
Genetically Engineered Extracellular Vesicles Harboring Transmembrane Scaffolds Exhibit Differences in Their Size, Expression Levels of Specific Surface Markers and Cell-Uptake

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Vectors and Fusion Genes
2.3. Cell Culture and Transfection
2.4. Isolation of EVs
2.5. Live Cell Microscopy
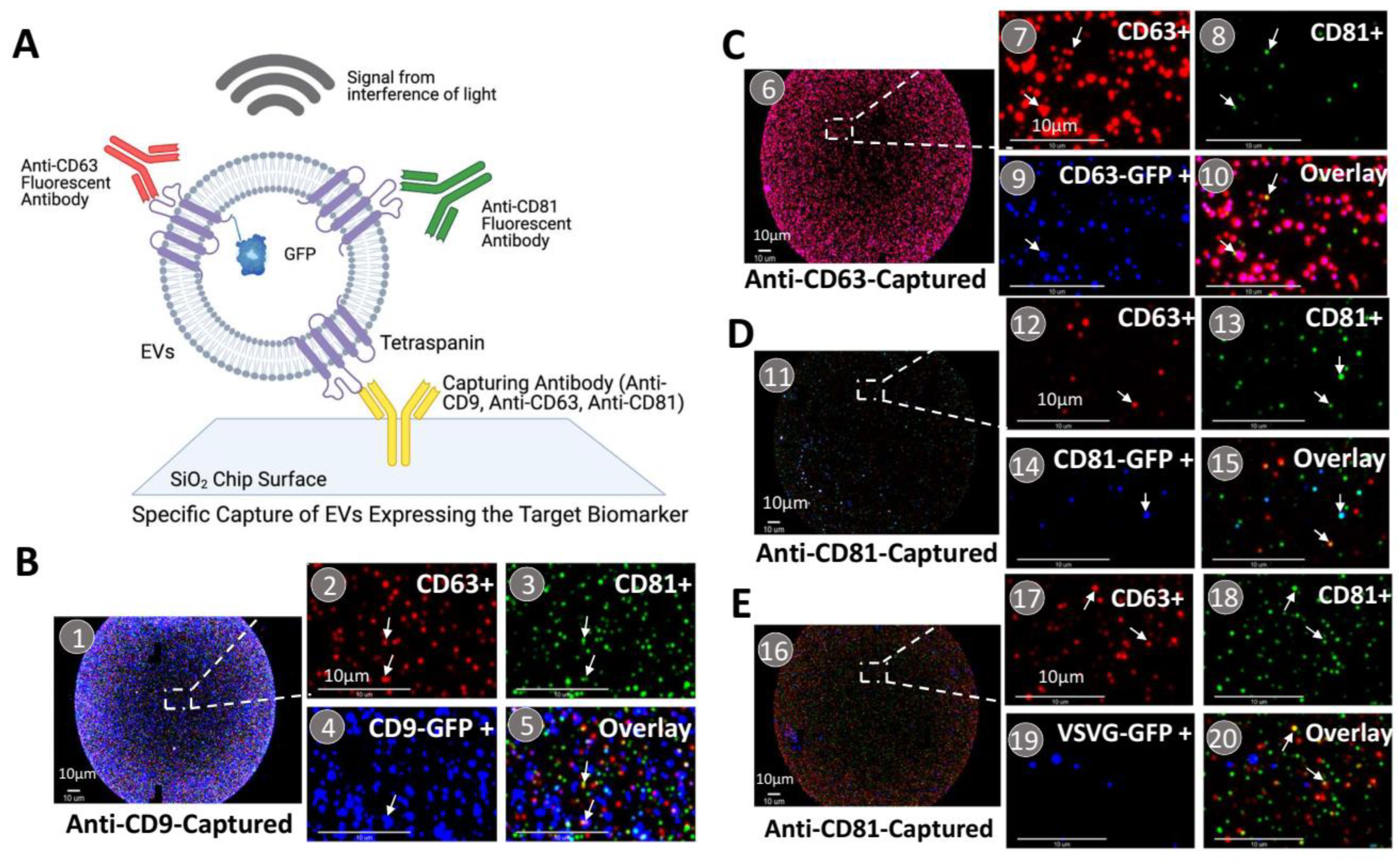
2.6. On-Chip Capture and Characterization of EVs
2.7. Fluorescein-Labeling of EVs
2.8. Cellular Uptake Assay
2.9. Flow Cytometry Analysis
2.10. Data Analysis and Statistics
3. Results
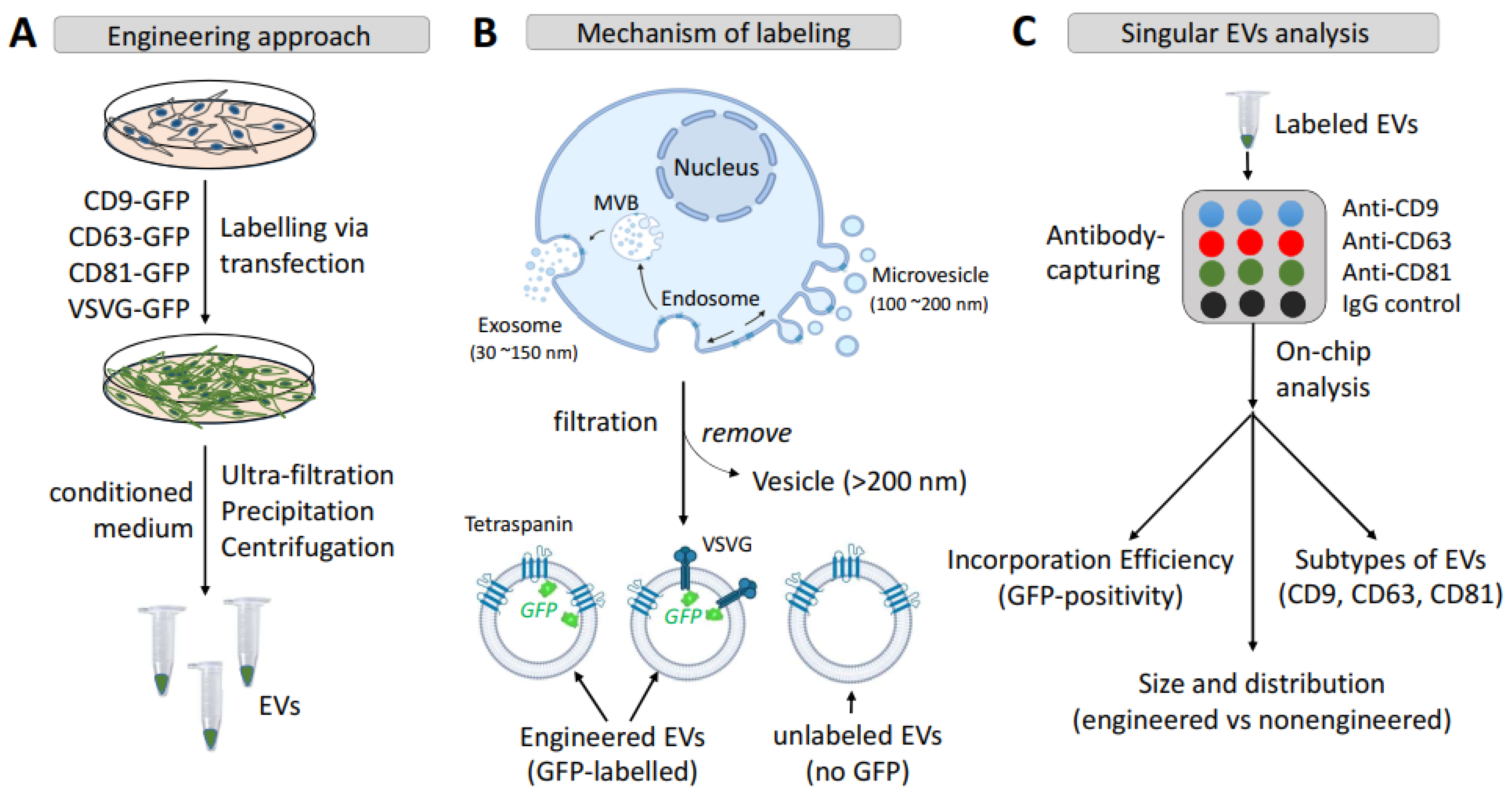
3.1. Engineering Strategy and Experimental Design
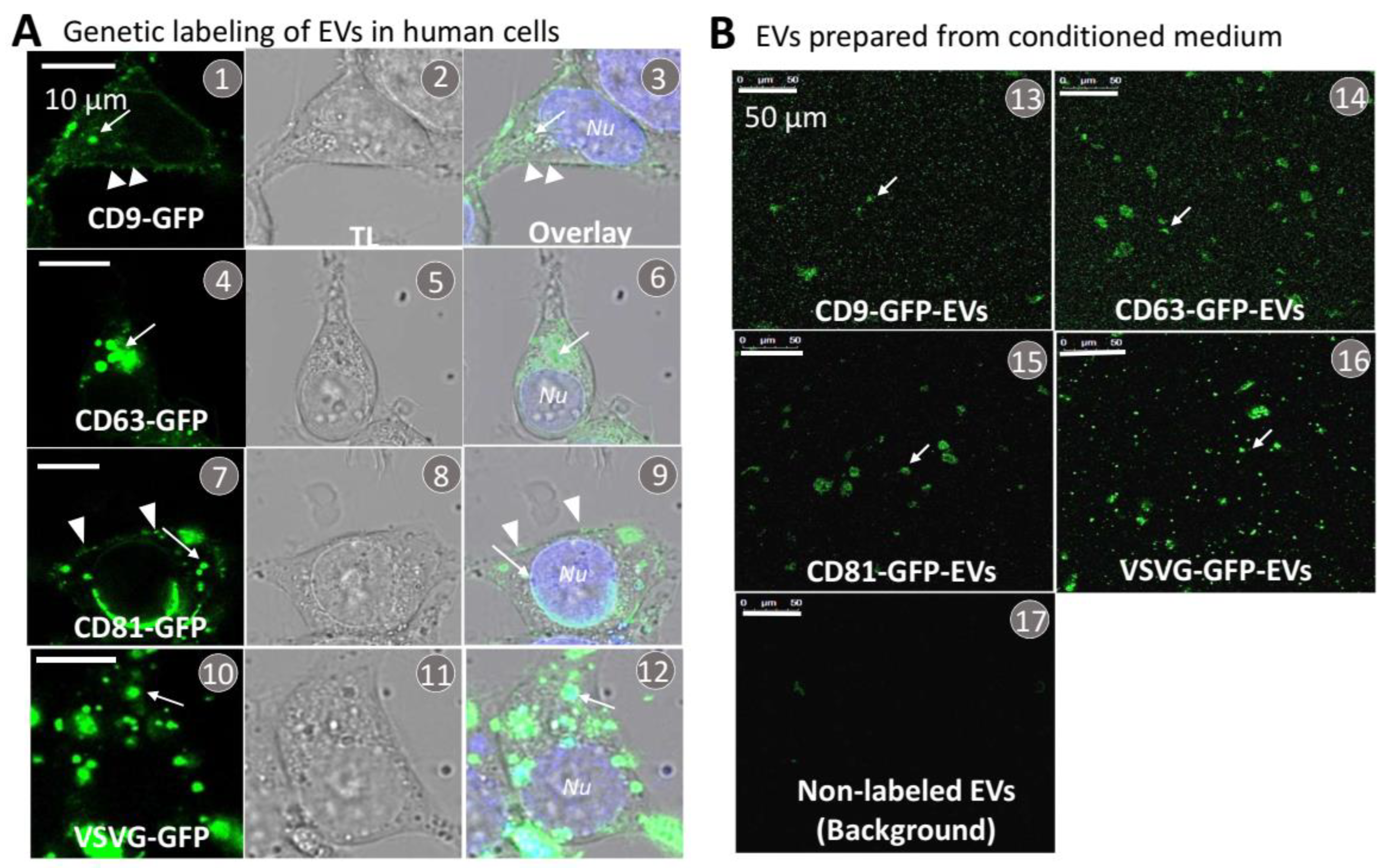
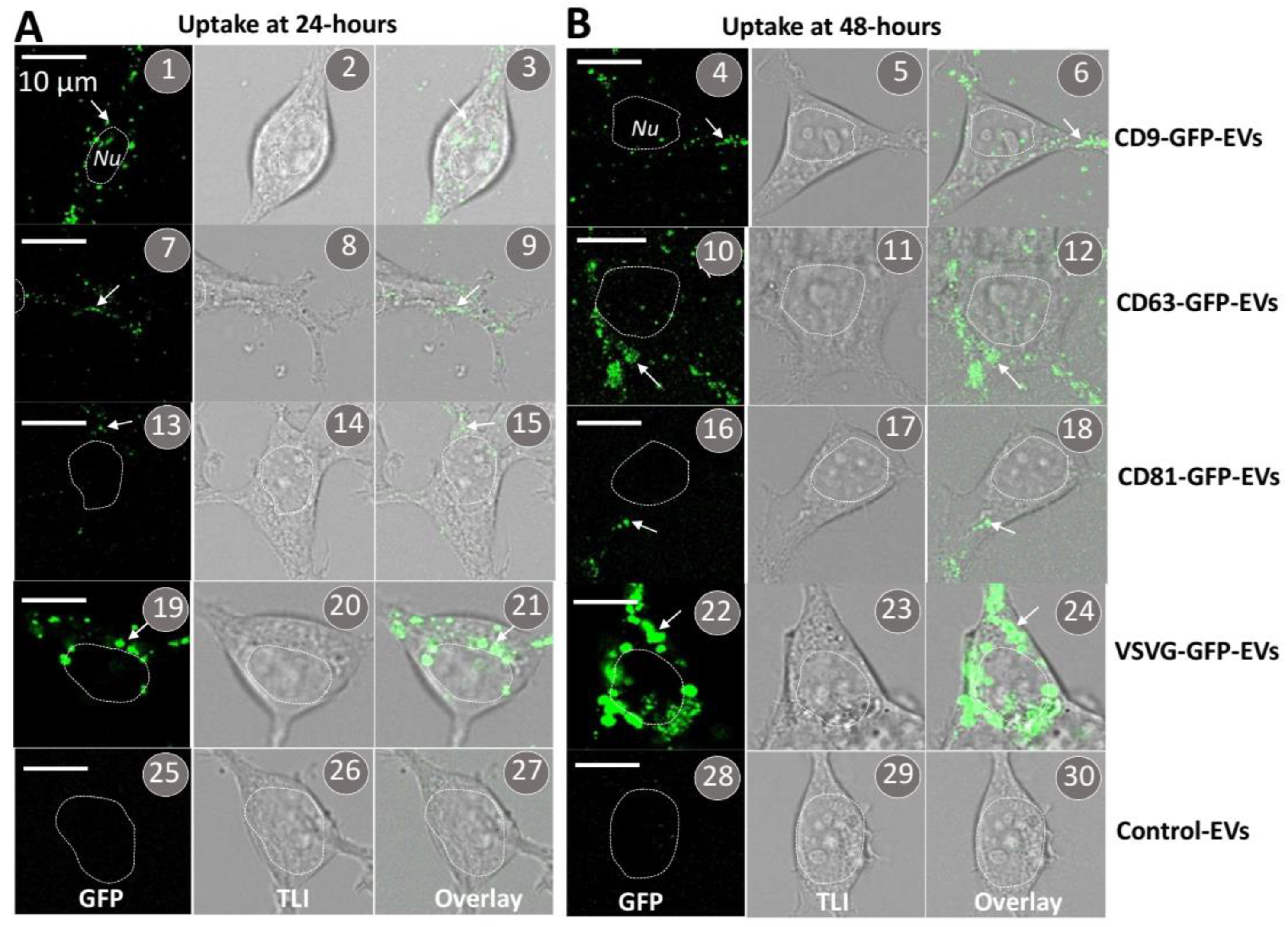
3.2. Transmembrane Scaffolds Incorporate into Biogenic Sites of EVs in Living Human Cells
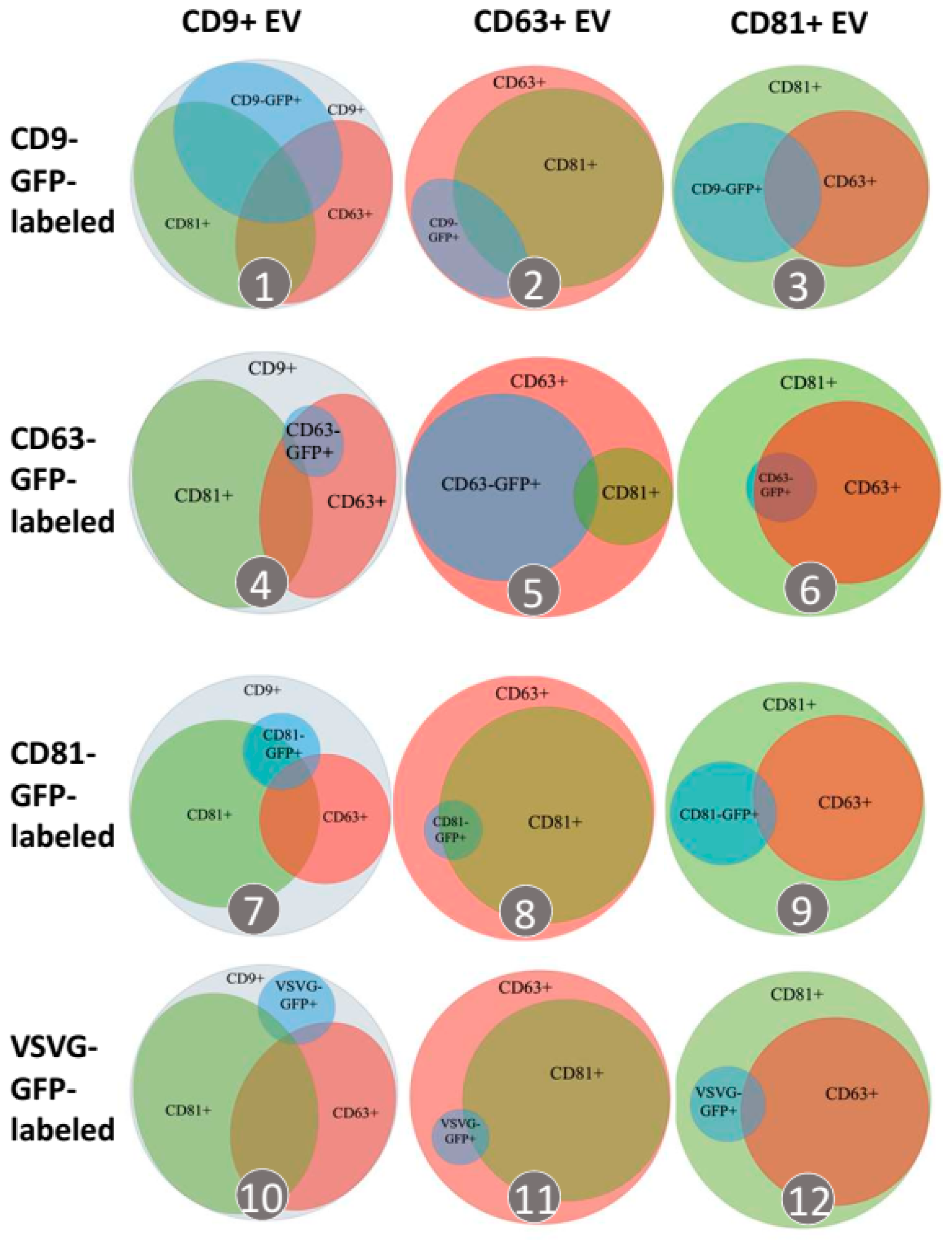
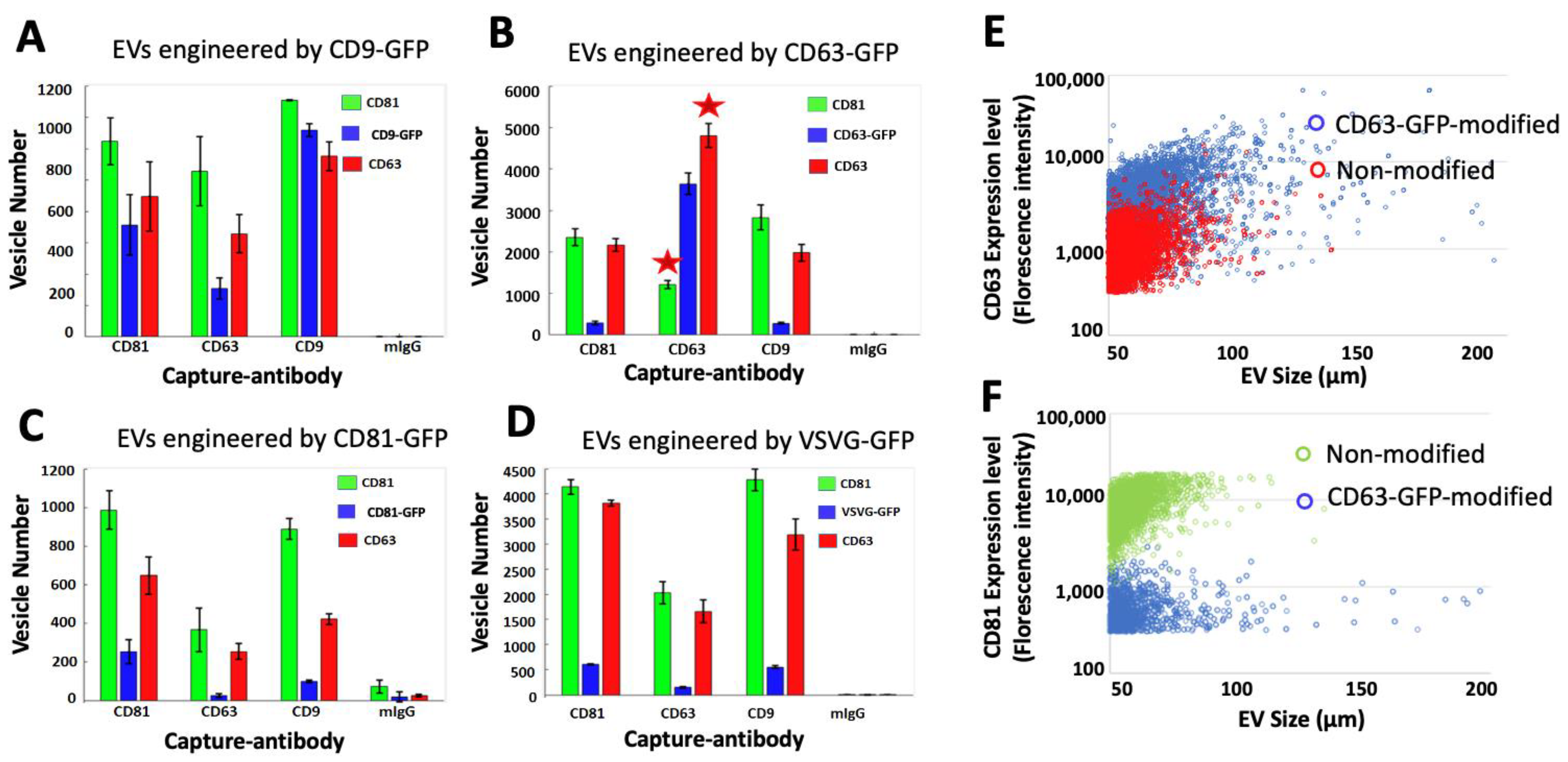
3.3. Each Transmembrane Scaffold Preferentially Incorporates into Different Subtypes of EVs with Various Efficiency
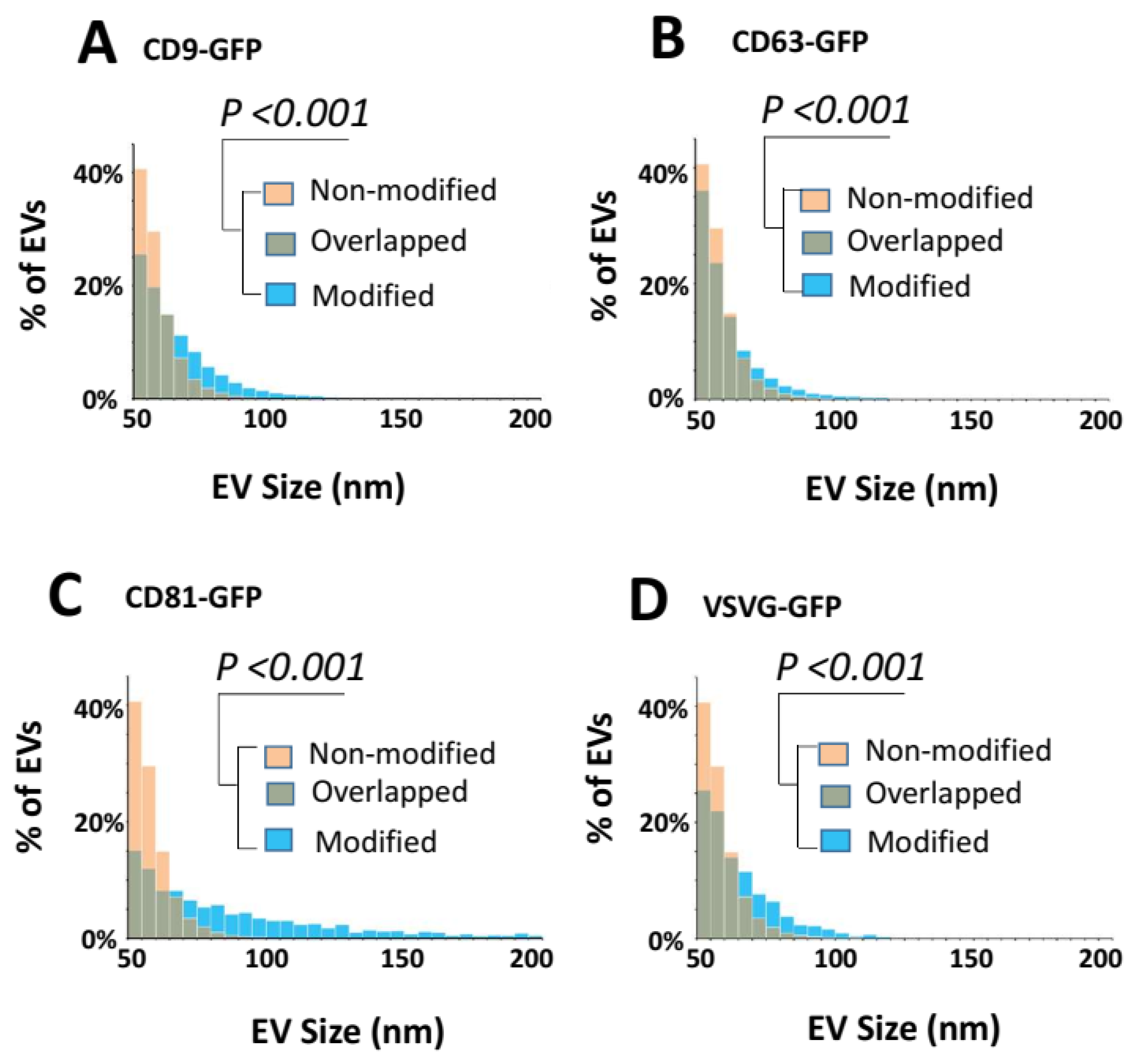
3.4. Surface Engineering Using Transmembrane Scaffolds Leads to an Increase in EV Size
3.5. CD63 Scaffold Decreases the Expression Levels of CD81 Markers on Modified EVs
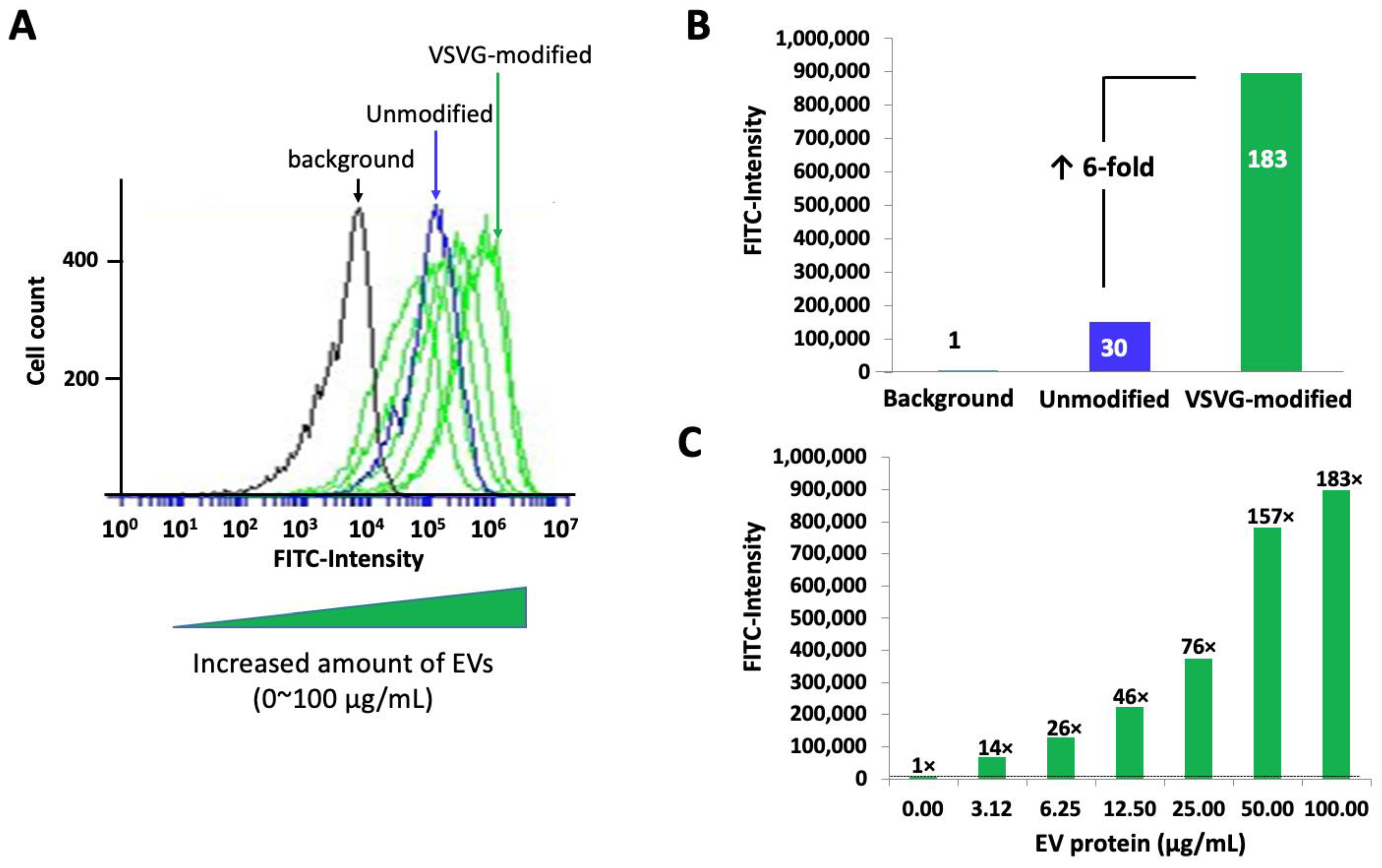
3.6. The VSVG Scaffold Increases the Cellular Uptake of EVs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gill, S.; Catchpole, R.; Forterre, P. Extracellular membrane vesicles in the three domains of life and beyond. FEMS Microbiol. Rev. 2019, 43, 273–303. [Google Scholar] [CrossRef] [PubMed]
- Woith, E.; Fuhrmann, G.; Melzig, M.F. Extracellular Vesicles-Connecting Kingdoms. Int. J. Mol. Sci. 2019, 20, 5695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Bordanaba-Florit, G.; Royo, F.; Kruglik, S.G.; Falcon-Perez, J.M. Using single-vesicle technologies to unravel the heterogeneity of extracellular vesicles. Nat. Protoc. 2021, 16, 3163–3185. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Escola, J.M.; Kleijmeer, M.J.; Stoorvogel, W.; Griffith, J.M.; Yoshie, O.; Geuze, H.J. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 1998, 273, 20121–20127. [Google Scholar] [CrossRef] [Green Version]
- Simpson, R.J.; Kalra, H.; Mathivanan, S. ExoCarta as a resource for exosomal research. J. Extracell. Vesicles 2012, 1, 18374. [Google Scholar] [CrossRef]
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.M.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627. [Google Scholar] [CrossRef]
- Srinivasan, S.; Vannberg, F.O.; Dixon, J.B. Lymphatic transport of exosomes as a rapid route of information dissemination to the lymph node. Sci. Rep. 2016, 6, 24436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asea, A.; Jean-Pierre, C.; Kaur, P.; Rao, P.; Linhares, I.M.; Skupski, D.; Witkin, S.S. Heat shock protein-containing exosomes in mid-trimester amniotic fluids. J. Reprod. Immunol. 2008, 79, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Zlotogorski-Hurvitz, A.; Dayan, D.; Chaushu, G.; Korvala, J.; Salo, T.; Sormunen, R.; Vered, M. Human saliva-derived exosomes: Comparing methods of isolation. J. Histochem. Cytochem. 2015, 63, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheruvanky, A.; Zhou, H.; Pisitkun, T.; Kopp, J.B.; Knepper, M.A.; Yuen, P.S.; Star, R.A. Rapid isolation of urinary exosomal biomarkers using a nanomembrane ultrafiltration concentrator. Am. J. Physiol. Renal. Physiol. 2007, 292, F1657–F1661. [Google Scholar] [CrossRef] [Green Version]
- Adriano, B.; Cotto, N.M.; Chauhan, N.; Jaggi, M.; Chauhan, S.C.; Yallapu, M.M. Milk exosomes: Nature’s abundant nanoplatform for theranostic applications. Bioact. Mater. 2021, 6, 2479–2490. [Google Scholar] [CrossRef]
- Vojtech, L.; Woo, S.; Hughes, S.; Levy, C.; Ballweber, L.; Sauteraud, R.P.; Strobl, J.; Westerberg, K.; Gottardo, R.; Tewari, M.; et al. Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic Acids Res. 2014, 42, 7290–7304. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.S.; Kim, D.K.; Kim, Y.K.; Gho, Y.S. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics 2013, 13, 1554–1571. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Sung, B.H.; Ketova, T.; Hoshino, D.; Zijlstra, A.; Weaver, A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015, 6, 7164. [Google Scholar] [CrossRef] [Green Version]
- McGough, I.J.; Vincent, J.P. Exosomes in developmental signalling. Development 2016, 143, 2482–2493. [Google Scholar] [CrossRef]
- Gutierrez-Vazquez, C.; Villarroya-Beltri, C.; Mittelbrunn, M.; Sanchez-Madrid, F. Transfer of extracellular vesicles during immune cell-cell interactions. Immunol. Rev. 2013, 251, 125–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Pisetsky, D.S. The role of microparticles in the pathogenesis of rheumatic diseases. Nat. Rev. Rheumatol. 2010, 6, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Hassanpour, M.; Rezaie, J.; Nouri, M.; Panahi, Y. The role of extracellular vesicles in COVID-19 virus infection. Infect. Genet. Evol. 2020, 85, 104422. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, H.M.; Fooladi, A.A.; Nourani, M.R.; Ghanezadeh, F. The role of exosomes in infectious diseases. Inflamm. Allergy Drug Targets 2013, 12, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Askenase, P.W. Ancient Evolutionary Origin and Properties of Universally Produced Natural Exosomes Contribute to Their Therapeutic Superiority Compared to Artificial Nanoparticles. Int. J. Mol. Sci. 2021, 22, 1429. [Google Scholar] [CrossRef]
- van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Koog, L.; Gandek, T.B.; Nagelkerke, A. Liposomes and Extracellular Vesicles as Drug Delivery Systems: A Comparison of Composition, Pharmacokinetics, and Functionalization. Adv. Healthc. Mater. 2022, 11, e2100639. [Google Scholar] [CrossRef] [PubMed]
- Rufino-Ramos, D.; Lule, S.; Mahjoum, S.; Ughetto, S.; Cristopher Bragg, D.; Pereira de Almeida, L.; Breakefield, X.O.; Breyne, K. Using genetically modified extracellular vesicles as a non-invasive strategy to evaluate brain-specific cargo. Biomaterials 2022, 281, 121366. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Hill, A.F. Therapeutically harnessing extracellular vesicles. Nat. Rev. Drug Discov. 2022, 21, 379–399. [Google Scholar] [CrossRef]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 148–156. [Google Scholar] [CrossRef]
- Burnouf, T.; Agrahari, V.; Agrahari, V. Extracellular Vesicles as Nanomedicine: Hopes And Hurdles In Clinical Translation. Int. J. Nanomed. 2019, 14, 8847–8859. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Garcia-Manrique, P.; Matos, M.; Gutierrez, G.; Pazos, C.; Blanco-Lopez, M.C. Therapeutic biomaterials based on extracellular vesicles: Classification of bio-engineering and mimetic preparation routes. J. Extracell. Vesicles 2018, 7, 1422676. [Google Scholar] [CrossRef] [Green Version]
- Stickney, Z.; Losacco, J.; McDevitt, S.; Zhang, Z.; Lu, B. Development of exosome surface display technology in living human cells. Biochem. Biophys. Res. Commun. 2016, 472, 53–59. [Google Scholar] [CrossRef]
- Vogt, S.; Bobbili, M.R.; Stadlmayr, G.; Stadlbauer, K.; Kjems, J.; Ruker, F.; Grillari, J.; Wozniak-Knopp, G. An engineered CD81-based combinatorial library for selecting recombinant binders to cell surface proteins: Laminin binding CD81 enhances cellular uptake of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12139. [Google Scholar] [CrossRef]
- Meyer, C.; Losacco, J.; Stickney, Z.; Li, L.; Marriott, G.; Lu, B. Pseudotyping exosomes for enhanced protein delivery in mammalian cells. Int. J. Nanomed. 2017, 12, 3153–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, M.A.; Levy, D.; Brown, A.; Marriott, G.; Lu, B. Targeted delivery of lysosomal enzymes to the endocytic compartment in human cells using engineered extracellular vesicles. Sci. Rep. 2019, 9, 17274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Andaloussi, S.; Lee, Y.; Lakhal-Littleton, S.; Li, J.; Seow, Y.; Gardiner, C.; Alvarez-Erviti, L.; Sargent, I.L.; Wood, M.J. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat. Protoc. 2012, 7, 2112–2126. [Google Scholar] [CrossRef] [PubMed]
- Delcayre, A.; Estelles, A.; Sperinde, J.; Roulon, T.; Paz, P.; Aguilar, B.; Villanueva, J.; Khine, S.; Le Pecq, J.B. Exosome Display technology: Applications to the development of new diagnostics and therapeutics. Blood Cells Mol. Dis. 2005, 35, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Forterre, A.V.; Zhao, J.; Frimannsson, D.O.; Delcayre, A.; Antes, T.J.; Efron, B.; Jeffrey, S.S.; Pegram, M.D.; Matin, A.C. Anti-HER2 scFv-Directed Extracellular Vesicle-Mediated mRNA-Based Gene Delivery Inhibits Growth of HER2-Positive Human Breast Tumor Xenografts by Prodrug Activation. Mol. Cancer Ther. 2018, 17, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Kooijmans, S.A.; Aleza, C.G.; Roffler, S.R.; van Solinge, W.W.; Vader, P.; Schiffelers, R.M. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J. Extracell. Vesicles 2016, 5, 31053. [Google Scholar] [CrossRef]
- Saludes, J.P.; Morton, L.A.; Coulup, S.K.; Fiorini, Z.; Cook, B.M.; Beninson, L.; Chapman, E.R.; Fleshner, M.; Yin, H. Multivalency amplifies the selection and affinity of bradykinin-derived peptides for lipid nanovesicles. Mol. Biosyst. 2013, 9, 2005–2009. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.; Do, M.A.; Zhang, J.; Brown, A.; Lu, B. Orchestrating Extracellular Vesicle With Dual Reporters for Imaging and Capturing in Mammalian Cell Culture. Front. Mol. Biosci. 2021, 8, 680580. [Google Scholar] [CrossRef]
- Levy, D.; Do, M.A.; Brown, A.; Asano, K.; Diebold, D.; Chen, H.; Zhang, J.; Lawler, B.; Lu, B. Genetic labeling of extracellular vesicles for studying biogenesis and uptake in living mammalian cells. Methods Enzymol. 2020, 645, 1–14. [Google Scholar] [CrossRef]
- Curley, N.; Levy, D.; Do, M.A.; Brown, A.; Stickney, Z.; Marriott, G.; Lu, B. Sequential deletion of CD63 identifies topologically distinct scaffolds for surface engineering of exosomes in living human cells. Nanoscale 2020, 12, 12014–12026. [Google Scholar] [CrossRef]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, F.; Fussenegger, M. Shedding Light on Extracellular Vesicle Biogenesis and Bioengineering. Adv. Sci. 2020, 8, 2003505. [Google Scholar] [CrossRef] [PubMed]
- Thery, C. Exosomes: Secreted vesicles and intercellular communications. F1000 Biol. Rep. 2021, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Daaboul, G.G.; Gagni, P.; Benussi, L.; Bettotti, P.; Ciani, M.; Cretich, M.; Freedman, D.S.; Ghidoni, R.; Ozkumur, A.Y.; Piotto, C.; et al. Digital Detection of Exosomes by Interferometric Imaging. Sci. Rep. 2016, 6, 37246. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S.; Anderson, R.G.; Russell, D.W.; Schneider, W.J. Receptor-mediated endocytosis: Concepts emerging from the LDL receptor system. Annu. Rev. Cell Biol. 1985, 1, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [Green Version]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, X.; Wei, M.; Gao, X.; Zhao, L.; Shi, R.; Sun, W.; Duan, Y.; Yang, G.; Yuan, L. In Vitro and in Vivo RNA Inhibition by CD9-HuR Functionalized Exosomes Encapsulated with miRNA or CRISPR/dCas9. Nano Lett. 2019, 19, 19–28. [Google Scholar] [CrossRef]
- Lu, B.; Ku, J.; Flojo, R.; Olson, C.; Bengford, D.; Marriott, G. Exosome- and extracellular vesicle-based approaches for the treatment of lysosomal storage disorders. Adv. Drug Deliv. Rev. 2022, 188, 114465. [Google Scholar] [CrossRef]
- Silva, A.M.; Lazaro-Ibanez, E.; Gunnarsson, A.; Dhande, A.; Daaboul, G.; Peacock, B.; Osteikoetxea, X.; Salmond, N.; Friis, K.P.; Shatnyeva, O.; et al. Quantification of protein cargo loading into engineered extracellular vesicles at single-vesicle and single-molecule resolution. J. Extracell. Vesicles 2021, 10, e12130. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; McKay, P.F.; Samnuan, K.; Najer, A.; Blakney, A.K.; Che, J.; O’Driscoll, G.; Cihova, M.; Stevens, M.M.; Shattock, R.J. Presentation of antigen on extracellular vesicles using transmembrane domains from viral glycoproteins for enhanced immunogenicity. J. Extracell. Vesicles 2022, 11, e12199. [Google Scholar] [CrossRef] [PubMed]
- Segel, M.; Lash, B.; Song, J.; Ladha, A.; Liu, C.C.; Jin, X.; Mekhedov, S.L.; Macrae, R.K.; Koonin, E.V.; Zhang, F. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science 2021, 373, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811. [Google Scholar] [CrossRef]
- Presley, J.F.; Cole, N.B.; Schroer, T.A.; Hirschberg, K.; Zaal, K.J.; Lippincott-Schwartz, J. ER-to-Golgi transport visualized in living cells. Nature 1997, 389, 81–85. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Barreiro, O.; Gordon-Alonso, M.; Sala-Valdes, M.; Sanchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell Biol. 2009, 19, 434–446. [Google Scholar] [CrossRef]
- Abache, T.; Le Naour, F.; Planchon, S.; Harper, F.; Boucheix, C.; Rubinstein, E. The transferrin receptor and the tetraspanin web molecules CD9, CD81, and CD9P-1 are differentially sorted into exosomes after TPA treatment of K562 cells. J. Cell Biochem. 2007, 102, 650–664. [Google Scholar] [CrossRef]
- Fitter, S.; Sincock, P.M.; Jolliffe, C.N.; Ashman, L.K. Transmembrane 4 superfamily protein CD151 (PETA-3) associates with beta 1 and alpha IIb beta 3 integrins in haemopoietic cell lines and modulates cell-cell adhesion. Biochem. J. 1999, 338 Pt 1, 61–70. [Google Scholar] [CrossRef]
- Nikolic, J.; Belot, L.; Raux, H.; Legrand, P.; Gaudin, Y.; Albertini, A.A. Structural basis for the recognition of LDL-receptor family members by VSV glycoprotein. Nat. Commun. 2018, 9, 1029. [Google Scholar] [CrossRef] [Green Version]
- Dalton, K.P.; Rose, J.K. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology 2001, 279, 414–421. [Google Scholar] [CrossRef]
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Sullivan, M.L.; Stolz, D.B.; Papworth, G.D.; Zahorchak, A.F.; Logar, A.J.; Wang, Z.; Watkins, S.C.; et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004, 104, 3257–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal, J.; Tkach, M.; Thery, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsac, D.; Loirat, D.; Petit, C.; Schwartz, O.; Michel, M.L. Enhanced presentation of major histocompatibility complex class I-restricted human immunodeficiency virus type 1 (HIV-1) Gag-specific epitopes after DNA immunization with vectors coding for vesicular stomatitis virus glycoprotein-pseudotyped HIV-1 Gag particles. J. Virol. 2002, 76, 7544–7553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temchura, V.V.; Tenbusch, M.; Nchinda, G.; Nabi, G.; Tippler, B.; Zelenyuk, M.; Wildner, O.; Uberla, K.; Kuate, S. Enhancement of immunostimulatory properties of exosomal vaccines by incorporation of fusion-competent G protein of vesicular stomatitis virus. Vaccine 2008, 26, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Kuate, S.; Cinatl, J.; Doerr, H.W.; Uberla, K. Exosomal vaccines containing the S protein of the SARS coronavirus induce high levels of neutralizing antibodies. Virology 2007, 362, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanuma, T.; Yamamoto, T.; Kobiyama, K.; Moriishi, E.; Masuta, Y.; Kusakabe, T.; Ozasa, K.; Kuroda, E.; Jounai, N.; Ishii, K.J. CD63-Mediated Antigen Delivery into Extracellular Vesicles via DNA Vaccination Results in Robust CD8(+) T Cell Responses. J. Immunol. 2017, 198, 4707–4715. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, M.; Nevo, N.; Jouve, M.; Valenzuela, J.I.; Maurin, M.; Verweij, F.J.; Palmulli, R.; Lankar, D.; Dingli, F.; Loew, D.; et al. Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9. Nat. Commun. 2021, 12, 4389. [Google Scholar] [CrossRef]
- Yim, N.; Ryu, S.W.; Choi, K.; Lee, K.R.; Lee, S.; Choi, H.; Kim, J.; Shaker, M.R.; Sun, W.; Park, J.H.; et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun. 2016, 7, 12277. [Google Scholar] [CrossRef] [Green Version]
- Yee, J.K.; Friedmann, T.; Burns, J.C. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol. 1994, 43 Pt A, 99–112. [Google Scholar] [CrossRef]
- Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee, J.K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8033–8037. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, X.; Wei, X.; Li, J.; Yang, J.; Tan, H.; Zhu, J.; Zhang, Q.; Wu, J.; Liu, L. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020, 92, 595–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemler, M.E. Targeting of tetraspanin proteins—potential benefits and strategies. Nat. Rev. Drug Discov. 2008, 7, 747–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassuna, N.; Monk, P.N.; Moseley, G.W.; Partridge, L.J. Strategies for targeting tetraspanin proteins: Potential therapeutic applications in microbial infections. BioDrugs 2009, 23, 341–359. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD9+ EVs | CD63+ EVs | CD81+ EVs | |
|---|---|---|---|
| CD9-GFP-EVs | 47.2% ± 0.8% | 19.7% ± 5.1% | 32.4% ± 4.0% |
| CD63-GFP-EVs | 6.7% ± 0.1% | 58.7% ± 1.5% | 7.3% ± 0.5% |
| CD81-GFP-EVs | 12.3% ± 6.3% | 4.9% ± 0.6% | 16.6% ± 2.0% |
| VSVG-GFP-EVs | 8.3% ± 0.2% | 4.7% ± 0.8% | 8.5% ± 0.1% |
| Name of Scaffold | Unmodified EVs (nm, size) | Modified EVs (nm, size) |
|---|---|---|
| CD9-GFP | 58.40 ± 8.07 | 66.13 ± 16.38 |
| CD63-GFP | 62.07 ± 14.20 | |
| CD81-GFP | 86.02 ± 34.18 | |
| VSVG-GFP | 65.42 ± 15.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Brown, A.; Johnson, B.; Diebold, D.; Asano, K.; Marriott, G.; Lu, B. Genetically Engineered Extracellular Vesicles Harboring Transmembrane Scaffolds Exhibit Differences in Their Size, Expression Levels of Specific Surface Markers and Cell-Uptake. Pharmaceutics 2022, 14, 2564. https://doi.org/10.3390/pharmaceutics14122564
Zhang J, Brown A, Johnson B, Diebold D, Asano K, Marriott G, Lu B. Genetically Engineered Extracellular Vesicles Harboring Transmembrane Scaffolds Exhibit Differences in Their Size, Expression Levels of Specific Surface Markers and Cell-Uptake. Pharmaceutics. 2022; 14(12):2564. https://doi.org/10.3390/pharmaceutics14122564
Chicago/Turabian StyleZhang, Jiayi, Annie Brown, Brendan Johnson, David Diebold, Kyle Asano, Gerard Marriott, and Biao Lu. 2022. "Genetically Engineered Extracellular Vesicles Harboring Transmembrane Scaffolds Exhibit Differences in Their Size, Expression Levels of Specific Surface Markers and Cell-Uptake" Pharmaceutics 14, no. 12: 2564. https://doi.org/10.3390/pharmaceutics14122564