
Therapeutic Effect of a Latent Form of Cortistatin in Experimental Inflammatory and Fibrotic Disorders

Abstract
:1. Introduction
2. Materials and Methods
2.1. The Animals and Ethic Statement
2.2. Reagents
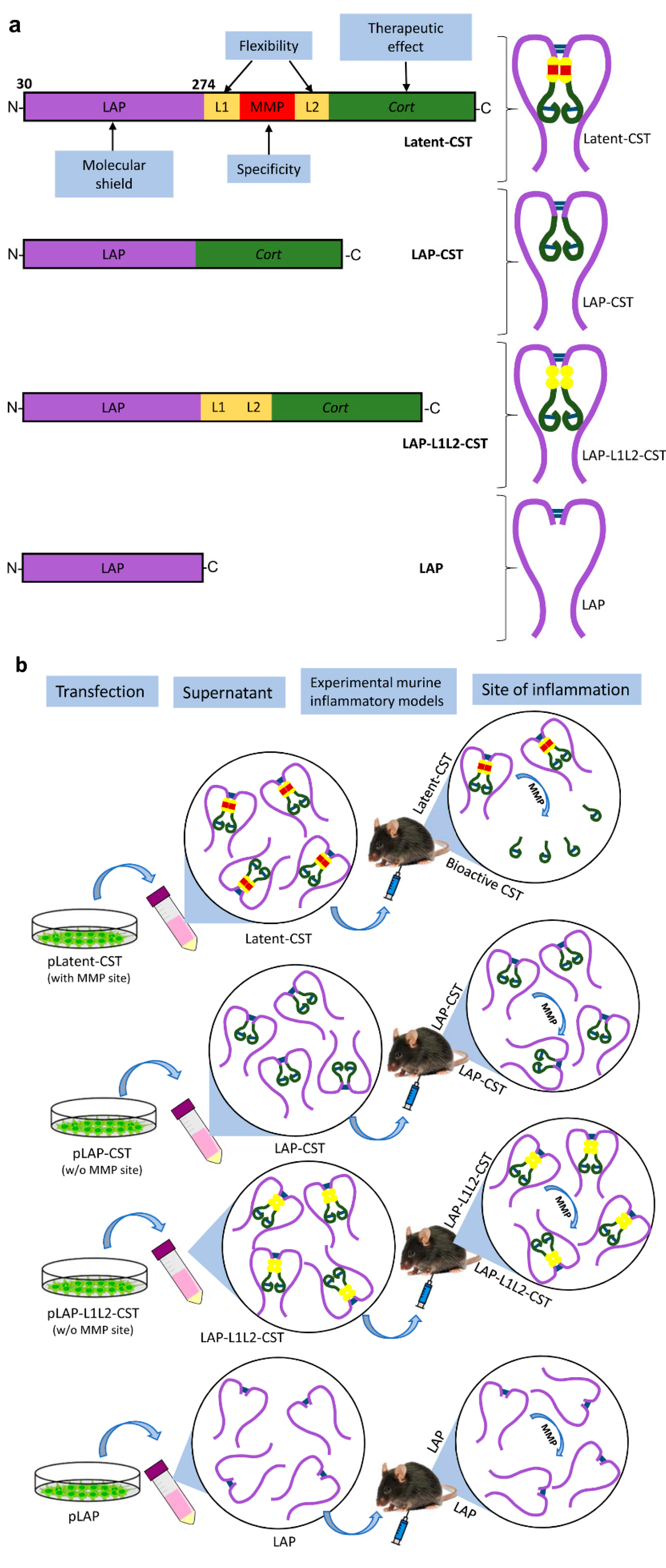
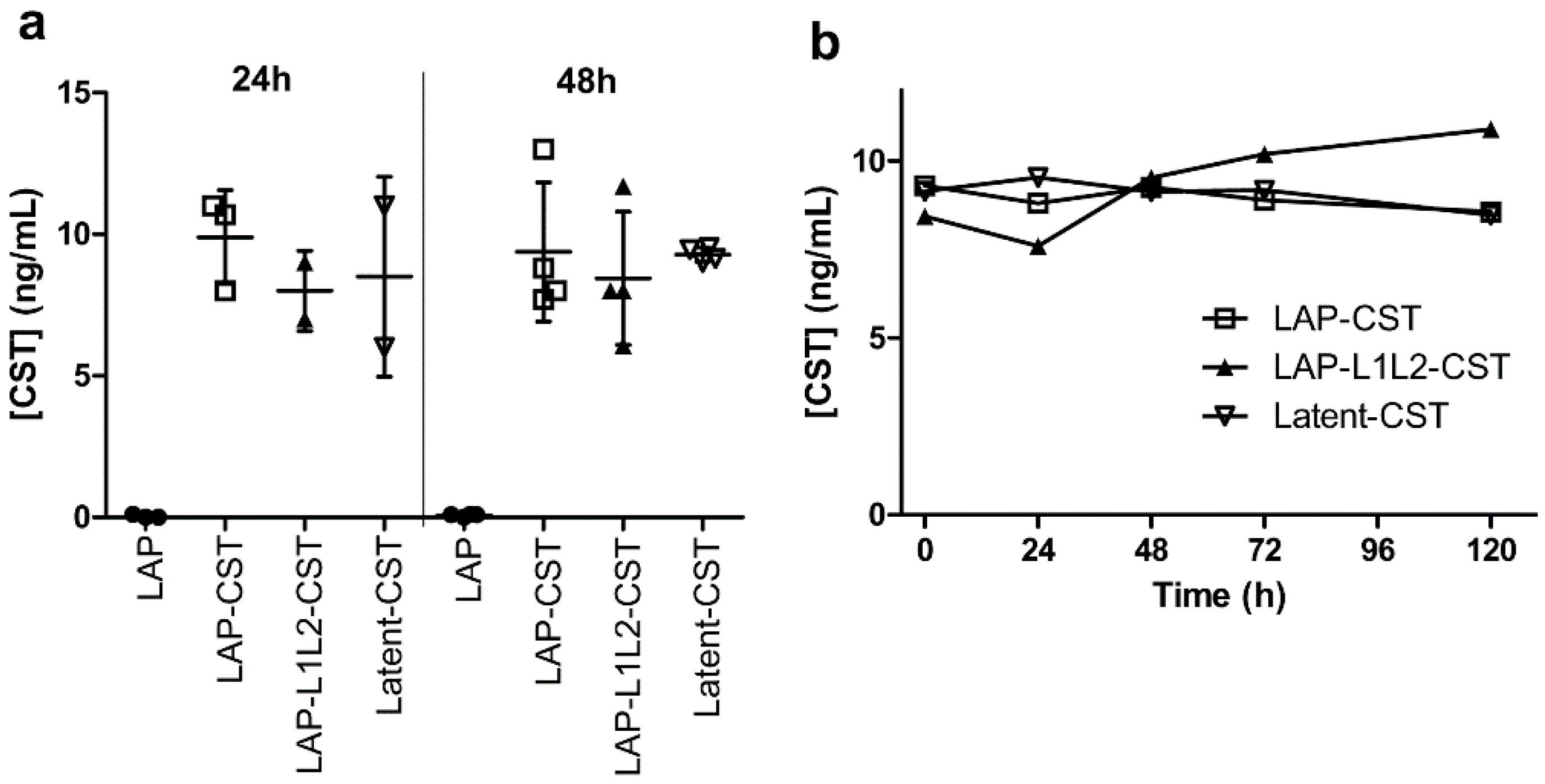
2.3. Cloning and Expression of Latent-CST
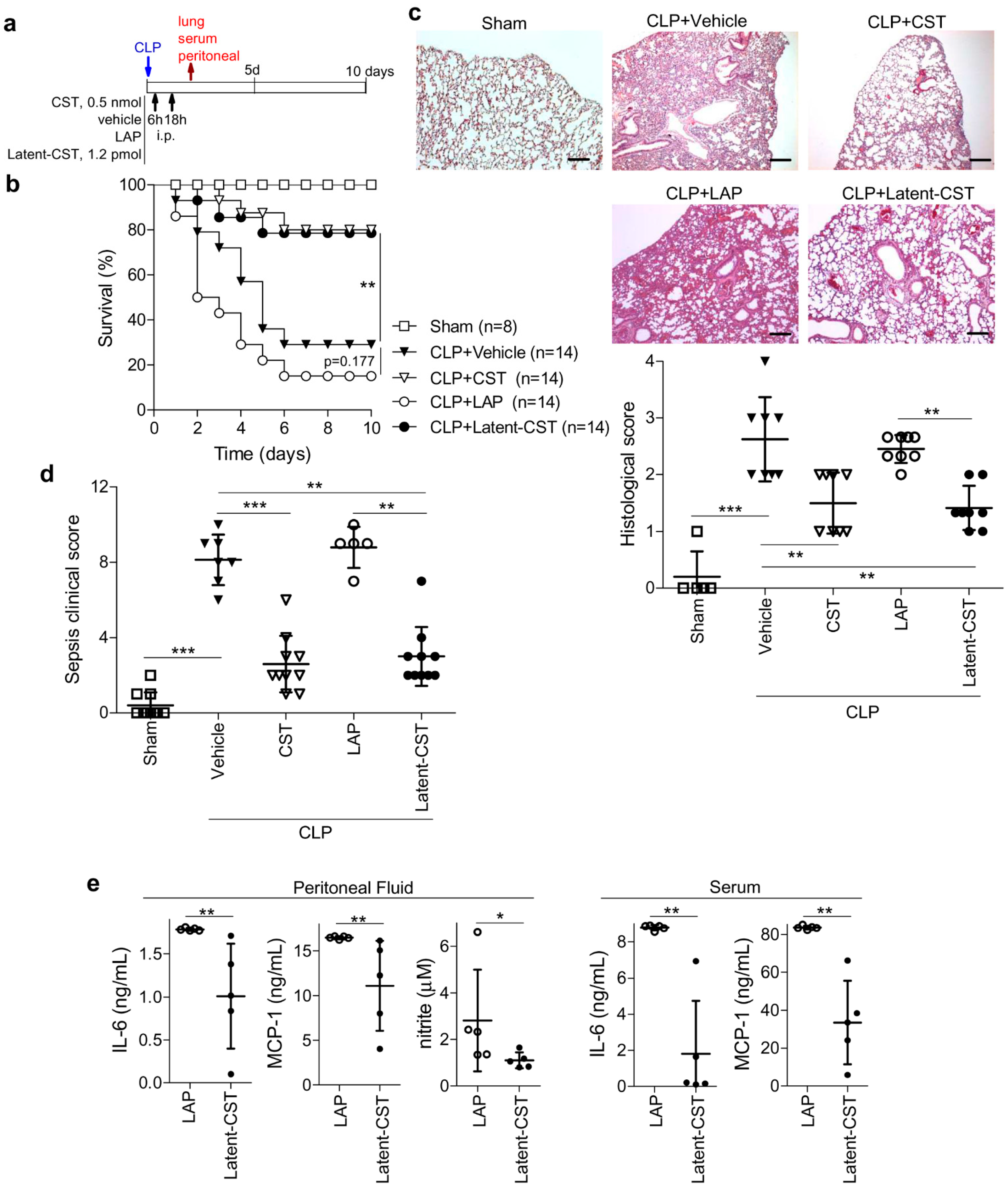
2.4. Induction and Treatment of Sepsis
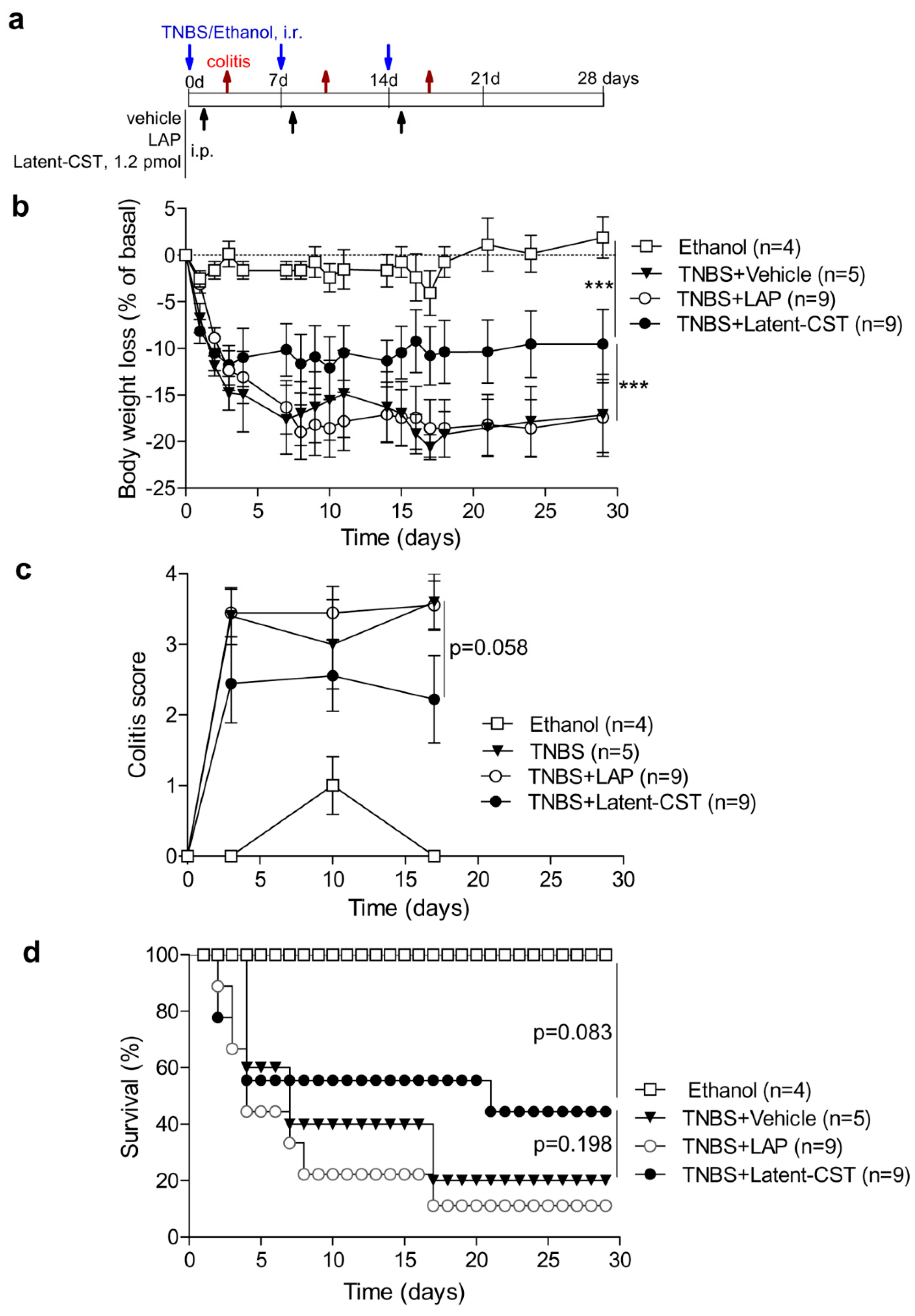
2.5. Induction and Treatment of IBD
2.6. Induction and Treatment of Experimental Scleroderma
2.7. Induction and Treatment of Experimental Lung Fibrosis
2.8. Data and Statistical Analysis
3. Results
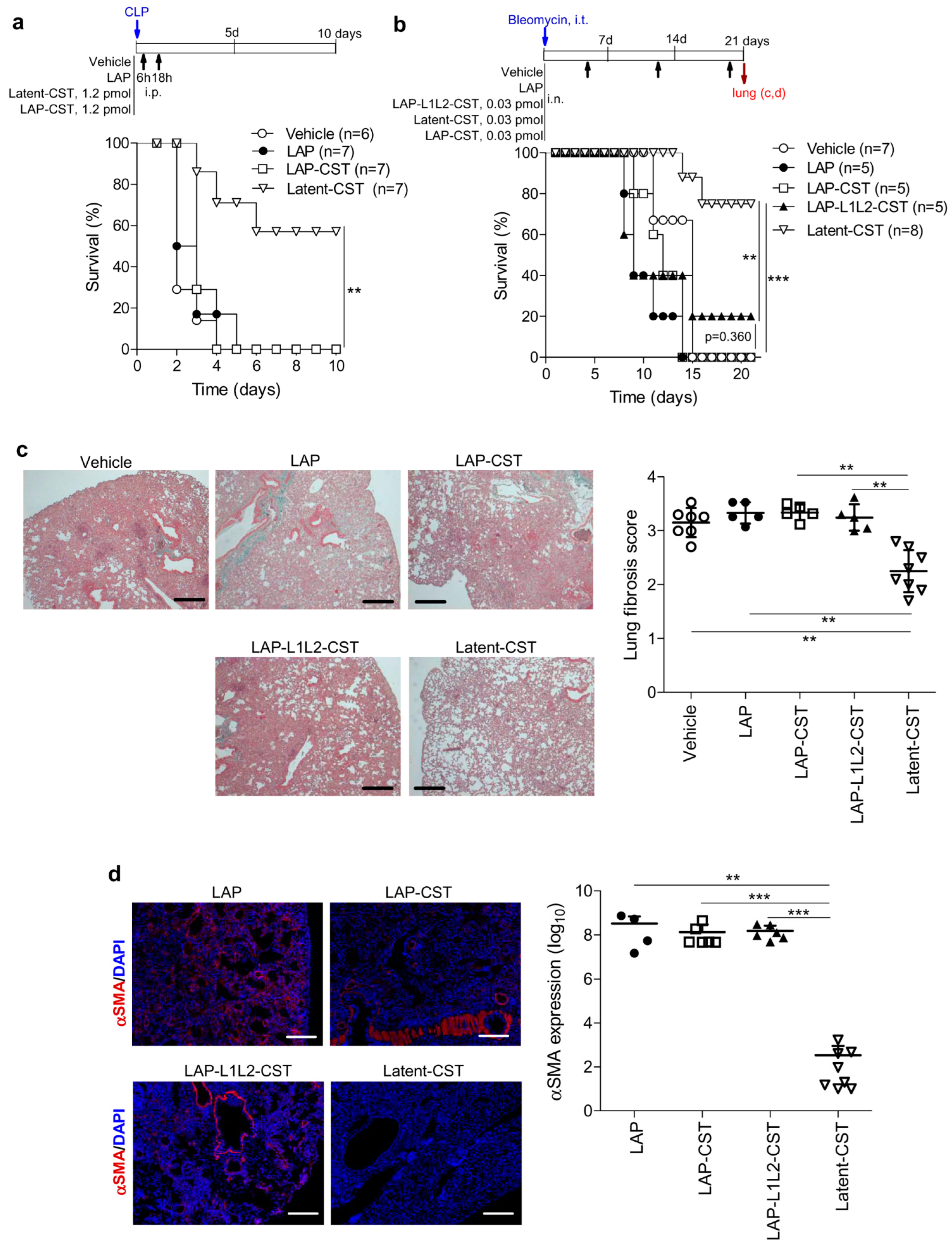
3.1. Treatment of Experimental Sepsis and IBD with Latent Cortistatin
3.2. Protective Effect of Latent-CST in Experimental Fibrosis
3.3. The MMP-Cleavage Site Is Essential for Latent-CST to Be Active in Ameliorating Inflammation and Fibrosis
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Lecea, L.; Criado, J.R.; Prospero-Garcia, Ó.; Gautvik, K.M.; Schweitzer, P.; Danielson, P.E.; Dunlop, C.L.; Siggins, G.R.; Henriksen, S.J.; Sutcliffe, G.J. A cortical neuropeptide with neuronal depressant and sleep-modulating properties. Nature 1996, 381, 242–245. [Google Scholar] [CrossRef] [PubMed]
- de Lecea, L.; Ruiz-Lozano, P.; Danielson, P.; Peelle-Kirley, J.; Foye, P.; Frankel, W.; Sutcliffe, G. Cloning, mRNA expression, and chromosomal mapping of mouse and human preprocortistatin. Genomics 1997, 42, 499–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spier, A.D.; De Lecea, L. Cortistatin: A member of the somatostatin neuropeptide family with distinct physiological functions. Brain Res. Rev. 2000, 33, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Dalm, V.A.; van Hagen, P.M.; van Koetsveld, P.M.; Achilefu, S.; Houtsmuller, A.B.; Pols, D.H.; van der Lely, A.J.; Lamberts, S.W.; Hofland, L.J. Expression of somatostatin, cortistatin, and somatostatin receptors in human monocytes, macrophages, and dendritic cells. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E344–E353. [Google Scholar] [CrossRef]
- Gonzalez-Rey, E.; Pedreño, M.; Delgado-Maroto, V.; Souza-Moreira, L.; Delgado, M. Lulling immunity, pain, and stress to sleep with cortistatin. Ann. N. Y. Acad. Sci. 2015, 1351, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Duran-Prado, M.; Morell, M.; Delgado-Maroto, V.; Castaño, J.P.; Aneiros-Fernandez, J.; de Lecea, L.; Culler, M.D.; Hernandez-Cortes, P.; O’Valle, F.; Delgado, M. Cortistatin inhibits migration and proliferation of human vascular smooth muscle cells and decreases neointimal formation on carotid artery ligation. Circ. Res. 2013, 112, 1444–1455. [Google Scholar] [CrossRef] [Green Version]
- Günther, T.; Tulipano, G.; Dournaud, P.; Bousquet, C.; Csaba, Z.; Kreienkamp, H.J.; Lupp, A.; Korbonits, M.; Castaño, J.P.; Wester, H.J.; et al. International Union of Basic and Clinical Pharmacology. CV. Somatostatin receptors: Structure, function, ligands, and new nomenclature. Pharmacol. Rev. 2018, 70, 763–835. [Google Scholar] [CrossRef] [Green Version]
- Deghenghi, R.; Papotti, M.; Ghigo, E.; Muccioli, G. Cortistatin, but not somatostatin, binds to growth hormone secretagogue (GHS) receptors of human pituitary gland. J. Endocrinol. Investig. 2001, 24, RC1–RC3. [Google Scholar] [CrossRef]
- de Lecea, L.; Castaño, J.P. Cortistatin: Not just another somatostatin analog. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 356–357. [Google Scholar] [CrossRef]
- Robas, N.; Mead, E.; Fidock, M. MrgX2 is a high potency cortistatin receptor expressed in dorsal root ganglion. J. Biol. Chem. 2003, 278, 44400–44404. [Google Scholar] [CrossRef]
- Córdoba-Chacón, J.; Gahete, M.D.; Durán-Prado, M.; Luque, R.M.; Castaño, J.P. Truncated somatostatin receptors as new players in somatostatin-cortistatin pathophysiology. Ann. N. Y. Acad. Sci. 2011, 1220, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rey, E.; Chorny, A.; Delgado, M. Regulation of immune tolerance by anti-inflammatory neuropeptides. Nat. Rev. Immunol. 2007, 7, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Maroto, V.; Falo, C.P.; Forte-Lago, I.; Adan, N.; Morell, M.; Maganto-Garcia, E.; Robledo, G.; O’Valle, F.; Lichtman, A.H.; Gonzalez-Rey, E.; et al. The neuropeptide cortistatin attenuates experimental autoimmune myocarditis via inhibition of cardiomyogenic T cell-driven inflammatory responses. Br. J. Pharmacol. 2017, 174, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, C.; Li, J.; Luo, D.; Chen, X.; Qu, R.; Liu, T.; Li, F.; Liu, Y. Cortistatin protects against inflammatory airway diseases through curbing CCL2 and antagonizing NF-κB signaling pathway. Biochem. Biophys. Res. Commun. 2020, 531, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Barriga, M.; Benitez, R.; Ferraz-de-Paula, V.; Garcia-Frutos, M.; Caro, M.; Robledo, G.; O’Valle, F.; Campos-Salinas, J.; Delgado, M. Protective role of cortistatin in pulmonary inflammation and fibrosis. Br. J. Pharmacol. 2021, 178, 4368–4388. [Google Scholar] [CrossRef]
- Wen, Q.; Ding, Q.; Wang, J.; Yin, Y.; Xu, S.; Ju, Y.; Ji, H.; Liu, B. Cortistatin-14 exerts neuroprotective effect against microglial activation, blood-brain barrier disruption, and cognitive impairment in sepsis-associated encephalopathy. J. Immunol. Res. 2022, 2022, 3334145. [Google Scholar] [CrossRef]
- Balbaba, M.; Dal, A.; Çolakoğlu, N.; Bulmuş, Ö.; Ulaş, F.; Yıldırım, H.; Aydemir, O.; Eröksüz, Y. Anti-inflammatory effect of cortistatin in rat endotoxin-induced uveitis model. Indian J. Ophthalmol. 2020, 68, 1920–1924. [Google Scholar] [CrossRef]
- Chiu, C.T.; Wen, L.L.; Pao, H.P.; Wang, J.Y. Cortistatin is induced in brain tissue and exerts neuroprotection in a rat model of bacterial meningoencephalitis. J. Infect. Dis. 2011, 204, 1563–1572. [Google Scholar] [CrossRef]
- Barriga, M.; Benitez, B.; Robledo, G.; Caro, M.; O’Valle, F.; Campos-Salinas, J.; Delgado, M. Neuropeptide cortistatin regulates dermal and pulmonary fibrosis in and experimental model of systemic sclerosis. Neuroendocrinology 2022, 112, 784–795. [Google Scholar] [CrossRef]
- Benitez, R.; Caro, M.; Andres-Leon, E.; O’Valle, F.; Delgado, M. Cortistatin regulates fibrosis and myofibroblast activation in experimental hepatotoxic- and cholestatic-induced liver injury. Br. J. Pharmacol. 2022, 179, 2275–2296. [Google Scholar] [CrossRef]
- Grottoli, S.; Gasco, V.; Broglio, F.; Baldelli, R.; Ragazzoni, F.; Gallenca, F.; Mainolfi, A.; Prodam, F.; Muccioli, G.; Ghigo, E. Cortistatin-17 and somatostatin-14 display the same effects on growth hormone, prolactin, and insulin secretion in patients with acromegaly or prolactinoma. J. Clin. Endocrinol. Metab. 2006, 91, 1595–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, R.; Picu, A.; Bonelli, L.; Broglio, F.; Prodam, F.; Grottoli, S.; Muccioli, G.; Ghigo, E.; Arvat, E. The activation of somatostatinergic receptors by either somatostatin-14 or cortistatin-17 often inhibits ACTH hypersecretion in patients with Cushing’s disease. Eur. J. Endocrinol. 2007, 157, 393–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rol, A.; Todorovski, T.; Martin-Malpartida, P.; Escola, A.; González-Rey, E.; Aragon, E.; Verdaguer, X.; Valles-Miret, M.; Farrera-Sinfreu, J.; Puig, E.; et al. Structure-based design of a cortistatin analogue with immunoregulatory activity in models of inflammatory bowel disease. Nat. Commun. 2021, 12, 1869. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Montesinos, R.; Castillo, P.M.; Klippstein, R.; Gonzalez-Rey, E.; Mejias, J.A.; Zaderenko, A.P.; Pozo, D. Chemical synthesis and characterization of silver-protected vasoactive intestinal peptide nanoparticles. Nanomedicine 2009, 4, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Athari, S.S.; Pourpak, Z.; Folkerts, G.; Garssen, J.; Moin, M.; Adcock, I.M.; Movassaghi, M.; Ardestani, M.S.; Moazzeni, S.M.; Mortaz, E. Conjugated Alpha-Alumina nanoparticle with vasoactive intestinal peptide as a Nano-drug in treatment of allergic asthma in mice. Eur. J. Pharmacol. 2016, 791, 811–820. [Google Scholar] [CrossRef]
- Jayawardena, D.; Anbazhagan, A.N.; Guzman, G.; Dudeja, P.K.; Onyuksel, H. Vasoactive intestinal peptide nanomedicine for the management of inflammatory bowel disease. Mol. Pharm. 2017, 14, 3698–3708. [Google Scholar] [CrossRef]
- Delgado, M.; Toscano, M.G.; Benabdellah, K.; Cobo, M.; O’Valle, F.; Gonzalez-Rey, E.; Martín, F. In vivo delivery of lentiviral vectors expressing vasoactive intestinal peptide complementary DNA as gene therapy for collagen-induced arthritis. Arthritis Rheum. 2008, 58, 1026–1037. [Google Scholar] [CrossRef]
- Misaka, S.; Aoki, Y.; Karaki, S.; Kuwahara, A.; Mizumoto, T.; Onoue, S.; Yamada, S. Inhalable powder formulation of a stabilized vasoactive intestinal peptide (VIP) derivative: Anti-inflammatory effect in experimental asthmatic rats. Peptides 2010, 31, 72–78. [Google Scholar] [CrossRef]
- Kolkhir, P.; Pyatilova, P.; Ashry, T.; Jiao, Q.; Abad-Perez, A.T.; Altrichter, S.; Vera Ayala, C.E.; Church, M.K.; He, J.; Lohse, K.; et al. Mast cells, cortistatin, and its receptor, MRGPRX2, are linked to the pathogenesis of chronic prurigo. J. Allergy Clin. Immunol. 2022, 149, 1998–2009. [Google Scholar] [CrossRef]
- Cao, C.; Kang, H.J.; Singh, I.; Chen, H.; Zhang, C.; Ye, W.; Hayes, B.W.; Liu, J.; Gumpper, R.H.; Bender, B.J.; et al. Structure, function and pharmacology of human itch GPCRs. Nature 2021, 600, 170–175. [Google Scholar] [CrossRef]
- Adams, G.; Vessillier, S.; Dreja, H.; Chernajovsky, Y. Targeting cytokines to inflammation sites. Nat. Biotechnol. 2003, 21, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Vessillier, S.; Adams, G.; Montero-Melendez, T.; Jones, R.; Seed, S.; Perretti, M.; Chernajovsky, Y. Molecular engineering of short half-life small peptides (VIP, αMSH and γ₃MSH) fused to Latency-Associated Peptide results in improved anti-inflammatory therapeutics. Ann. Rheum. Dis. 2012, 71, 143–149. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.T.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE Guidelines 2.0: Updated guidelines for reporting animal research. Br. J. Pharmacol. 2020, 177, 3617–3624. [Google Scholar] [CrossRef] [PubMed]
- Dumont, J.; Euwart, D.; Mei, B.; Estes, S.; Kshirsagar, R. Human cell lines for biopharmaceutical manufacturing: History, status, and future perspectives. Crit. Rev. Biotechnol. 2016, 36, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Mullen, L.; Rigby, A.; Sclanders, M.; Adams, G.; Mittal, G.; Colston, J.; Fatah, R.; Subang, C.; Foste, J.; Francis-West, P.; et al. Latency can be conferred to a variety of cytokines by fusion with latency-associated peptide from TGF-β. Expert. Opin. Drug. Deliv. 2014, 11, 5–16. [Google Scholar] [CrossRef]
- Chorny, A.; Delgado, M. Neuropeptides rescue mice from lethal sepsis by down- regulating secretion of the late-acting inflammatory mediator high mobility group box 1. Am. J. Pathol. 2008, 172, 1297–1307. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M. Inhibition of interferon (IFN) gamma-induced Jak-STAT1 activation in microglia by vasoactive intestinal peptide: Inhibitory effect on CD40, IFN-induced protein-10, and inducible nitric-oxide synthase expression. J. Biol. Chem. 2003, 278, 27620–27629. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rey, E.; Chorny, A.; Robledo, G.; Delgado, M. Cortistatin, a new antiinflammatory peptide with therapeutic effect on lethal endotoxemia. J. Exp. Med. 2006, 203, 563–571. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, E. Animal models of IBD: Linkage to human disease. Curr. Opin. Pharmacol. 2010, 10, 578–587. [Google Scholar] [CrossRef]
- Gonzalez-Rey, E.; Varela, N.; Sheibanie, A.F.; Chorny, A.; Ganea, D.; Delgado, M. Cortistatin, an antiinflammatory peptide with therapeutic action in inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2006, 103, 4228–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T. Animal model of systemic sclerosis. J. Dermatol. 2010, 37, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Perelas, A.; Silver, R.M.; Arrossi, V.; Highland, K.B. Systemic sclerosis-associated interstitial lung disease. Lancet Respir. Med. 2020, 8, 304–320. [Google Scholar] [CrossRef] [PubMed]
- Dalm, V.A.; van Hagen, P.M.; van Koetsveld, P.M.; Langerak, A.W.; van der Lely, A.J.; Lamberts, S.W.; Hofland, L.J. Cortistatin rather than somatostatin as a potential endogenous ligand for somatostatin receptors in the human immune system. J. Clin. Endocrinol. Metab. 2003, 88, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovics, A.; Szoke, É.; Sándor, K.; Börzsei, R.; Bagoly, T.; Kemény, Á.; Elekes, K.; Pintér, E.; Szolcsányi, J.; Helyes, Z. Comparison of the anti-inflammatory and antinociceptive effects of cortistatin-14 and somatostatin-14 in distinct in vitro and in vivo model systems. J. Mol. Neurosci. 2012, 46, 40–50. [Google Scholar] [CrossRef]
- Morell, M.; Camprubí-Robles, M.; Culler, M.D.; de Lecea, L.; Delgado, M. Cortistatin attenuates inflammatory pain via spinal and peripheral actions. Neurobiol. Dis. 2014, 63, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Falo, C.P.; Benitez, R.; Caro, M.; Morell, M.; Forte-Lago, I.; Hernandez-Cortes, P.; Sanchez-Gonzalez, C.; O’Valle, F.; Delgado, M.; Gonzalez-Rey, E. The neuropeptide cortistatin alleviates neuropathic pain in experimental models of peripheral nerve injury. Pharmaceutics 2021, 13, 947. [Google Scholar] [CrossRef]
- Zhao, W.; Han, S.; Qiu, N.; Feng, W.; Lu, M.; Zhang, W.; Wang, M.; Zhou, Q.; Chen, S.; Xu, W.; et al. Structural insights into ligand recognition and selectivity of somatostatin receptors. Cell Res. 2022, 32, 761–772. [Google Scholar] [CrossRef]
- Itoh, Y. Metalloproteinases in rheumatoid arthritis: Potential therapeutic targets to improve current therapies. Prog. Mol. Biol. Transl. Sci. 2017, 148, 327–338. [Google Scholar]
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 2015, 53, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Yong, V.W.; Power, C.; Forsyth, P.; Edwards, D.R. Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2001, 2, 502–511. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pLAP-CST | |
| Forward Primer | 5′TCTGCAAAGCGAATTCCAGGAAAGACCACCCCTCCAGCAGCCCCCACACCGGGATAAAAAGCCCTGCAAGAACTTCTTCTGGAAAACCTTCTCCTCGTGCAAGTAGGAATTCTGCAGATATC3′ |
| Reverse Primer | 5′GATATCTGCAGAATTCCTACTTGCACGAGGAGAAGGTTTTCCAGAAGAAGTTCTTGCAGGGCTTTTTATCCCGGTGTGGGGGCTGCTGGAGGGGTGGTCTTTCCTGGAATTCGCTTTGCAGA3′ |
| pLAP-L1L2-CST | |
| Forward Primer | 5′TCTGCAAAGCGAATTCGGGGGAGGCGGATCCGGGGGAGGGGGCTCAGCGGCCGCCCAGGAAAGACCACCCCTCCAGCAGCCCCCACACCGGGATAAAAAGCCCTGCAAGAACTTCTTCTGGAAAACCTTCTCCTCGTGCAAGTAGGAATTCTGCAGATATC3′ |
| Reverse Primer | 5′GATATCTGCAGAATTCCTACTTGCACGAGGAGAAGGTTTTCCAGAAGAAGTTCTTGCAGGGCTTTTTATCCCGGTGTGGGGGCTGCTGGAGGGGTGGTCTTTCCTGGGCGGCCGCTGAGCCCCCTCCCCCGGATCCGCCTCCCCCGAATTCGCTTTGCAGA3′ |
| pLatent-CST | |
| Forward Primer | 5′TCTGCAAAGCGAATTCGGGGGAGGCGGATCCCCGCTCGGGCTTTGGGCGGGGGGAGGGGGCTCAGCGGCCGCCCAGGAAAGACCACCCCTCCAGCAGCCCCCACACCGGGATAAAAAGCCCTGCAAGAACTTCTTCTGGAAAACCTTCTCCTCGTGCAAGTAGGAATTCTGCAGATATC3′ |
| Reverse Primer | 5′GATATCTGCAGAATTCCTACTTGCACGAGGAGAAGGTTTTCCAGAAGAAGTTCTTGCAGGGCTTTTTATCCCGGTGTGGGGGCTGCTGGAGGGGTGGTCTTTCCTGGGCGGCCGCTGAGCCCCCTCCCCCCGCCCAAAGCCCGAGCGGGGATCCGCCTCCCCCGAATTCGCTTTGCAGA3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos-Salinas, J.; Barriga, M.; Delgado, M. Therapeutic Effect of a Latent Form of Cortistatin in Experimental Inflammatory and Fibrotic Disorders. Pharmaceutics 2022, 14, 2785. https://doi.org/10.3390/pharmaceutics14122785
Campos-Salinas J, Barriga M, Delgado M. Therapeutic Effect of a Latent Form of Cortistatin in Experimental Inflammatory and Fibrotic Disorders. Pharmaceutics. 2022; 14(12):2785. https://doi.org/10.3390/pharmaceutics14122785
Chicago/Turabian StyleCampos-Salinas, Jenny, Margarita Barriga, and Mario Delgado. 2022. "Therapeutic Effect of a Latent Form of Cortistatin in Experimental Inflammatory and Fibrotic Disorders" Pharmaceutics 14, no. 12: 2785. https://doi.org/10.3390/pharmaceutics14122785