
Chemical Attachment of 5-Nitrosalicylaldimine Motif to Silatrane Resulting in an Organic–Inorganic Structure with High Medicinal Significance

 , , , , , , , , and
, , , , , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Spectral Analysis
2.2.2. X-ray Crystallography
2.2.3. Cell Culture and Treatment
2.2.4. Cell Viability and Cytotoxicity Assays
XTT Assay
Live/Dead Cell Assay
2.2.5. Bio- and Mucoadhesivity Tests
2.2.6. Antimicrobial Assay
2.2.7. Molecular Docking Computations
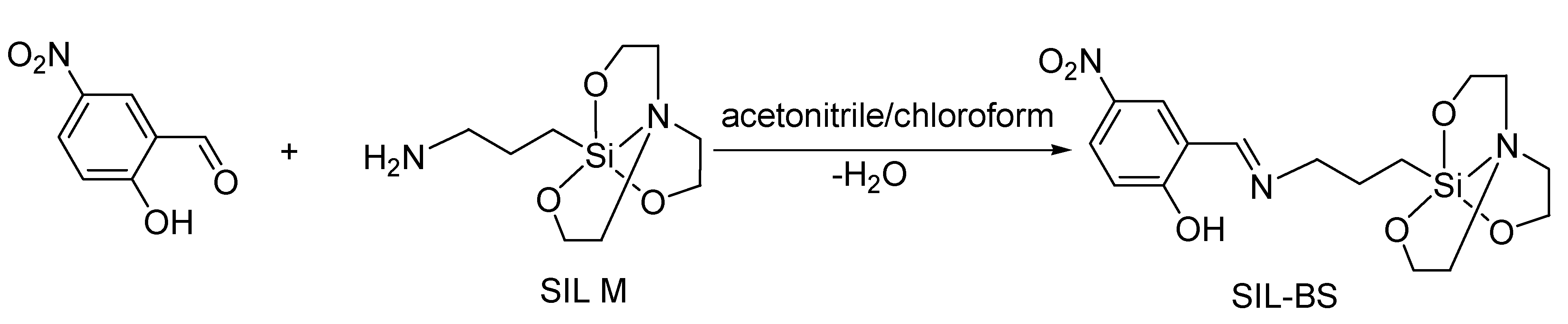
2.3. Synthesis of 1-(3-{[(2-hydroxy-5-nitrophenyl)methylidene]amino}propyl)silatrane, SIL- BS
3. Results and Discussion
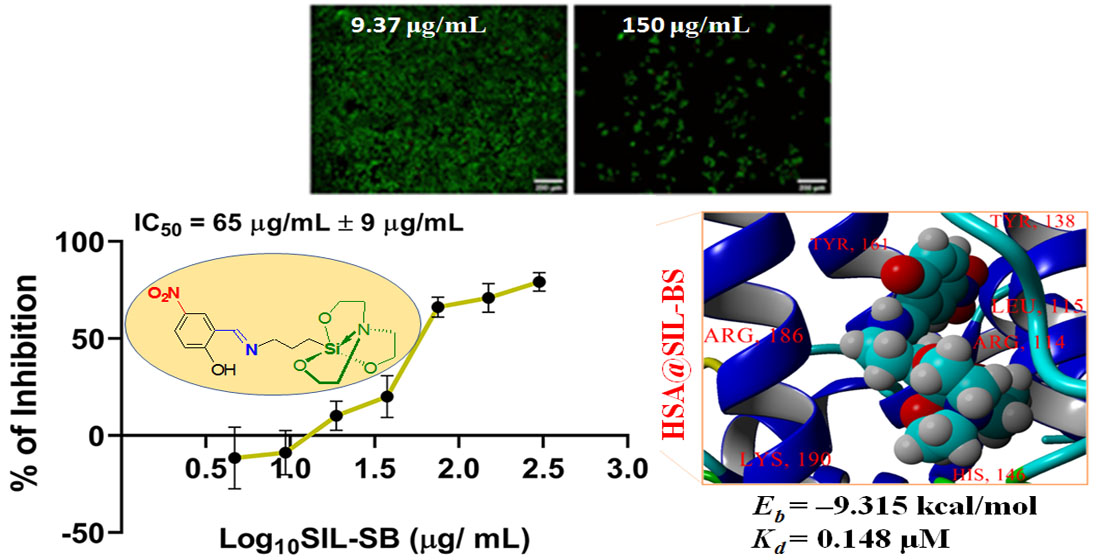
3.1. Structural Analysis
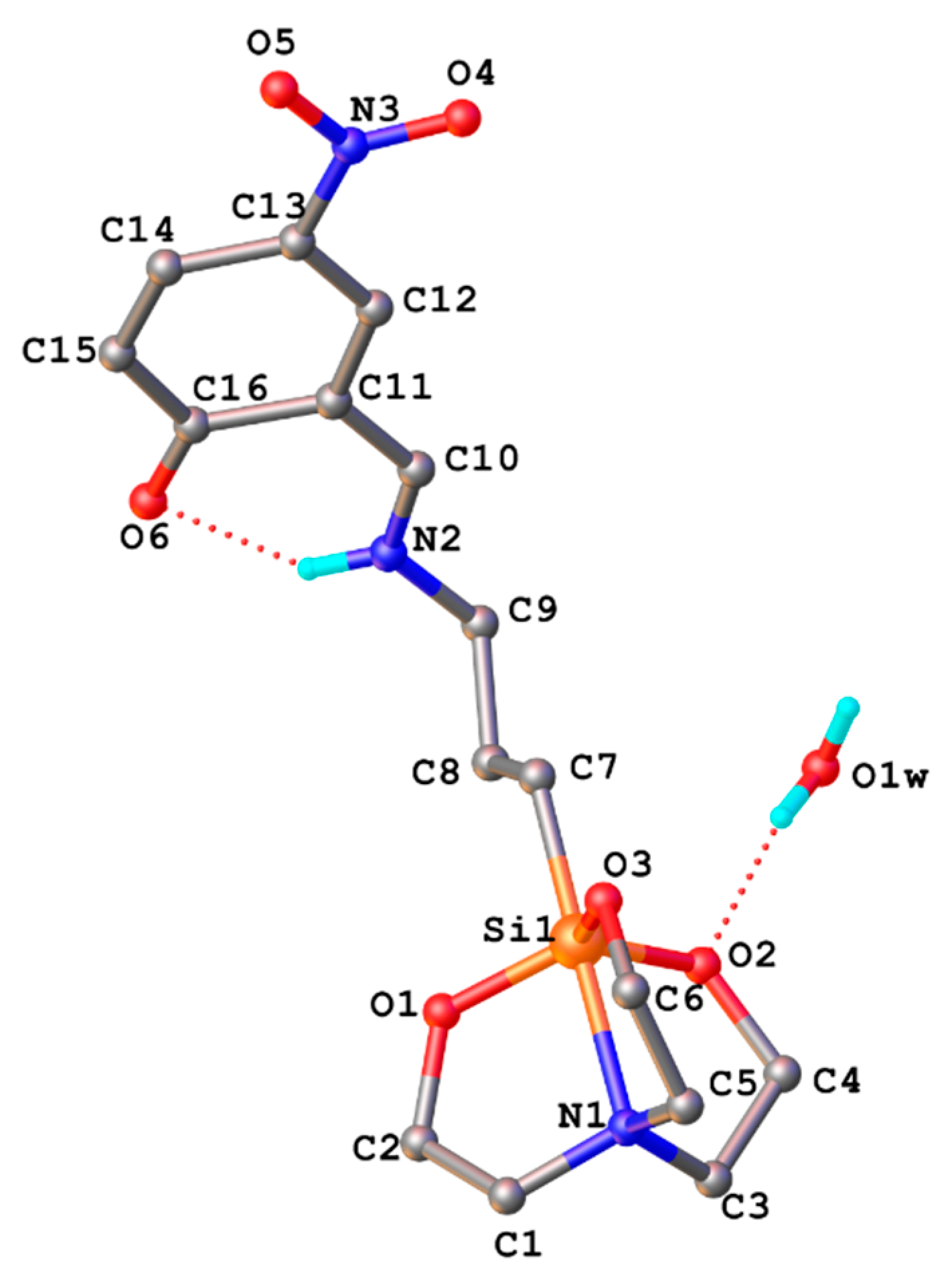
3.2. X-ray Crystallography
3.3. Hydrolytic Stability of SIL BS in PBS Media by Mimicking the Physiological Conditions
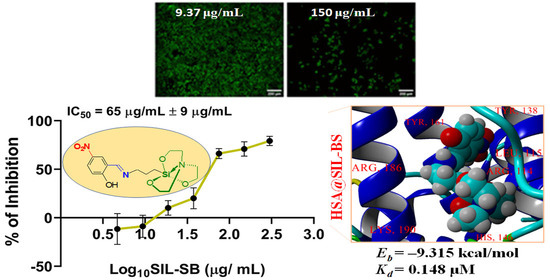
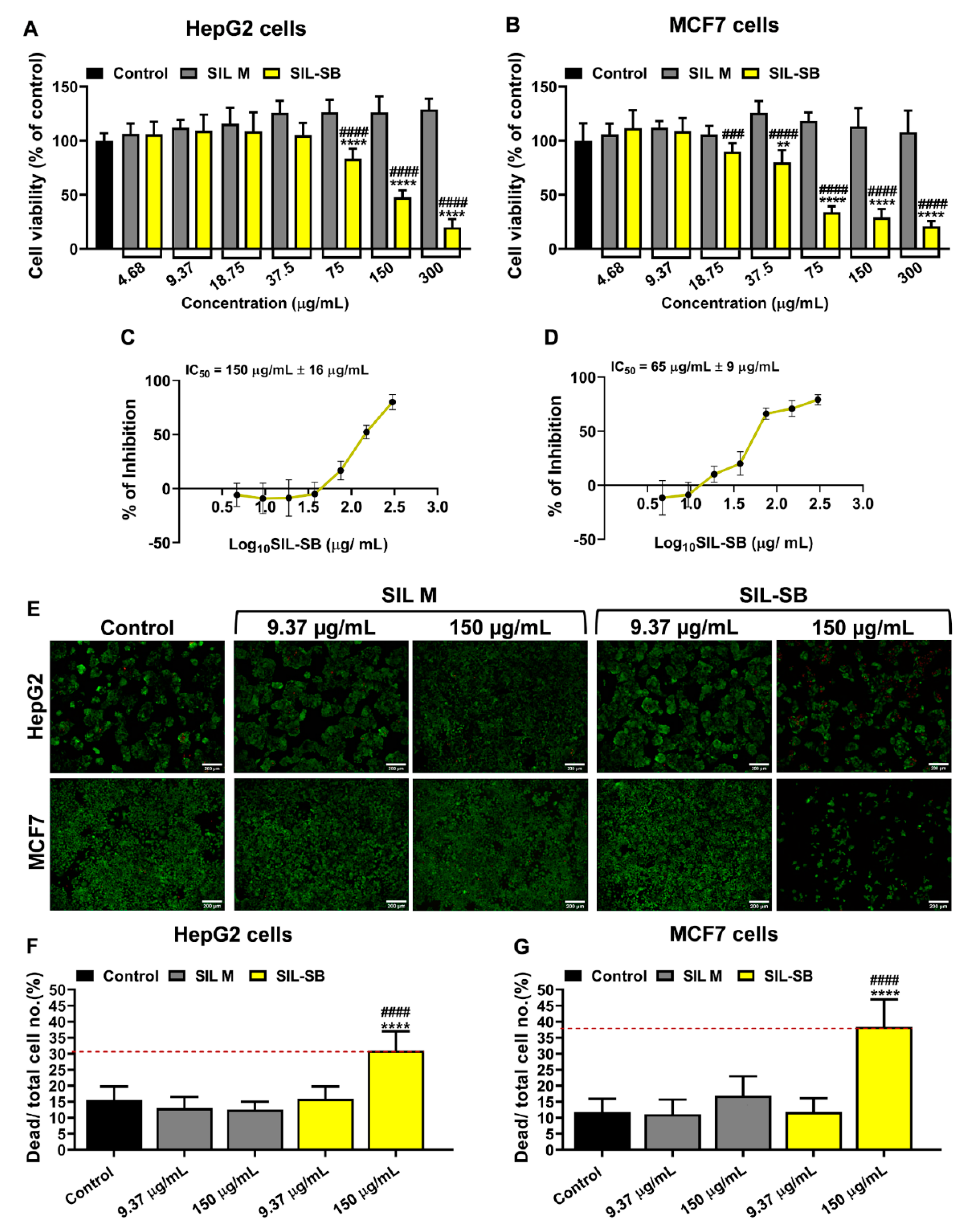
3.4. Evaluation of SIL-SB Effect on Cell Viability
3.5. Protein Binding Ability
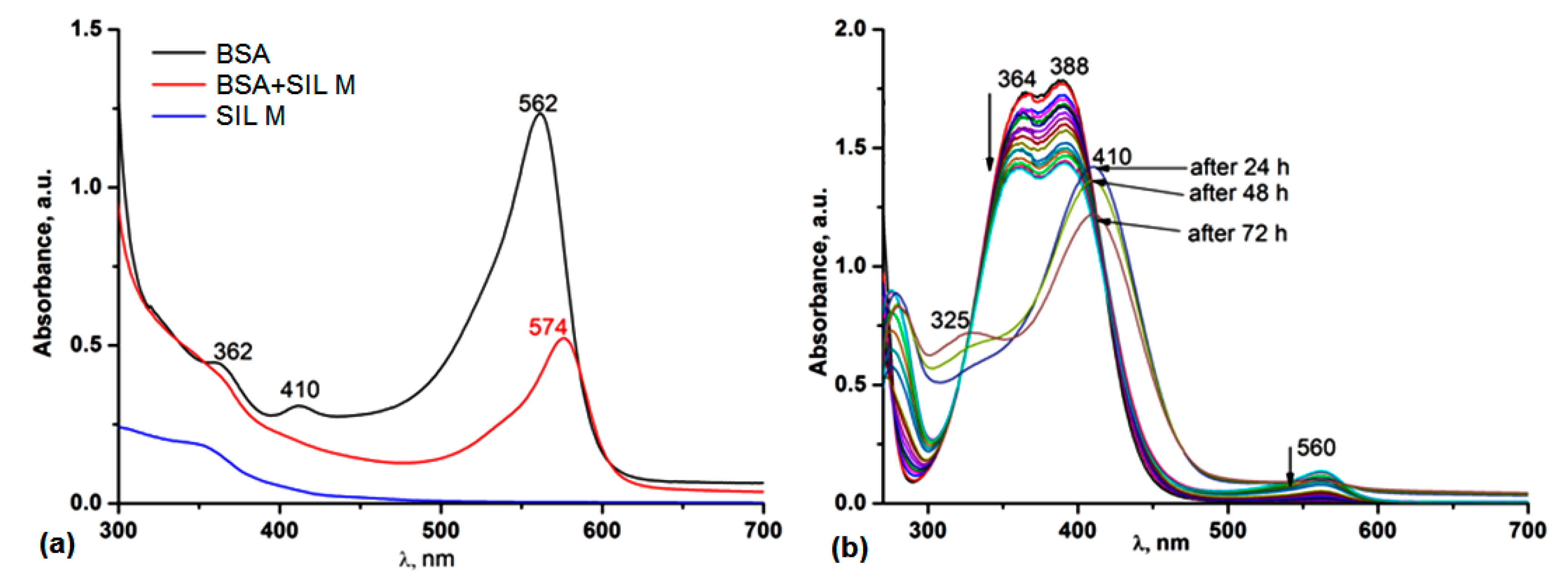
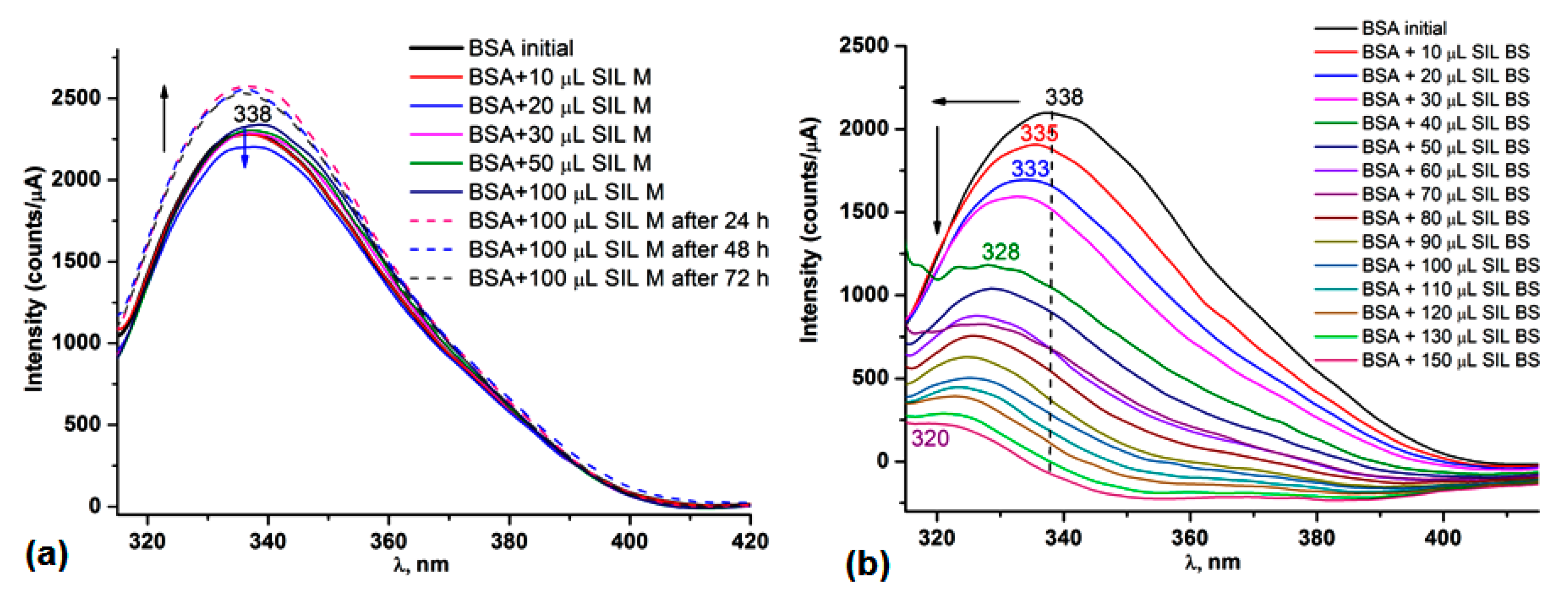
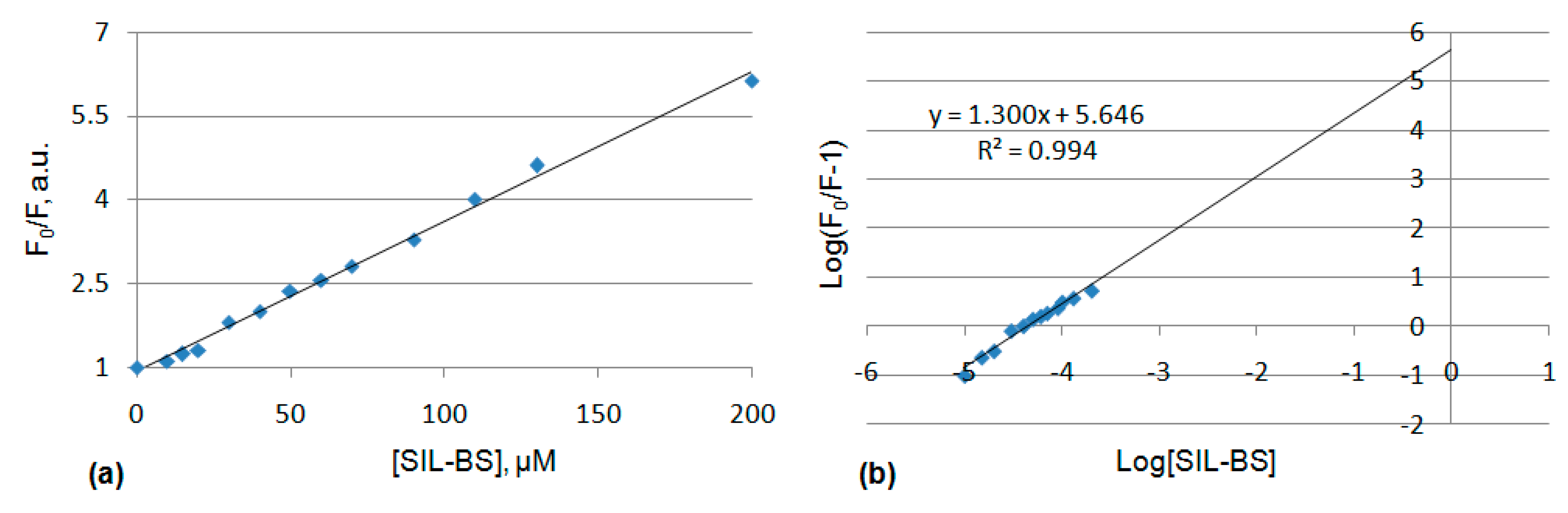
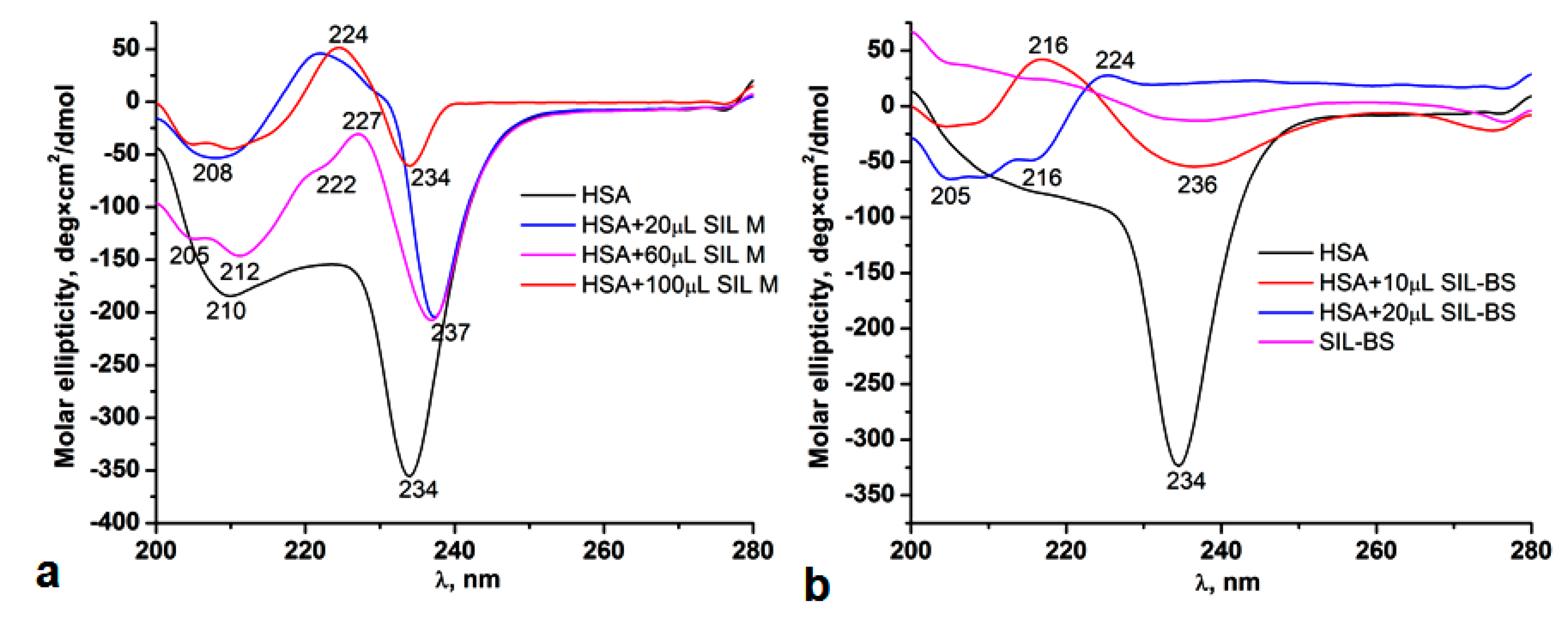
3.5.1. UV-Vis and Fluorescence Spectroscopy
3.5.2. NMR Spectroscopy
3.5.3. Circular Dichroism
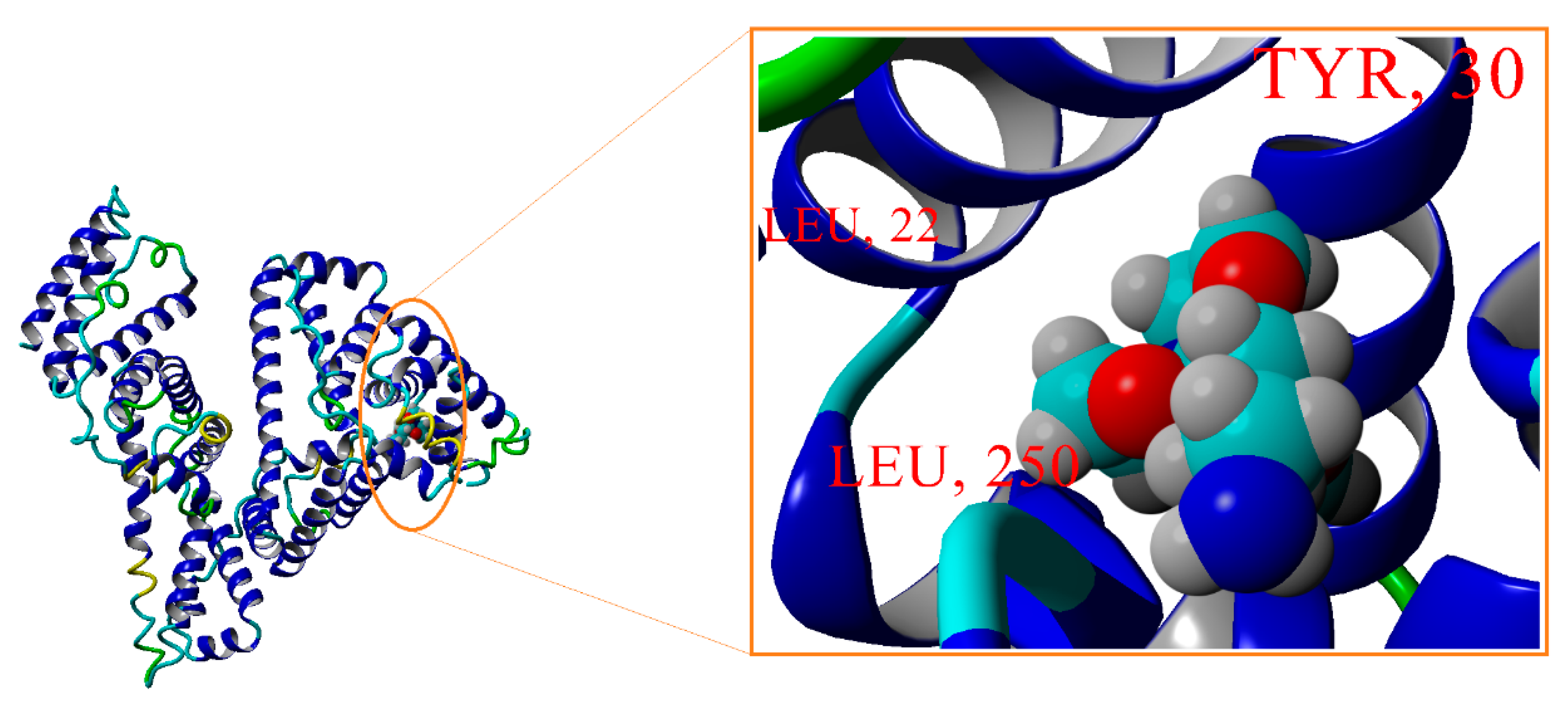
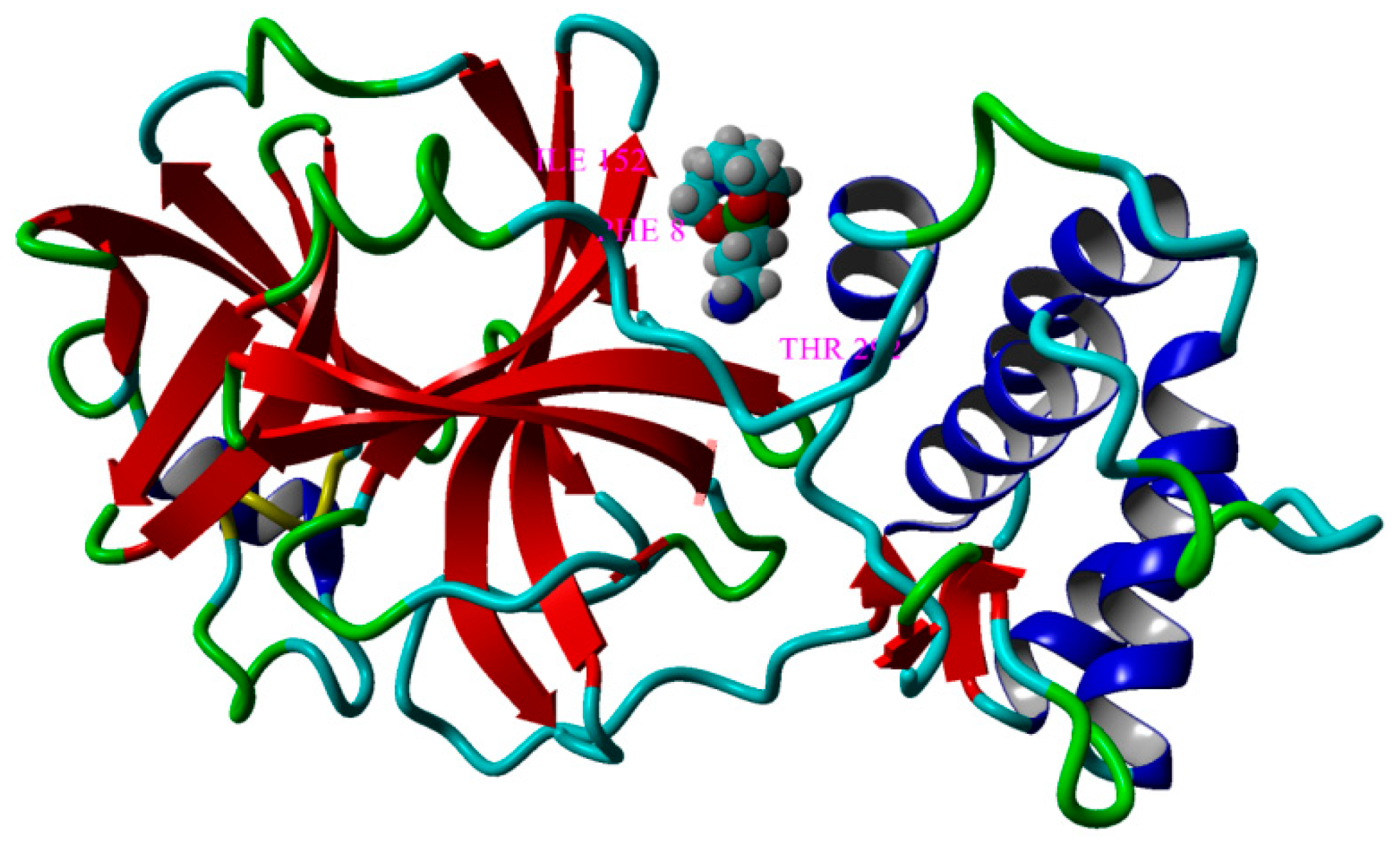
3.5.4. Molecular Docking Computations
3.6. The Bio- and Mucoadhesiveness
3.7. Antimicrobial Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mills, J.S.; Graham, A.S. Exploitation of silicon medicinal chemistry in drug discovery. Expert Opin. Investig. Drugs 2004, 13, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Sieburth, S.M.; Chen, C.-A. Silanediol Protease Inhibitors: From Conception to Validation. Eur. J. Org. Chem. 2006, 2, 311–322. [Google Scholar] [CrossRef]
- Pooni, P.K.; Showell, G.A. Silicons witches of marketed drugs. Mini Rev. Med. Chem. 2006, 6, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D. Silicon in Drug Molecules, Revisited. Science, Chemical New. Available online: https://www.science.org/content/blog-post/silicon-drug-molecules-revisited (accessed on 27 May 2022).
- Barnes, M.J.; Conroy, R.; Miller, D.J.; Mills, J.S.; Montana, J.G.; Pooni, P.K.; Showell, G.A.; Walsh, L.M.; Warneck, J.B. Trimethylsilylpyrazoles as novel inhibitors of p38 MAP kinase: A new use of silicon bioisosteres in medicinal chemistry. Bioorg. Med. Chem. Lett. 2007, 17, 354–357. [Google Scholar] [CrossRef]
- Adamovich, S.N.; Sadykov, E.K.; Ushakov, I.A.; Oborina, E.N.; Belovezhets, L.A. Antibacterial activity of new silatrane pyrrole-2-carboxamide hybrids. Mendeleev Commun. 2021, 31, 204–206. [Google Scholar] [CrossRef]
- Adamovich, S.N.; Oborina, E.N.; Nalibayeva, A.M.; Rozentsveig, I.B. 3-Aminopropylsilatrane and Its Derivatives: A Variety of Applications. Molecules 2022, 27, 3549. [Google Scholar] [CrossRef]
- Puri, J.K.; Singh, R.; Chahal, V.K. Silatranes: A review on their synthesis, structure, reactivity and applications. Chem. Soc. Rev. 2011, 40, 1791–1840. [Google Scholar] [CrossRef]
- Singh, G.; Arora, A.; Mangat, S.S.; Rani, S.; Kaur, H.; Goyal, K.; Sehgal, R.; Maurya, I.K.; Tewari, R.; Choquesillo-Lazarte, D.; et al. Design, Synthesis and biological evaluation of chalconyl blended triazole allied organosilatranes as giardicidal and trichomonacidal agents. Eur. J. Med. Chem. 2016, 108, 287–300. [Google Scholar] [CrossRef]
- Voronkov, M.G.; Baryshok, V.P. Antitumor activity of silatranes—A review. Pharm. Chem. J. 2004, 38, 3–7. Available online: https://link.springer.com/article/10.1023/B%3APHAC.0000027635.41154.0d (accessed on 30 May 2022). [CrossRef]
- Dumitriu, A.-M.-C.; Cazacu, M.; Shova, S.; Turta, C.; Simionescu, B.C. Synthesis and structural characterization of 1-(3-aminopropyl)silatrane and some new derivatives. Polyhedron 2012, 33, 119–126. [Google Scholar] [CrossRef]
- Bargan, A.; Zaltariov, M.F.; Vlad, A.; Dumitriu, A.-M.-C.; Soroceanu, A.; Macsim, A.-M.; Dascalu, M.; Varganici, C.D.; Cazacu, M.; Shova, S. Keto-enol tautomerism in new silatranes Schiff bases tailed with different substituted salicylicaldehyde. Arab. J. Chem. 2020, 13, 3100–3111. [Google Scholar] [CrossRef]
- Singh, G.; Singh, J.; Singh, A.; Singh, J.; Kumar, M.; Gupta, K.; Chhibber, S. Synthesis, characterization and antibacterial studies of schiff based 1,2,3-triazole bridged silatranes. J. Organomet. Chem. 2018, 871, 21–27. [Google Scholar] [CrossRef]
- Adamovich, S.N.; Vchislo, N.V.; Oborina, E.N.; Ushakovb, I.A.; Rozentsvei, I.B. Novel α,β-unsaturated imine derivatives of 3-aminopropylsilatrane. Mendeleev Commun. 2017, 27, 443–445. [Google Scholar] [CrossRef]
- Adamovich, S.N.; Ushakov, I.A.; Afonin, A.V.; Vchislo, N.V.; Oborina, E.N.; Pavlov, D.V. O-and S-containing 1-azadiene derivatives of 3-aminopropylsilatrane. Russ. Chem. Bull. 2021, 70, 406–411. [Google Scholar] [CrossRef]
- Singh, G.; Arora, A.; Kalra, P.; Maurya, I.K.; Ruizc, C.E.; Estebanc, M.A.; Sinha, S.; Goyal, K.; Sehgal, R. A strategic approach to the synthesis of ferrocene appended chalcone linked triazole allied organosilatranes: Antibacterial, Antifungal, Antiparasitic and Antioxidant studies. Bioorg. Med. Chem. 2019, 27, 188–195. [Google Scholar] [CrossRef]
- Li, Z.; Song, X.; Su, H.; Chen, J. Synthesis of 1-substituted benzoyl aminopropylsilatranes and their biological activities. Heterocycl. Commun. 2005, 11, 475–478. [Google Scholar] [CrossRef] [Green Version]
- Materna, K.L.; Brennan, B.J.; Brudvig, G.W. Silatranes for binding inorganic complexes to metaloxide surfaces. Dalton Trans. 2015, 44, 20312–20315. [Google Scholar] [CrossRef] [Green Version]
- Bratasz, A.; Weir, N.M.; Parinandi, N.L.; Zweier, J.L.; Sridhar, R.; Ignarro, L.J.; Kuppusamy, P. Reversal to cisplatin sensitivity in recurrent human ovarian cancer cells by NCX-4016, a nitro derivative of aspirin. Proc. Natl. Acad. Sci. USA 2006, 103, 3914–3919. [Google Scholar] [CrossRef] [Green Version]
- Güngör, T.; Tokay, E.; Güven Gülhan, Ü.; Hacıoğlu, N.; Çelik, A.; Köçkar, F.; Ay, M. Prodrugs for nitroreductase based cancer therapy- 4: Towards prostate cancer targeting: Synthesisof N-heterocyclic nitro prodrugs, Ssap-NtrB enzymatic activation and anticancer evaluation. Bioorg. Chem. 2020, 105, 104450. [Google Scholar] [CrossRef]
- Xu, G.; McLeod, H.L. Strategies for enzyme/prodrug cancer therapy. Clin. Cancer Res. 2001, 7, 3314–3324. [Google Scholar]
- Huerta, S. Nitric oxide for cancer therapy. Future Sci. OA 2015, 1, FSO55. [Google Scholar] [CrossRef]
- Kim, T.; Suh, J.; Kim, J.; Kim, W.J. Lymph-Directed Self-Immolative Nitric Oxide Prodrug for Inhibition of Intractable Metastatic. Cancer Adv. Sci. 2022, 9, 2101935. [Google Scholar] [CrossRef]
- Lee, S.M.; Lo, K.M.; Liew, L.Y.; Tan, C.H.; Sim, J.S.; Tiekink, E.R.T. Synthesis, characterization and biological activity of diorganotin compounds of (E)-N′-(5-nitro-2-hydroxybenzylidene)-3-hydroxy-2-naphthohydrazide. Polyhedron 2022, 223, 115955. [Google Scholar] [CrossRef]
- Devi, J.; Pachwania, S.; Kumar, D.; Jindal, D.K.; Jan, S.; Dash, A.K. Diorganotin(IV) complexes derived from thiazole Schiffbases: Synthesis, characterization, antimicrobial and cytotoxic studies. Res. Chem. Intermed. 2022, 48, 267–289. [Google Scholar] [CrossRef]
- Marri, S.; Kakkerla, R.; Krishna, M.P.S.M.; Rajam, M.V. Synthesis and antimicrobial evaluation of isoxazole-substituted 1,3,4-oxadiazoles. Heterocycl. Commun. 2018, 24, 285–292. [Google Scholar] [CrossRef]
- CrysAlis RED; Version 1.171.40.53; Oxford Diffraction Ltd.: Oxford, UK, 2003.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Turtoi, M.; Anghelache, M.; Patrascu, A.A.; Maxim, C.; Manduteanu, I.; Calin, M.; Popescu, D.-L. Synthesis, Characterization, and InVitro Insulin-Mimetic Activity Evaluation of Valine Schiff Base Coordination Compounds of Oxidovanadium(V). Biomedicines 2021, 9, 562. [Google Scholar] [CrossRef]
- Turtoi, M.; Anghelache, M.; Bucatariu, S.M.; Deleanu, M.; Voicu, G.; Safciuc, F.; Manduteanu, I.; Fundueanu, G.; Simionescu, M.; Calin, M. A novel platform for drug testing: Biomimetic three-dimensional hyaluronic acid-based scaffold seeded withhuman hepato carcinoma cells. Int. J. Biol. Macromol. 2021, 185, 604–619. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smart phones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Official Web-Site of YASARA Software. Available online: www.yasara.org/ (accessed on 27 May 2022).
- Herzfeld, R.; Nagy, P. Studies of the Solvent Effect Observed in the Absorption Spectra of Certain Types of Schiff Bases. Curr. Org. Chem. 2001, 5, 373–394. [Google Scholar] [CrossRef]
- Ziółek, M.; Kubicki, J.; Maciejewski, A.; Naskrcki, R.; Grabowska, A. Enol-keto tautomerism of aromatic photochromic Schiffbase N,N′-bis(salicylidene)-p-phenylenediamine: Ground state equilibrium and excited state deactivation studied by solvatochromic measurements on ultrafast time scale. J. Chem. Phys. 2006, 124, 124518. [Google Scholar] [CrossRef] [PubMed]
- Khalaji, A.D.; Foroghnia, A.; Khalilzadeh, M.A.; Fejfarova, K.; Dušek, M. 2-(3,4-Dimethoxyphenyl)-1H-benzimidazole. Acta Cryst. 2011, 67, o3255. [Google Scholar] [CrossRef] [Green Version]
- Brennan, B.J.; Gust, D.; Brudvig, G.W. Organosilatrane building blocks. Tetrahedron Lett. 2014, 55, 1062–1064. [Google Scholar] [CrossRef]
- Franz, A.K.; Wilson, S.O. Organosilicon Molecules with Medicinal Applications. J. Med. Chem. 2013, 56, 388–405. [Google Scholar] [CrossRef]
- Babgi, B.A.; Alzahrani, A. Optical Sensing Properties of Pyrene-Schiff Bases toward Different Acids. J. Fluoresc. 2016, 26, 1415–1419. [Google Scholar] [CrossRef]
- Metzler, C.M.; Cahill, A.; Metzler, D.E. Equilibria and Absorption Spectra of Schiff Bases. J. Am. Chem. Soc. 1980, 102, 6075–6082. [Google Scholar] [CrossRef]
- Voronkov, M.G.; Toryashinova, D.-S.D.; Baryshok, V.P.; Shainyan, B.A.; Brodskaya, E.I. Kinetics of hydrolysis of silatranes in a neutral medium. Bull. Acad. Sci. USSR Div. Chem. Sci. 1984, 33, 2447–2450. Available online: https://link.springer.com/article/10.1007/BF00960256 (accessed on 13 December 2022). [CrossRef]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepali, K.; Lee, H.-Y.; Liou, J.-P. Nitro Group Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Spada, A.; Emami, J.; Tuszynski, J.A.; Lavasanifar, A. The Uniqueness of Albumin as a Carrier in Nanodrug Delivery. Mol. Pharm. 2021, 18, 1862–1894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, F.; Xiong, X.; Ge, Y.; Liu, Y. Spectroscopic and molecular docking studies on the interaction of Dimetridazole with human serum albumin. J. Chil. Chem. Soc. 2013, 58, 1717–1721. [Google Scholar] [CrossRef] [Green Version]
- Suryawanshi, V.D.; Walekar, L.S.; Gore, A.H.; Anbhule, P.V.; Kolekar, G.B. S pectroscopic analysis on the binding interaction of biologically active pyrimidine derivative with bovine serum albumin. J. Pharm. Anal. 2016, 6, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Mishra, V.; Heath, R.J. Structural andB iochemical Features of Human Serum Albumin Essential for Eukaryotic Cell Culture. Int. J. Mol. Sci. 2021, 2, 8411. [Google Scholar] [CrossRef]
- Barreca, D.; Laganà, G.; Ficarra, S.; Tellone, E.; Leuzzi, U.; Magazù, S.; Galtieri, A.; Bellocco, E. Anti-aggregation properties of trehalose on heat-induced secondary structure and conformation changes of bovine serum albumin. Biophys. Chem. 2010, 147, 146–152. [Google Scholar] [CrossRef]
- Cojocaru, C.; Clima, L. Binding assessment of methylene blue to human serum albumin and poly(acrylic acid): Experimental and computer-aided modeling studies. J. Mol. Liq. 2019, 285, 811–821. [Google Scholar] [CrossRef]
- Wei, Y.; Thyparambil, A.A.; Latour, R.A. Protein Helical Structure Determination Using CD Spectroscopy for Solutions with Strong Background Absorbance from 190–230 nm. Biochim. Biophys. Acta 2014, 1844, 2331–2337. [Google Scholar] [CrossRef] [Green Version]
- Ahanger, I.A.; Parray, Z.A.; Nasreen, K.; Ahmad, F.; Hassan, M.I.; Islam, A.; Sharma, A. Heparin Accelerates the Protein Aggregation via the Downhill Polymerization Mechanism: Multi-Spectroscopic Studies to Delineate the Implications on Proteinopathies. ACS Omega 2021, 6, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Batool, A.; Arshad, R.; Razzaq, S.; Nousheen, K.; Kiani, M.H.; Shahnaz, G. Formulation and evaluation of hyaluronic acid-based mucoadhesive self nanoemulsifying drug delivery system (SNEDDS) of tamoxifen for targeting breast cancer. Int. J. Biol. Macromol. 2020, 152, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, S.A.; Smart, J.D. An investigation into the role of water movement and mucus gel dehydration in mucoadhesion. J. Control Release 1993, 25, 197–203. [Google Scholar] [CrossRef]
- Yang, N.J.; Hinner, M.J. Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Methods Mol Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkade, J.G. Main group atranes: Chemical and structural features. Coord. Chem. Rev. 1994, 137, 233–295. [Google Scholar] [CrossRef]
- Garabadzhiu, A.V.; Voronkov, M.G.; Nyanikova, G.G.; Samokhin, G.S.; Vrazhnov, D.V.; Kochina, T.A. The Influence of Silatranes, Germatranes, Protatranes, and Triethanolamine on Vital Functions of Microorganisms. Dokl. Biol. Sci. 2011, 439, 264–266. [Google Scholar] [CrossRef]
- Bonnet, M.; Lagier, J.C.; Raoult, D.; Khelaifia, S. Bacterial culture through selective andnon-selective conditions:The evolution of culture media in clinical microbiology. New Microbe New Infect. 2020, 34, 100622. Available online: https://jglobal.jst.go.jp/en/detail?JGLOBAL_ID=202002250908961004 (accessed on 14 December 2022). [CrossRef]
- Kabara, J.J.; Conley, A.J.; Truant, J.P. Relationship of Chemical Structure and Antimicrobial Activity of Alkyl Amides and Amines. Antimicrob. Agents Chemother. 1972, 6, 492–498. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, K.; Caputo, G.A.; DeGrado, W.F. The Role of Hydrophobicity in the Antimicrobial and Hemolytic Activities of Polymethacrylate Derivatives. Chem. Eur. J. 2009, 15, 123–1133. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Song, X.; Liu, J.; Xu, X.; Wang, Y.; Hu, L.; Wang, Y.; Liang, G.; Guo, P.; Xie, Z. Design, Synthesis, and Biological Evaluation of γ-Aminopropyl Silatrane—Acyclovir Hybrids with Immunomodulatory Effects. Chem. Biol. Drug Des. 2015, 86, 905–910. [Google Scholar] [CrossRef]
- Palanisamy, K.; Rubavathy, S.M.E.; Prakash, M.; Thilagavathi, R.; Hosseini-Zare, M.S.; Selvam, C. Antiviral activities of natural compounds and ionic liquids to inhibit the Mpro of SARS-CoV-2: A computational approach. RSC Adv. 2022, 12, 3687–3695. [Google Scholar] [CrossRef] [PubMed]
- Thurakkal, L.; Singh, S.; Roy, R.; Kar, P.; Sadhukhan, S.; Porel, M. An in-silico study on selected organosulfur compounds as potential drugs for SARS-CoV-2 infection via binding multiple drug targets. Chem. Phys. Lett. 2021, 763, 138193. [Google Scholar] [CrossRef] [PubMed]
- Haribabu, J.; Garisetti, V.; Malekshah, R.E.; Srividya, S.; Gayathri, D.; Bhuvanesh, N.; Mangalaraja, R.V.; Echeverria, C.; Karvembu, R. Design and synthesis of heterocyclicazole based bioactive compounds: Molecular structures, quantum simulation, and mechanistic studies through docking as multi-target inhibitors of SARS-CoV-2 and cytotoxicity. J. Mol. Struct. 2022, 15, 131782. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Solvent Characteristics | SIL-BS | ||
|---|---|---|---|---|
| Relative Permittivity | Polarity Index (Pi) | λmaxEnol (ε) | λmaxKeto (ε) | |
| DMSO | 47.24 | 7.2 | 368 (27,500) | 410 (33,796) |
| ACN | 36.64 | 5.8 | 358 (18,450) | 404 (20,708) |
| MeOH | 33.00 | 5.1 | 352 (16,300) | 392 (16,426) |
| DCM | 8.93 | 3.1 | 356 (28,594) | 408 (28,784) |
| THF | 7.52 | 4 | 354 (20,436) | 406 (21,148) |
| Compound | SIL-BS |
|---|---|
| empirical formula | C16H25N3O7Si |
| Fw | 399.48 |
| space group | C2/c |
| a [Å] | 23.484(2) |
| b [Å] | 18.2616(18) |
| c [Å] | 9.4033(10) |
| α [°] | 90 |
| β[°] | 99.431(9) |
| γ [°] | 90 |
| V [Å3] | 3978.2(7) |
| Z | 8 |
| ρcalcd[g cm−3] | 1.334 |
| Crystal size [mm] | 0.45 × 0.15 × 0.1 |
| T [K] | 293(2) |
| μ [mm−1] | 0.160 |
| 2Θ range | 3.516 to 50.048 |
| Reflections collected | 8645 |
| Independent reflections | 3513 [Rint = 0.0516] |
| Data/restraints/parameters | 3513/0/247 |
| R1 [a] | 0.0944 |
| wR2 [b] | 0.2847 |
| GOF [c] | 1.042 |
| Largest diff. peak/hole / e Å−3 | 0.66/−0.36 |
| D-H-A | d(D-H)/Å | d(H-A)/Å | d(D-A)/Å | D-H-A/° |
|---|---|---|---|---|
| N2-H2-O6 1 | 0.86 | 2.13 | 2.839(7) | 139.9 |
| N2-H2-O6 | 0.86 | 2.05 | 2.698(7) | 131.2 |
| O1w-H1wA-O2 | 0.85 | 2.03 | 2.866(6) | 167 |
| O1w-H1wB-O32 | 0.85 | 2.1 | 2.910(6) | 157.7 |
| Docked complex (Receptor@Ligand) | No. of Contacting Residues | Contacting Residues in the Receptor (HSA) | Eb (kcal/mol) | Kd (µM) |
|---|---|---|---|---|
| HSA@SIL M | 14 | LEU22, VAL23, ALA26, PHE27, TYR30, VAL46, LEU66, HIS67, PHE70, ASN99, ASP249, LEU250, LEU251, GLU252 | −7.470 | 3.345 |
| HSA@SIL-BS | 12 | ARG114, LEU115, LEU135, TYR138, LEU139, ILE142, ARG145, HIS146, ALA158, TYR161, ARG186, LYS190 | −9.315 | 0.148 |
| Sample | Bio-Adhesion | Muco-Adhesion | ||
|---|---|---|---|---|
| Adhesion Force (N) | Work of Adhesion (mJ) | Adhesion Force (N) | Work of Adhesion (mJ) | |
| SIL M | 0.11539 ± 0.00911 | 0.00857 ± 0.00085049 | 0.04778 ± 0.00247 | 0.00623 ± 0.00215 |
| SIL-BS | 0.16367 ± 0.02607 | 0.03117 ± 0.0024 | 0.09283 ± 0.01178 | 0.013 ± 0.000655744 |
| Sample | MIC a (µg/mL) | ||||
|---|---|---|---|---|---|
| Fungi | Bacteria | ||||
| Aspergillus fumigatus | Penicillium chrysogenum | Fusarium | Bacillus sp. | Pseudomonas sp. | |
| SIL M | 1.20 ± 0.02 | 1.20 ± 0.01 | 1.21 ± 0.01 | 2.80 ± 0.01 | 2.90 ± 0.11 |
| SIL-BS 1% | 2.08 ± 0.21 | 1.90 ± 0.12 | 2.08 ± 0.11 | 4.04 ± 0.21 | 4.08 ± 0.24 |
| SIL-BS 25% | >32 | >32 | >32 | >256 | >256 |
| Caspofungin b | 0.72 ± 0.01 | 0.72 ± 0.01 | 0.72 ± 0.01 | - | - |
| Kanamycin b | - | - | - | 1.8 ± 1.11 | 1.6 ± 0.98 |
| Docked Complex (Receptor@Ligand) | No. of Contacting Residues | Contacting Residues in the Receptor (MPRO) | Eb (kcal/mol) | Kd (µM) |
|---|---|---|---|---|
| MPRO@SIL M | 14 | PHE8, LYS102, GLN110, THR111, PHE112, GLN127, ASN151, ILE152, ASP153, SER158, THR292, PHE294, ASP295, ARG298 | −5.794 | 56.61 |
| Remdesivir * | −7.17 | |||
| Hidroxychloroquine * | −6.68 | |||
| Chloroquine * | −6.47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaltariov, M.-F.; Turtoi, M.; Peptanariu, D.; Macsim, A.-M.; Clima, L.; Cojocaru, C.; Vornicu, N.; Ciubotaru, B.-I.; Bargan, A.; Calin, M.; et al. Chemical Attachment of 5-Nitrosalicylaldimine Motif to Silatrane Resulting in an Organic–Inorganic Structure with High Medicinal Significance. Pharmaceutics 2022, 14, 2838. https://doi.org/10.3390/pharmaceutics14122838
Zaltariov M-F, Turtoi M, Peptanariu D, Macsim A-M, Clima L, Cojocaru C, Vornicu N, Ciubotaru B-I, Bargan A, Calin M, et al. Chemical Attachment of 5-Nitrosalicylaldimine Motif to Silatrane Resulting in an Organic–Inorganic Structure with High Medicinal Significance. Pharmaceutics. 2022; 14(12):2838. https://doi.org/10.3390/pharmaceutics14122838
Chicago/Turabian StyleZaltariov, Mirela-Fernanda, Mihaela Turtoi, Dragos Peptanariu, Ana-Maria Macsim, Lilia Clima, Corneliu Cojocaru, Nicoleta Vornicu, Bianca-Iulia Ciubotaru, Alexandra Bargan, Manuela Calin, and et al. 2022. "Chemical Attachment of 5-Nitrosalicylaldimine Motif to Silatrane Resulting in an Organic–Inorganic Structure with High Medicinal Significance" Pharmaceutics 14, no. 12: 2838. https://doi.org/10.3390/pharmaceutics14122838