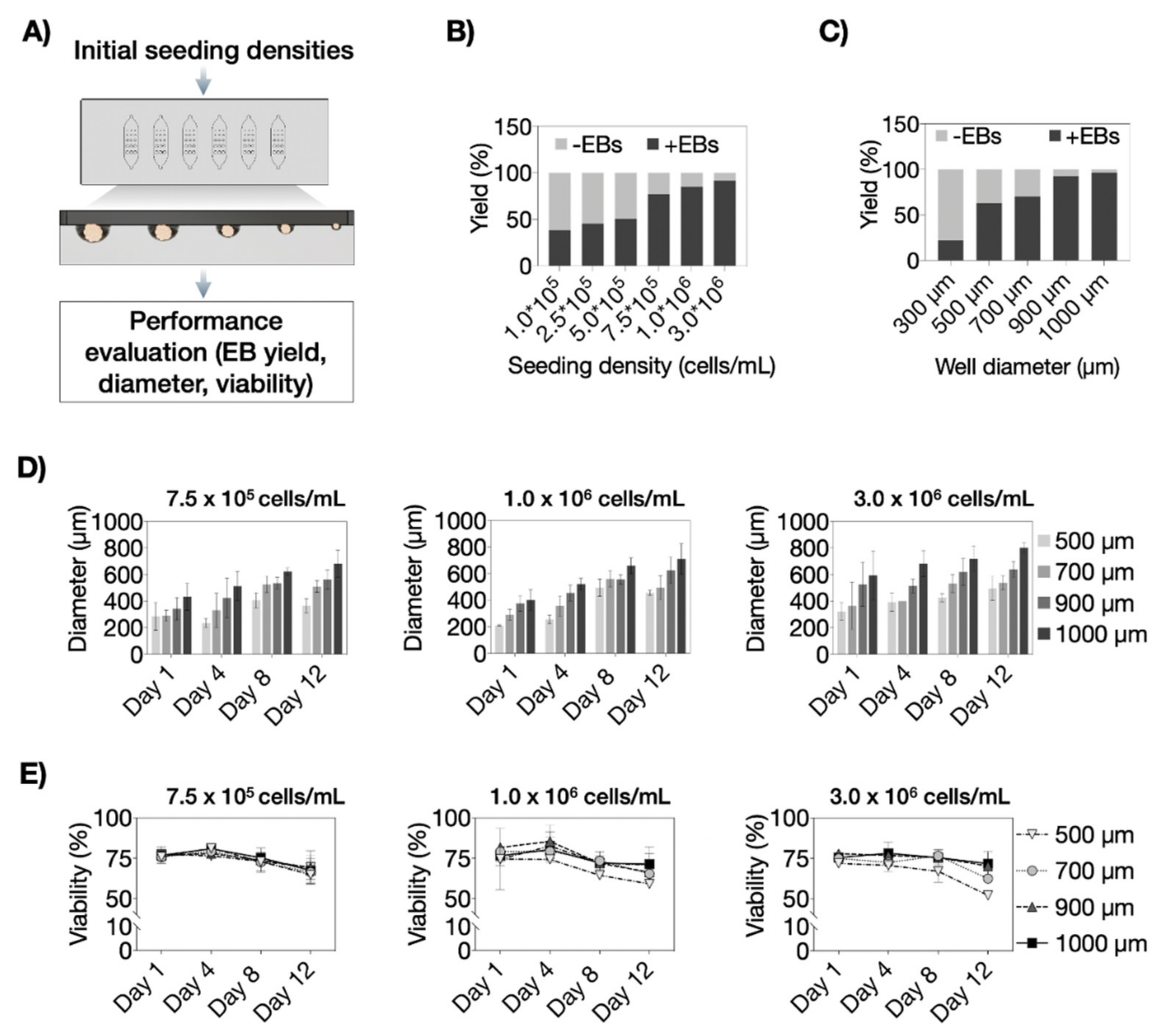
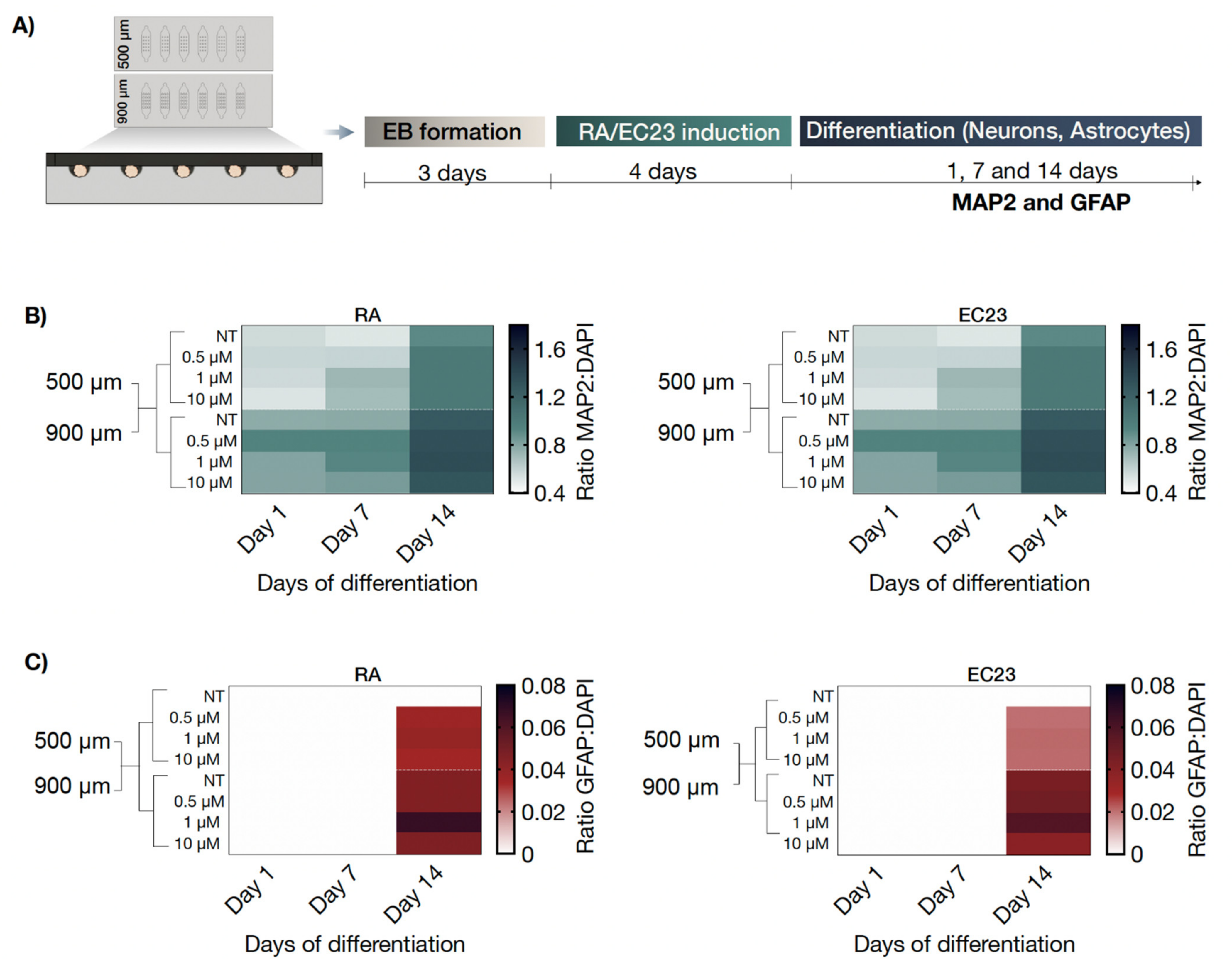
3.3. Optimization of P19 Cell Seeding Protocols for Size-Controllable On-Chip Embryoid Body Formation
3.4. On-Chip Screening of Embryoid Body Size Effects on Neuronal and Astrocytic Differentiation Capacity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jongkamonwiwat, N.; Noisa, P. Biomedical and clinical promises of human pluripotent stem cells for neurological disorders. BioMed Res. Int. 2013, 2013, 656531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Q.; Habegger, L.; Noisa, P.; Szekely, A.; Qiu, C.; Hutchison, S.; Raha, D.; Egholm, M.; Lin, H.; Weissman, S.; et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 5254–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wobus, A.M.; Boheler, K.R. Embryonic stem cells: Prospects for developmental biology and cell therapy. Physiol. Rev. 2005, 85, 635–678. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.E.; Melton, D. Turning straw into gold: Directing cell fate for regenerative medicine. Nat. Rev. Genet. 2011, 12, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Perrier, A.L.; Tabar, V.; Barberi, T.; Rubio, M.E.; Bruses, J.; Topf, N.; Harrison, N.L.; Studer, L. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2004, 101, 12543–12548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plachta, N.; Bibel, M.; Tucker, K.L.; Barde, Y.A. Developmental potential of defined neural progenitors derived from mouse embryonic stem cells. Development 2004, 131, 5449–5456. [Google Scholar] [CrossRef] [Green Version]
- Reubinoff, B.E.; Itsykson, P.; Turetsky, T.; Pera, M.F.; Reinhartz, E.; Itzik, A.; Ben-Hur, T. Neural progenitors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1134–1140. [Google Scholar] [CrossRef]
- Zeevaert, K.; Elsafi Mabrouk, M.H.; Wagner, W.; Goetzke, R. Cell Mechanics in Embryoid Bodies. Cells 2020, 9, 2270. [Google Scholar] [CrossRef]
- Odorico, J.S.; Kaufman, D.S.; Thomson, J.A. Multilineage differentiation from human embryonic stem cell lines. Stem Cells 2001, 19, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Mummery, C.; Ward-van Oostwaard, D.; Doevendans, P.; Spijker, R.; van den Brink, S.; Hassink, R.; van der Heyden, M.; Opthof, T.; Pera, M.; de la Riviere, A.B.; et al. Differentiation of human embryonic stem cells to cardiomyocytes: Role of coculture with visceral endoderm-like cells. Circulation 2003, 107, 2733–2740. [Google Scholar] [CrossRef] [Green Version]
- Osafune, K.; Caron, L.; Borowiak, M.; Martinez, R.J.; Fitz-Gerald, C.S.; Sato, Y.; Cowan, C.A.; Chien, K.R.; Melton, D.A. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol. 2008, 26, 313–315. [Google Scholar] [CrossRef]
- Hamazaki, T.; Oka, M.; Yamanaka, S.; Terada, N. Aggregation of embryonic stem cells induces Nanog repression and primitive endoderm differentiation. J. Cell Sci. 2004, 117, 5681–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pompe, S.; Bader, M.; Tannert, C. Stem-cell research: The state of the art. Future regulations of embryonic-stem-cell research will be influenced more by economic interests and cultural history than by ethical concerns. EMBO Rep. 2005, 6, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Keller, G.M. In vitro differentiation of embryonic stem cells. Curr. Opin. Cell Biol. 1995, 7, 862–869. [Google Scholar] [CrossRef]
- Schuldiner, M.; Yanuka, O.; Itskovitz-Eldor, J.; Melton, D.A.; Benvenisty, N. Effects of eight growth factors on the differentiation of cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 11307–11312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, R.; Fuller, B.; Li, J.; Tavana, H. Statistical analysis of multi-dimensional, temporal gene expression of stem cells to elucidate colony size-dependent neural differentiation. Mol. Omics 2018, 14, 109–120. [Google Scholar] [CrossRef]
- Zarei Fard, N.; Talaei-Khozani, T.; Bahmanpour, S.; Esmaeilpour, T. Comparison of cell viability and embryoid body size of two embryonic stem cell lines after different exposure times to bone morphogenetic protein 4. Iran. J. Med. Sci. 2015, 40, 110–117. [Google Scholar] [PubMed]
- Moon, S.H.; Ju, J.; Park, S.J.; Bae, D.; Chung, H.M.; Lee, S.H. Optimizing human embryonic stem cells differentiation efficiency by screening size-tunable homogenous embryoid bodies. Biomaterials 2014, 35, 5987–5997. [Google Scholar] [CrossRef]
- Volpato, V.; Webber, C. Addressing variability in iPSC-derived models of human disease: Guidelines to promote reproducibility. Dis. Models Mech. 2020, 13, dmm042317. [Google Scholar] [CrossRef] [Green Version]
- Itskovitz-Eldor, J.; Schuldiner, M.; Karsenti, D.; Eden, A.; Yanuka, O.; Amit, M.; Soreq, H.; Benvenisty, N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 2000, 6, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.Y.; Chung, B.G.; Lee, D.H.; Khademhosseini, A.; Kim, J.H.; Lee, S.H. Controlled-size embryoid body formation in concave microwell arrays. Biomaterials 2010, 31, 4296–4303. [Google Scholar] [CrossRef] [PubMed]
- Mohr, J.C.; Zhang, J.; Azarin, S.M.; Soerens, A.G.; de Pablo, J.J.; Thomson, J.A.; Lyons, G.E.; Palecek, S.P.; Kamp, T.J. The microwell control of embryoid body size in order to regulate cardiac differentiation of human embryonic stem cells. Biomaterials 2010, 31, 1885–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, J.M.; Yeh, J.; Eng, G.; Fukuda, J.; Blumling, J.; Suh, K.Y.; Cheng, J.; Mahdavi, A.; Borenstein, J.; Langer, R.; et al. Controlling size, shape and homogeneity of embryoid bodies using poly(ethylene glycol) microwells. Lab Chip 2007, 7, 786–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, T.; Wang, Y.; Chen, W.; Li, Z.; Su, W.; Guo, Y.; Deng, P.; Qin, J. Engineering human islet organoids from iPSCs using an organ-on-chip platform. Lab Chip 2019, 19, 948–958. [Google Scholar] [CrossRef]
- Lee, L.H.; Peerani, R.; Ungrin, M.; Joshi, C.; Kumacheva, E.; Zandstra, P. Micropatterning of human embryonic stem cells dissects the mesoderm and endoderm lineages. Stem Cell Res. 2009, 2, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Peerani, R.; Rao, B.M.; Bauwens, C.; Yin, T.; Wood, G.A.; Nagy, A.; Kumacheva, E.; Zandstra, P.W. Niche-mediated control of human embryonic stem cell self-renewal and differentiation. EMBO J. 2007, 26, 4744–4755. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, H.; Deng, P.; Chen, W.; Guo, Y.; Tao, T.; Qin, J. In situ differentiation and generation of functional liver organoids from human iPSCs in a 3D perfusable chip system. Lab Chip 2018, 18, 3606–3616. [Google Scholar] [CrossRef]
- Semrau, S.; van Oudenaarden, A. Studying lineage decision-making in vitro: Emerging concepts and novel tools. Annu. Rev. Cell Dev. Biol. 2015, 31, 317–345. [Google Scholar] [CrossRef]
- Khademhosseini, A.; Langer, R.; Borenstein, J.; Vacanti, J.P. Microscale technologies for tissue engineering and biology. Proc. Natl. Acad. Sci. USA 2006, 103, 2480–2487. [Google Scholar] [CrossRef] [Green Version]
- Whitesides, G.M.; Ostuni, E.; Takayama, S.; Jiang, X.; Ingber, D.E. Soft lithography in biology and biochemistry. Annu. Rev. Biomed. Eng. 2001, 3, 335–373. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Musso, N.; Grasso, M.; Costantino, A.; Lazzarino, G.; Tascedda, F.; Gulisano, M.; Lunte, S.M.; Caraci, F. Microfluidics as a Novel Tool for Biological and Toxicological Assays in Drug Discovery Processes: Focus on Microchip Electrophoresis. Micromachines 2020, 11, 593. [Google Scholar] [CrossRef]
- Williamson, A.; Singh, S.; Fernekorn, U.; Schober, A. The future of the patient-specific Body-on-a-chip. Lab Chip 2013, 13, 3471–3480. [Google Scholar] [CrossRef]
- Mofazzal Jahromi, M.A.; Abdoli, A.; Rahmanian, M.; Bardania, H.; Bayandori, M.; Moosavi Basri, S.M.; Kalbasi, A.; Aref, A.R.; Karimi, M.; Hamblin, M.R. Microfluidic Brain-on-a-Chip: Perspectives for Mimicking Neural System Disorders. Mol. Neurobiol. 2019, 56, 8489–8512. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Lie, D.C.; DeCicco, K.L.; Shi, Y.; DeLuca, L.M.; Gage, F.H.; Evans, R.M. Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc. Natl. Acad. Sci. USA 2006, 103, 3902–3907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosher, K.I.; Schaffer, D.V. Proliferation versus Differentiation: Redefining Retinoic Acid’s Role. Stem Cell Rep. 2018, 10, 1673–1675. [Google Scholar] [CrossRef] [PubMed]
- McBurney, M. P19 embryonal carcinoma cells. Int. J. Dev. Biol. 1993, 140, 135–140. [Google Scholar]
- Taneja, R.; Roy, B.; Plassat, J.L.; Zusi, C.F.; Ostrowski, J.; Reczek, P.R.; Chambon, P. Cell-type and promoter-context dependent retinoic acid receptor (RAR) redundancies for RAR beta 2 and Hoxa-1 activation in F9 and P19 cells can be artefactually generated by gene knockouts. Proc. Natl. Acad. Sci. USA 1996, 93, 6197–6202. [Google Scholar] [CrossRef] [Green Version]
- Eilenberger, C.; Rothbauer, M.; Selinger, F.; Gerhartl, A.; Jordan, C.; Harasek, M.; Schädl, B.; Grillari, J.; Weghuber, J.; Neuhaus, W.; et al. A Microfluidic Multisize Spheroid Array for Multiparametric Screening of Anticancer Drugs and Blood-Brain Barrier Transport Properties. Adv. Sci. 2021, 8, e2004856. [Google Scholar] [CrossRef]
- Ulrich, H.; Majumder, P. Neurotransmitter receptor expression and activity during neuronal differentiation of embryonal carcinoma and stem cells: From basic research towards clinical applications. Cell Prolif. 2006, 39, 281–300. [Google Scholar] [CrossRef]
- Snodgrass, S.R. Vitamin neurotoxicity. Mol. Neurobiol. 1992, 6, 41–73. [Google Scholar] [CrossRef]
- Edwards, M.K.; McBurney, M.W. The concentration of retinoic acid determines the differentiated cell types formed by a teratocarcinoma cell line. Dev. Biol. 1983, 98, 187–191. [Google Scholar] [CrossRef]
- Liu, L.; Michowski, W.; Kolodziejczyk, A.; Sicinski, P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019, 21, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Russo, F.B.; Cugola, F.R.; Fernandes, I.R.; Pignatari, G.C.; Beltrão-Braga, P.C. Induced pluripotent stem cells for modeling neurological disorders. World J. Transplant. 2015, 5, 209–221. [Google Scholar] [CrossRef]
- Okada, Y.; Shimazaki, T.; Sobue, G.; Okano, H. Retinoic-acid-concentration-dependent acquisition of neural cell identity during in vitro differentiation of mouse embryonic stem cells. Dev. Biol. 2004, 275, 124–142. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.S.; Chung, B.G.; Ortmann, D.; Hattori, N.; Moeller, H.C.; Khademhosseini, A. Microwell-mediated control of embryoid body size regulates embryonic stem cell fate via differential expression of WNT5a and WNT11. Proc. Natl. Acad. Sci. USA 2009, 106, 16978–16983. [Google Scholar] [CrossRef] [Green Version]
- Bratt-Leal, A.M.; Carpenedo, R.L.; McDevitt, T.C. Engineering the embryoid body microenvironment to direct embryonic stem cell differentiation. Biotechnol. Prog. 2009, 25, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, D.; Nakazawa, K. Differentiation of mouse iPS cells is dependent on embryoid body size in microwell chip culture. J. Biosci. Bioeng. 2016, 122, 507–512. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eilenberger, C.; Rothbauer, M.; Brandauer, K.; Spitz, S.; Ehmoser, E.-K.; Küpcü, S.; Ertl, P. Screening for Best Neuronal-Glial Differentiation Protocols of Neuralizing Agents Using a Multi-Sized Microfluidic Embryoid Body Array. Pharmaceutics 2022, 14, 339. https://doi.org/10.3390/pharmaceutics14020339
Eilenberger C, Rothbauer M, Brandauer K, Spitz S, Ehmoser E-K, Küpcü S, Ertl P. Screening for Best Neuronal-Glial Differentiation Protocols of Neuralizing Agents Using a Multi-Sized Microfluidic Embryoid Body Array. Pharmaceutics. 2022; 14(2):339. https://doi.org/10.3390/pharmaceutics14020339
Chicago/Turabian StyleEilenberger, Christoph, Mario Rothbauer, Konstanze Brandauer, Sarah Spitz, Eva-Kathrin Ehmoser, Seta Küpcü, and Peter Ertl. 2022. "Screening for Best Neuronal-Glial Differentiation Protocols of Neuralizing Agents Using a Multi-Sized Microfluidic Embryoid Body Array" Pharmaceutics 14, no. 2: 339. https://doi.org/10.3390/pharmaceutics14020339