
Preparation of PLGA-Coated Porous Silica Nanofibers for Drug Release

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Self-Assembly of M-BTC Nanofibers
2.3. Preparation of Silica Nanofibers
2.4. Drug Loading on Silica Nanofibers
2.5. PLGA Coating on Silica Nanofibers
2.6. In Vitro Drug Release
2.7. Characterization
3. Results and Discussion
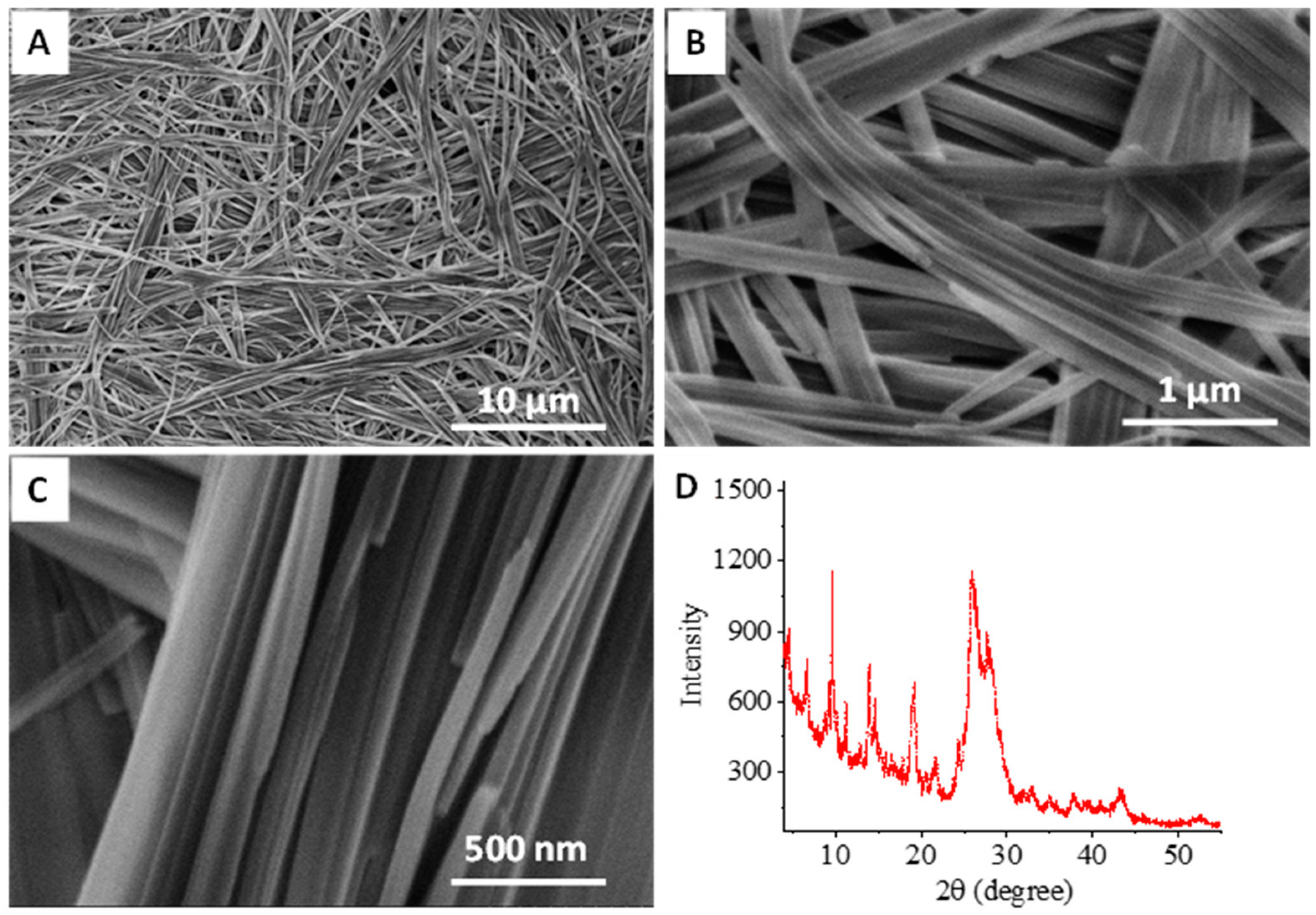
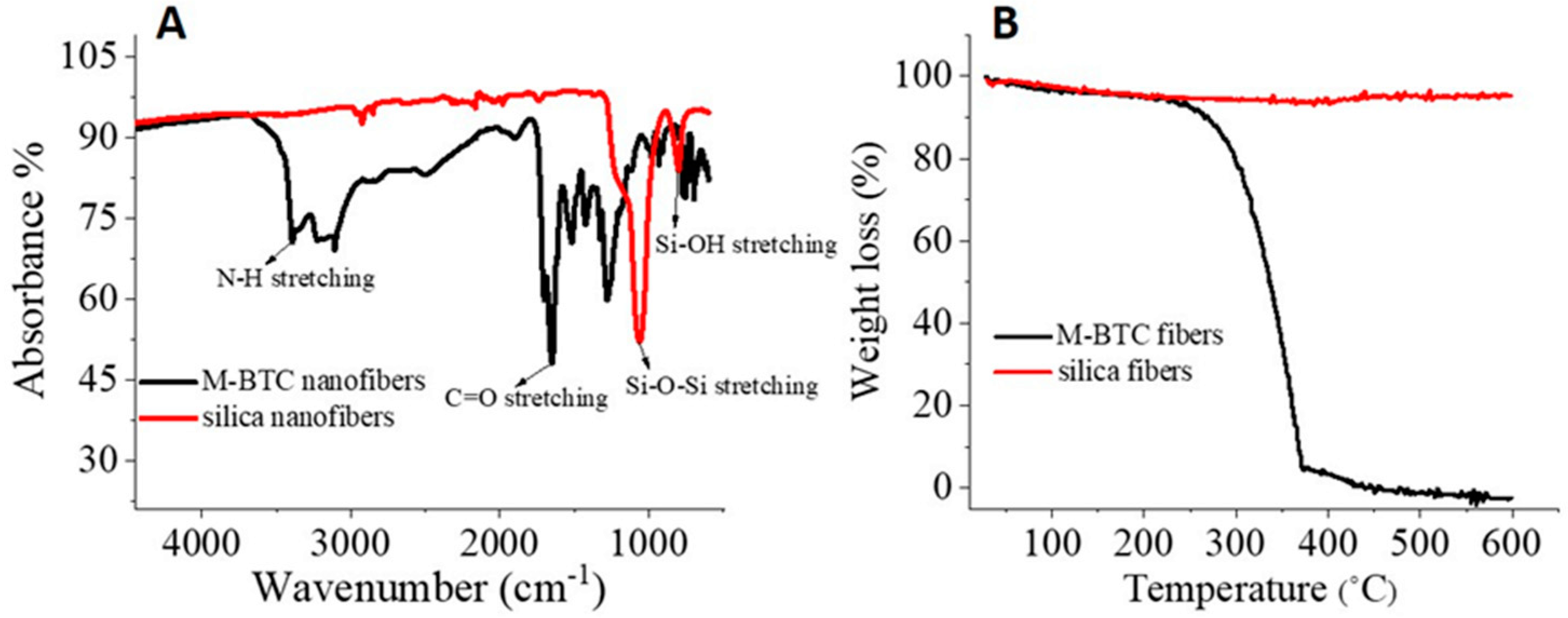
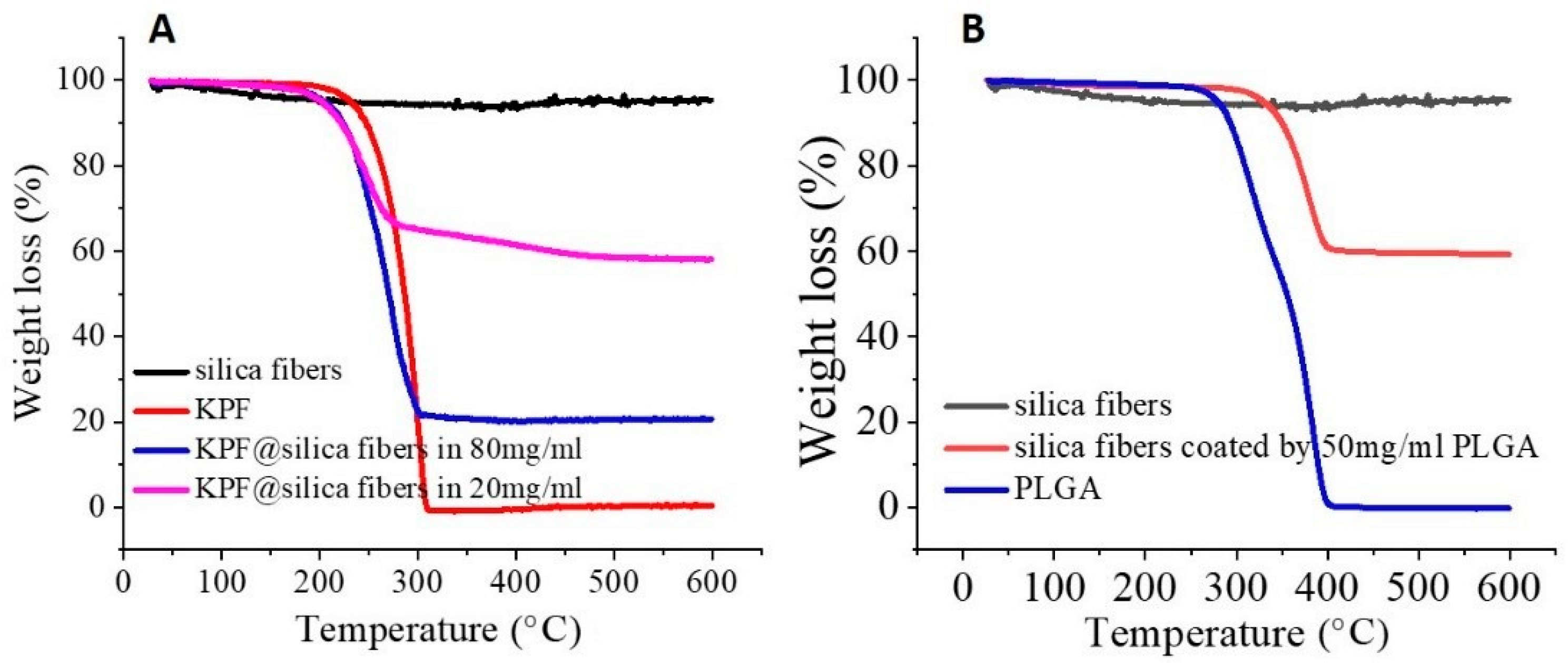
3.1. Organic Nanofibers to Silica Nanofibers
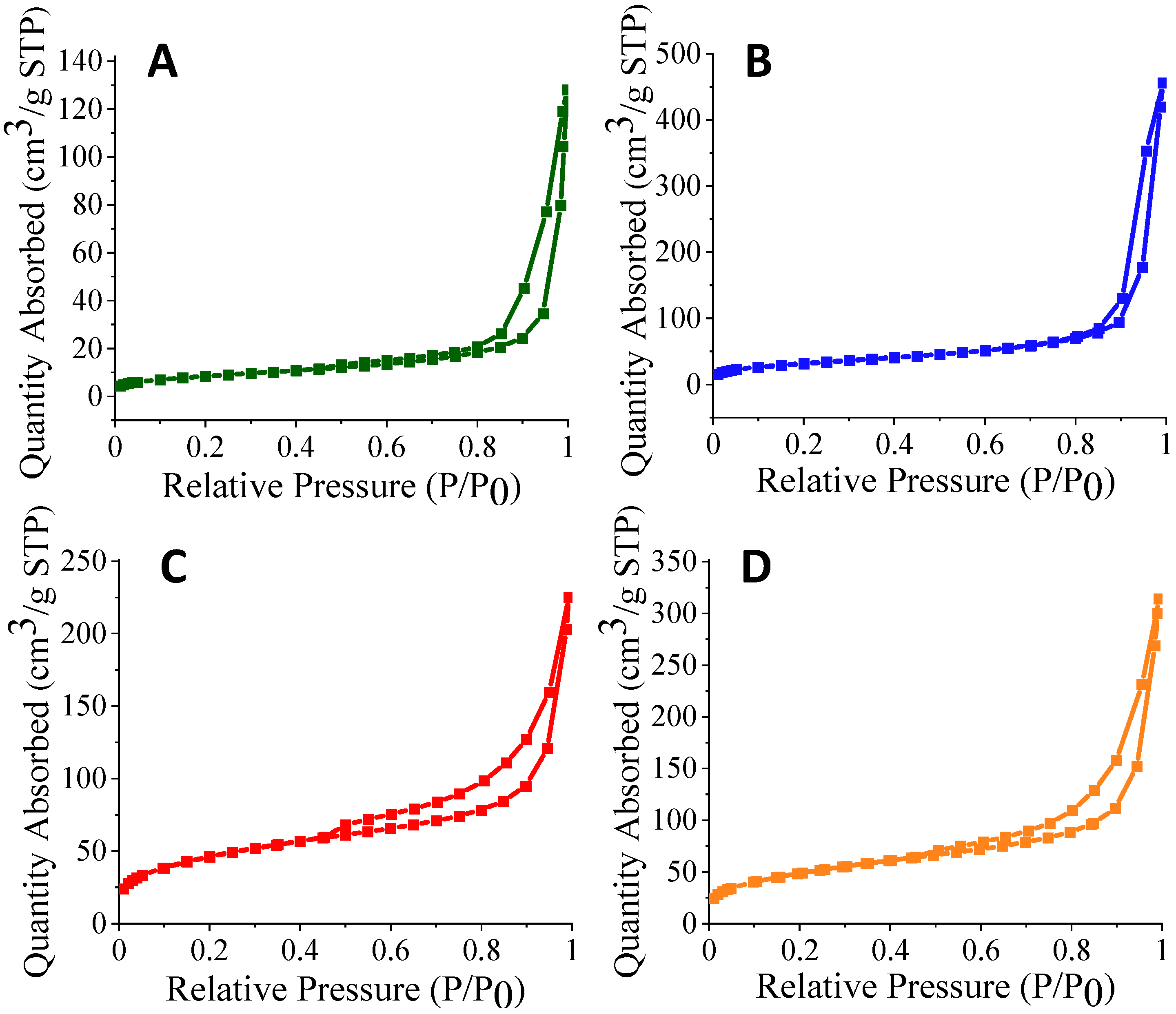
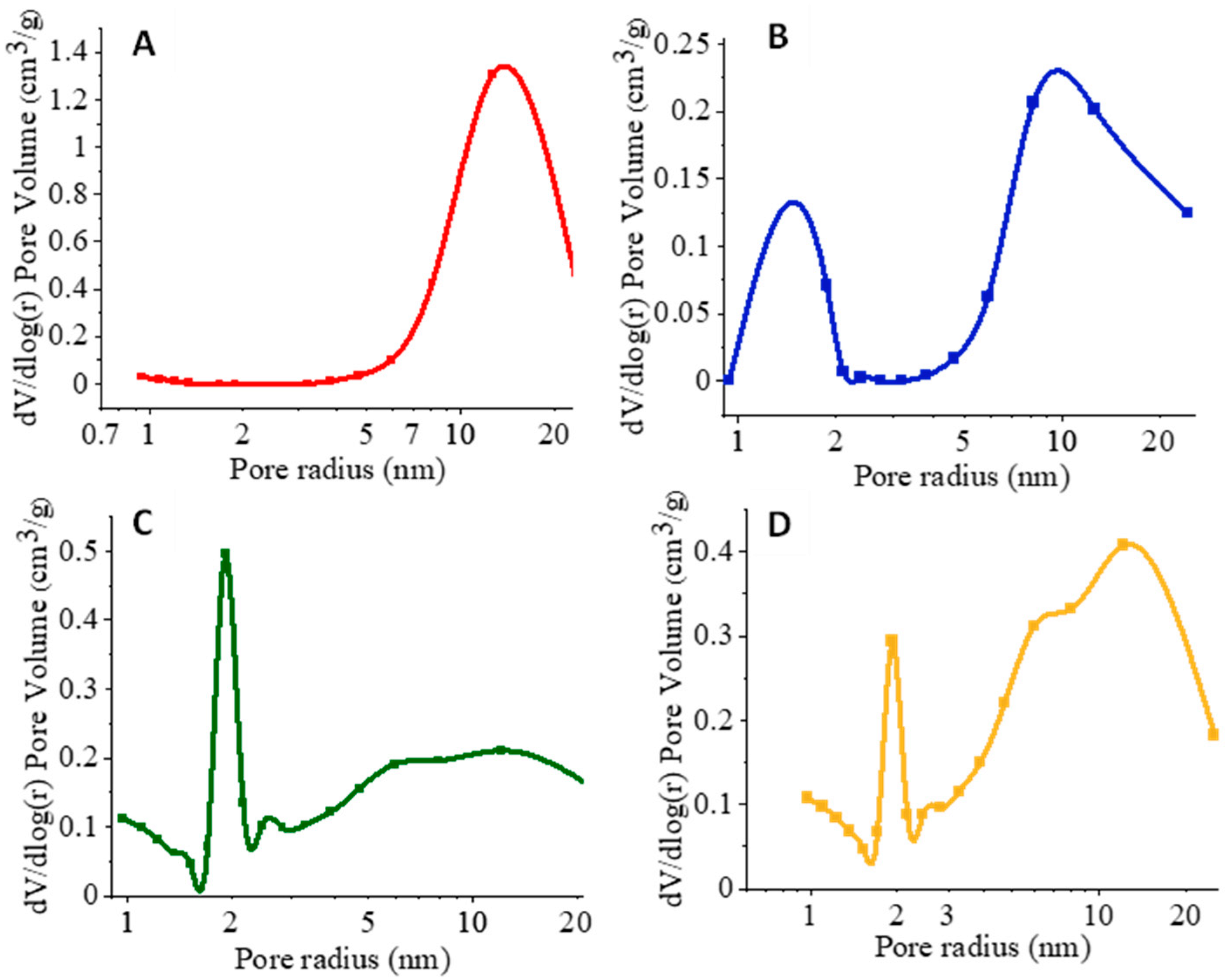
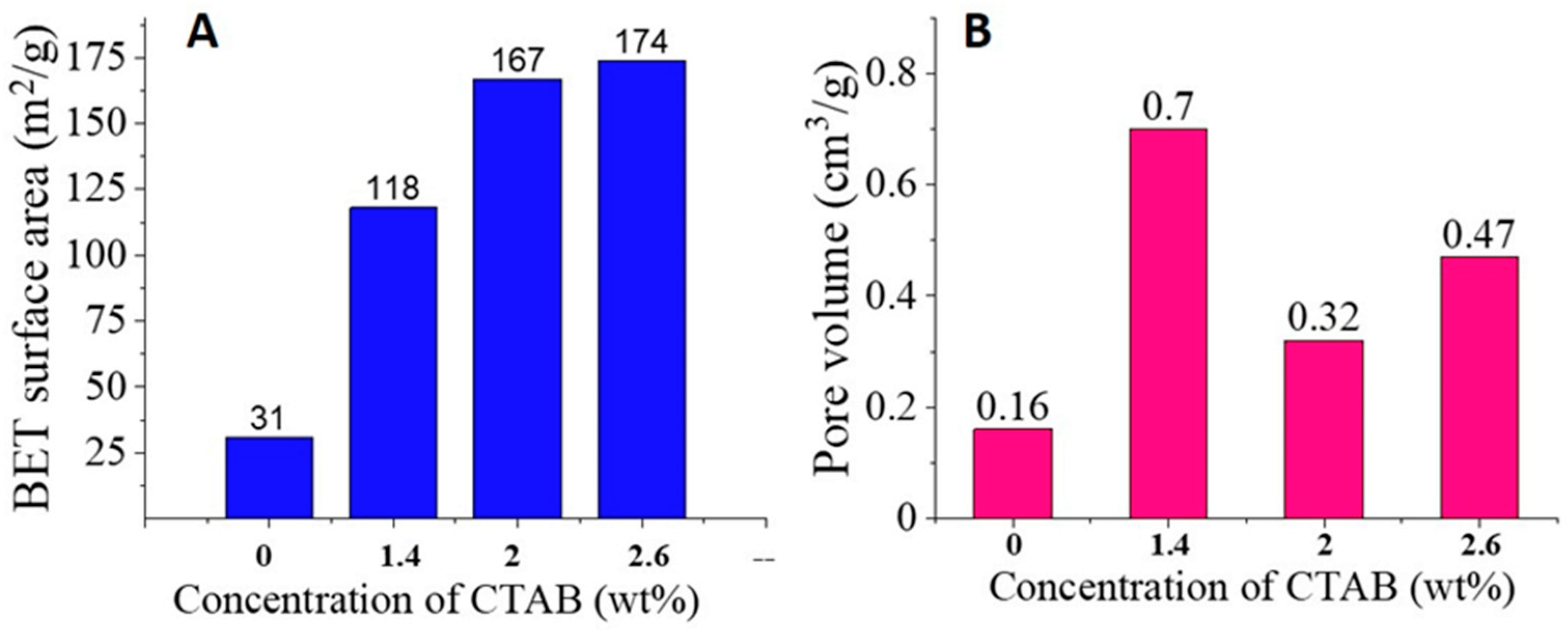
3.2. N2 Sorption Analysis of Silica Nanofibers
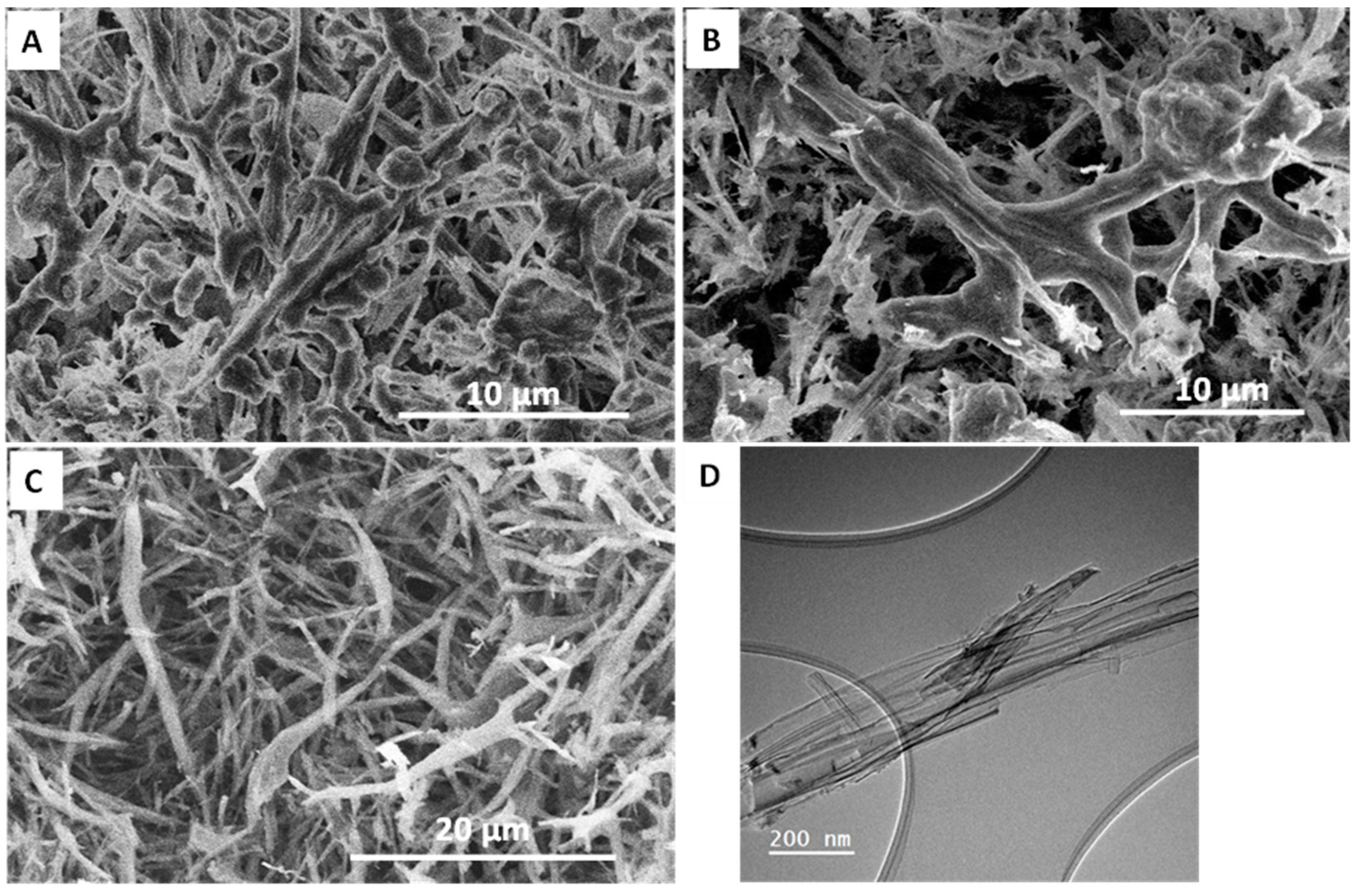
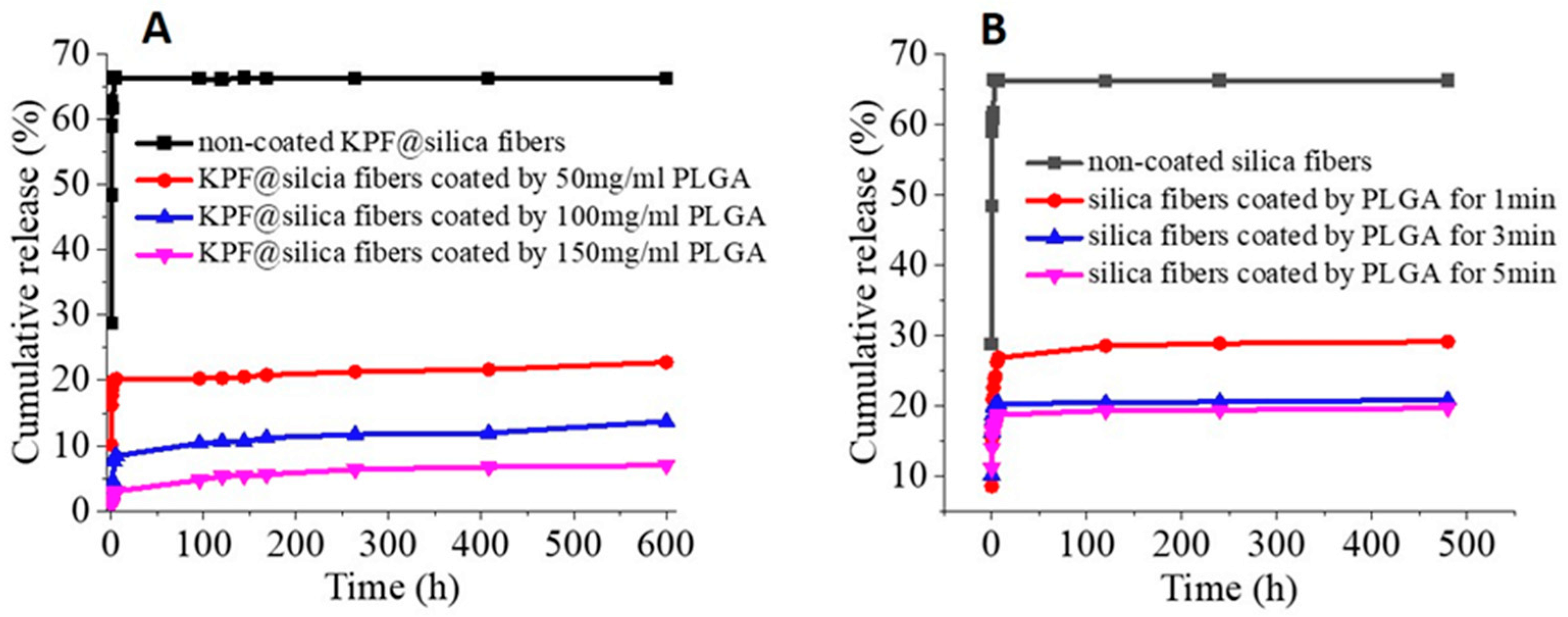
3.3. Drug Loading and Release Behavior from Silica Nanofibers
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, Y.K.; Kim, S.W. Recent advances in polymeric drug delivery systems. Biomater. Res. 2020, 24, 12. [Google Scholar] [CrossRef] [PubMed]
- Bansal, V.; Sharma, P.K.; Sharma, N.; Pal, O.P.; Malviya, R. Applications of chitosan and chitosan derivatives in drug delivery. Adv. Biol. Res. 2011, 5, 28–37. [Google Scholar]
- Horcajada, P.; Serre, C.; Vallet-Regí, M.; Sebban, M.; Taulelle, F.; Férey, G. Metal–organic frameworks as efficient materials for drug delivery. Angew. Chem. Int. Ed. 2006, 45, 5974–5978. [Google Scholar] [CrossRef] [PubMed]
- He, X.-T.; Luo, Y.-H.; Hong, D.-L.; Chen, F.-H.; Zheng, Z.-Y.; Wang, C.; Wang, J.-Y.; Chen, C.; Sun, B.-W. Atomically thin nanoribbons by exfoliation of hydrogen-bonded organic frameworks for drug delivery. ACS Appl. Nano Mater. 2019, 2, 2437–2445. [Google Scholar] [CrossRef]
- Kossovsky, N.; Gelman, A.; Sponsler, E.E.; Hnatyszyn, H.J.; Rajguru, S.; Torres, M.; Pham, M.; Crowder, J.; Zemanovich, J.; Chung, A. Surface-modified nanocrystalline ceramics for drug delivery applications. Biomaterials 1994, 15, 1201–1207. [Google Scholar] [CrossRef]
- Slowing, I.I.; Trewyn, B.G.; Giri, S.; Lin, V.Y. Mesoporous silica nanoparticles for drug delivery and biosensing applications. Adv. Funct. Mater. 2007, 17, 1225–1236. [Google Scholar] [CrossRef]
- Koosha, M.; Mirzadeh, H. Electrospinning, mechanical properties, and cell behavior study of chitosan/PVA nanofibers. J. Biomed. Mater. Res. A 2015, 103, 3081–3093. [Google Scholar] [CrossRef]
- Alavi, M.; Karimi, N.; Safaei, M. Application of various types of liposomes in drug delivery systems. Adv. Pharm. Bull. 2017, 7, 3. [Google Scholar] [CrossRef]
- Ashley, G.W.; Henise, J.; Reid, R.; Santi, D.V. Hydrogel drug delivery system with predictable and tunable drug release and degradation rates. Proc. Natl. Acad. Sci. USA 2013, 110, 2318–2323. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Li, L.; Chen, D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef]
- Lei, Q.; Guo, J.; Noureddine, A.; Wang, A.; Wuttke, S.; Brinker, C.J.; Zhu, W. Sol–gel-based advanced porous silica materials for biomedical applications. Adv. Funct. Mater. 2020, 30, 1909539. [Google Scholar] [CrossRef]
- García-González, C.A.; Sosnik, A.; Kalmár, J.; De Marco, I.; Erkey, C.; Concheiro, A.; Alvarez-Lorenzo, C. Aerogels in drug delivery: From design to application. J. Control. Release 2021, 332, 40–63. [Google Scholar] [CrossRef]
- Delasoie, J.; Zobi, F. Natural diatom biosilica as microshuttles in drug delivery systems. Pharmaceutics 2019, 11, 537. [Google Scholar] [CrossRef] [Green Version]
- García-Fernández, A.; Aznar, E.; Martínez-Máñez, R.; Sancenón, F. New advances in in vivo applications of gated mesoporous silica as drug delivery nanocarriers. Small 2020, 16, 1902242. [Google Scholar] [CrossRef]
- Esfahani, M.K.M.; Alavi, S.E.; Cabot, P.J.; Nazrul Islam, N.; Izake, E.L. Application of mesoporous silica nanoparticles in cancer therapy and delivery of repurposed anthelmintics for cancer therapy. Pharmaceutics 2022, 14, 1579. [Google Scholar] [CrossRef]
- Yuan, L.; Tang, Q.; Yang, D.; Zhang, J.Z.; Zhang, F.; Hu, J. Preparation of pH-responsive mesoporous silica nanoparticles and their application in controlled drug delivery. J. Phys. Chem. C 2011, 115, 9926–9932. [Google Scholar] [CrossRef]
- Stephen, S.; Gorain, B.; Choudhury, H.; Chatterjee, B. Exploring the role of mesoporous silica nanoparticle in the development of novel drug delivery systems. Drug Deliv. Transl. Res. 2022, 12, 105–123. [Google Scholar] [CrossRef]
- Xu, L.; Li, W.; Sadeghi-Soureh, S.; Amirsaadat, S.; Pourpirali, R.; Alijani, S. Dual drug release mechanisms through mesoporous silica nanoparticle/electrospun nanofiber for enhanced anticancer efficiency of curcumin. J. Biomed. Mater. Res. 2022, 110, 316–330. [Google Scholar] [CrossRef]
- Huang, X.; Tao, Z.; Praskavich Jr, J.C.; Goswami, A.; Al-Sharab, J.F.; Minko, T.; Polshettiwar, V.; Asefa, T. Dendritic silica nanomaterials (KCC-1) with fibrous pore structure possess high DNA adsorption capacity and effectively deliver genes in vitro. Langmuir 2014, 30, 10886–10898. [Google Scholar] [CrossRef]
- Hou, Z.; Zhang, C.; Li, C.; Xu, Z.; Cheng, Z.; Li, G.; Wang, W.; Peng, C.; Lin, J. Luminescent porous silica fibers as drug carriers. Eur. J. Chem. 2010, 16, 14513–14519. [Google Scholar] [CrossRef]
- Zhou, J.; Nie, Y.; Jin, C.; Zhang, J.X.J. Engineering biomimetic extracellular matrix with silica nanofibers: From 1D material to 3D network. ACS Biomater. Sci. Eng. 2022, 8, 2258–2280. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Li, J.; Zhang, X.; Liu, T.; Ding, J.; Chen, X. Electrospun polymer micro/nanofibers as pharmaceutical repositories for healthcare. J. Control. Release 2019, 302, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Yi, K.; Kuang, H.; You, Z.; Morsi, Y.; Mo, X. Electrospun nanofibers for tissue engineering with drug loading and release. Pharmaceutics 2019, 11, 182. [Google Scholar]
- Xue, J.; Wu, T.; Dai, Y.; Xia, Y. Electrospinning and electrospun nanofibers: Methods, materials, and applications. Chem. Rev. 2019, 119, 5298–5415. [Google Scholar] [CrossRef] [PubMed]
- Haider, A.; Haider, S.; Kang, I.-K. A comprehensive review summarizing the effect of electrospinning parameters and potential applications of nanofibers in biomedical and biotechnology. Arab. J. Chem. 2018, 11, 1165–1188. [Google Scholar] [CrossRef]
- Shahhosseininia, M.; Bazgir, S.; Joupari, M.D. Fabrication and investigation of silica nanofibers via electrospinning. Mater. Sci. Eng. C 2018, 91, 502–511. [Google Scholar] [CrossRef]
- Nagamine, S.; Kosaka, K.; Tohyama, S.; Ohshima, M. Silica nanofiber with hierarchical pore structure templated by a polymer blend nanofiber and surfactant micelle. Mater. Res. Bull. 2014, 50, 108–112. [Google Scholar] [CrossRef]
- Zhang, S. Emerging biological materials through molecular self-assembly. Biotechnol. Adv. 2002, 20, 321–339. [Google Scholar] [CrossRef]
- Jun, Y.S.; Lee, E.Z.; Wang, X.; Hong, W.H.; Stucky, G.D.; Thomas, A. From melamine-cyanuric acid supramolecular aggregates to carbon nitride hollow spheres. Adv. Funct. Mater. 2013, 23, 3661–3667. [Google Scholar] [CrossRef]
- Cong, H.-P.; Ren, X.-C.; Wang, P.; Yu, S.-H. Macroscopic multifunctional graphene-based hydrogels and aerogels by a metal ion induced self-assembly process. ACS Nano 2012, 6, 2693–2703. [Google Scholar] [CrossRef]
- Tang, J.; Liu, J.; Zheng, Q.; Li, W.; Sheng, J.; Mao, L.; Wang, M. In-situ encapsulation of protein into nanoscale hydrogen-bonded organic frameworks for intracellular biocatalysis. Angew. Chem. Int. Ed. 2021, 60, 22315–22321. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Li, B.; Dai, Y.; Chen, X. Melamine-induced novel MSONs heterostructured framework: Controlled-switching between MOF and SOF via a self-assembling approach for rapid uranium sequestration. J. Chem. Eng. 2020, 379, 122279. [Google Scholar] [CrossRef]
- Liu, J.; Shin, Y.; Nie, Z.; Chang, J.H.; Wang, L.-Q.; Fryxell, G.E.; Samuels, W.D.; Exarhos, G.J. Molecular assembly in ordered mesoporosity: A new class of highly functional nanoscale materials. J. Phys. Chem. A 2000, 104, 8328–8339. [Google Scholar] [CrossRef]
- Lee, C.C.; Grenier, C.; Meijer, E.W.; Schenning, A.P.H.J. Preparation and characterization of helical self-assembled nanofibers. Chem. Soc. Rev. 2009, 38, 671–683. [Google Scholar] [CrossRef]
- Mendes, A.C.; Baran, E.T.; Reis, R.L.; Azevedo, H.S. Self-assembly in nature: Using the principles of nature to create complex nanobiomaterials. WIREs Nanomed. Nanobiotechnol. 2013, 5, 582–612. [Google Scholar] [CrossRef]
- Wang, H.; Xu, X.; Johnson, N.M.; Dandala, N.K.R.; Ji, H.-F. High proton conductivity of water channels in a highly ordered nanowire. Angew. Chem. Int. Ed. 2011, 50, 12538–12541. [Google Scholar] [CrossRef]
- Wang, F.; Dou, L.; Dai, J.; Li, Y.; Huang, L.; Si, Y.; Yu, J.; Ding, B. In situ synthesis of biomimetic silica nanofibrous aerogels with temperature-invariant superelasticity over one million compressions. Angew. Chem. Int. Ed. 2020, 59, 8285–8292. [Google Scholar] [CrossRef]
- Smith, B.C. IR Spectral Interpretation Workshop. Organic nitrogen compounds, VII: Amides—The rest of the story. Spectroscopy 2020, 35, 10–15. [Google Scholar]
- Karadagli, I.; Schulz, B.; Schestakow, M.; Milow, B.; Gries, T.; Ratke, L. Production of porous cellulose aerogel fibers by an extrusion process. J. Supercrit. Fluids 2015, 106, 105–114. [Google Scholar] [CrossRef]
- Villarroel-Rocha, J.; Barrera, D.; Sapag, K. Introducing a self-consistent test and the corresponding modification in the Barrett, Joyner and Halenda method for pore-size determination. Microporous Mesoporous Mater. 2014, 200, 68–78. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Lidder, J.; Bahri, M.; Zhang, H. Preparation of PLGA-Coated Porous Silica Nanofibers for Drug Release. Pharmaceutics 2022, 14, 2660. https://doi.org/10.3390/pharmaceutics14122660
Zhang M, Lidder J, Bahri M, Zhang H. Preparation of PLGA-Coated Porous Silica Nanofibers for Drug Release. Pharmaceutics. 2022; 14(12):2660. https://doi.org/10.3390/pharmaceutics14122660
Chicago/Turabian StyleZhang, Meina, Jasmine Lidder, Mounib Bahri, and Haifei Zhang. 2022. "Preparation of PLGA-Coated Porous Silica Nanofibers for Drug Release" Pharmaceutics 14, no. 12: 2660. https://doi.org/10.3390/pharmaceutics14122660