

Evaluation of Different Thermoanalytical Methods for the Analysis of the Stability of Naproxen-Loaded Amorphous Solid Dispersions
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Amorphous Solid Dispersions
2.3. Scanning Electron Microscopy (SEM)
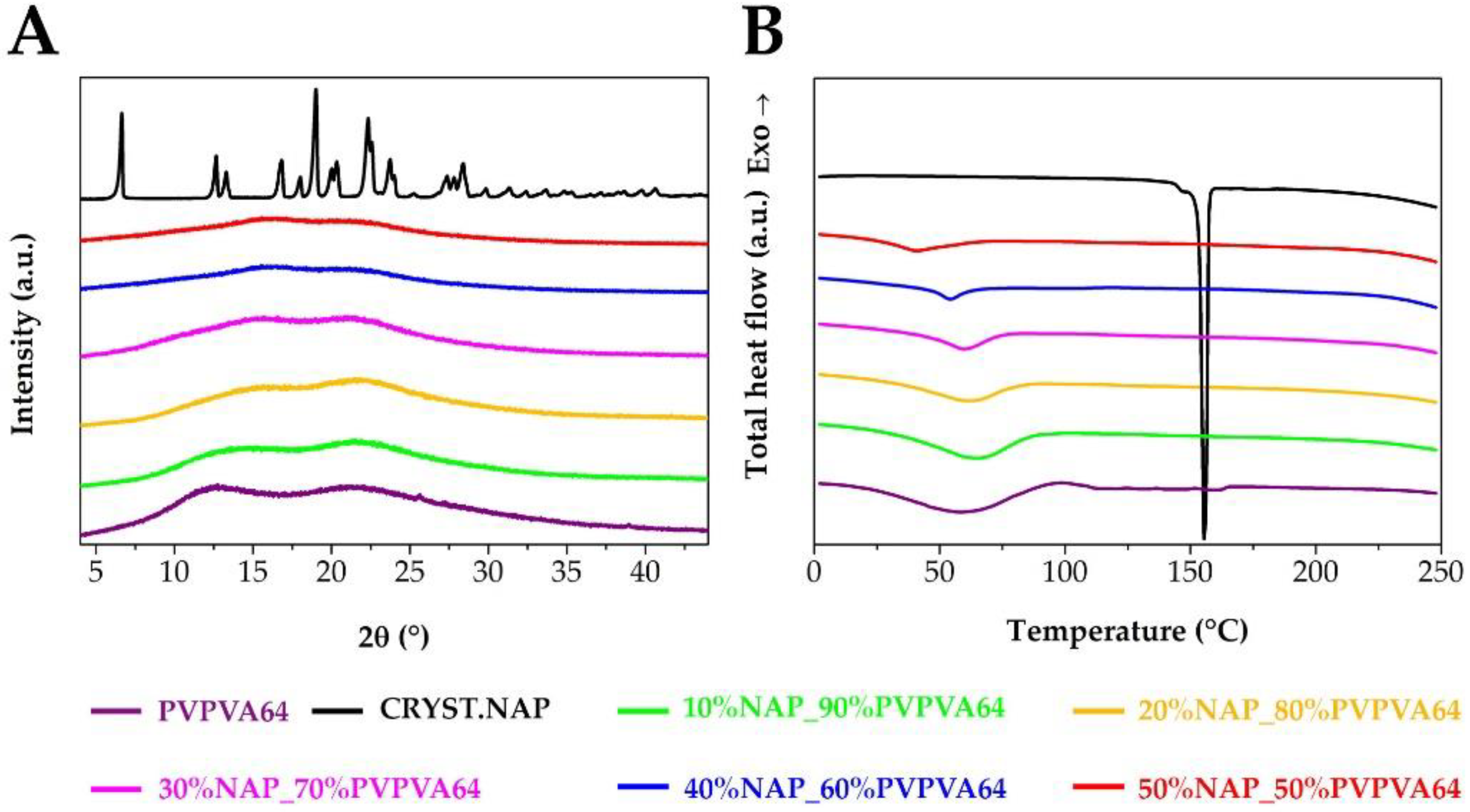
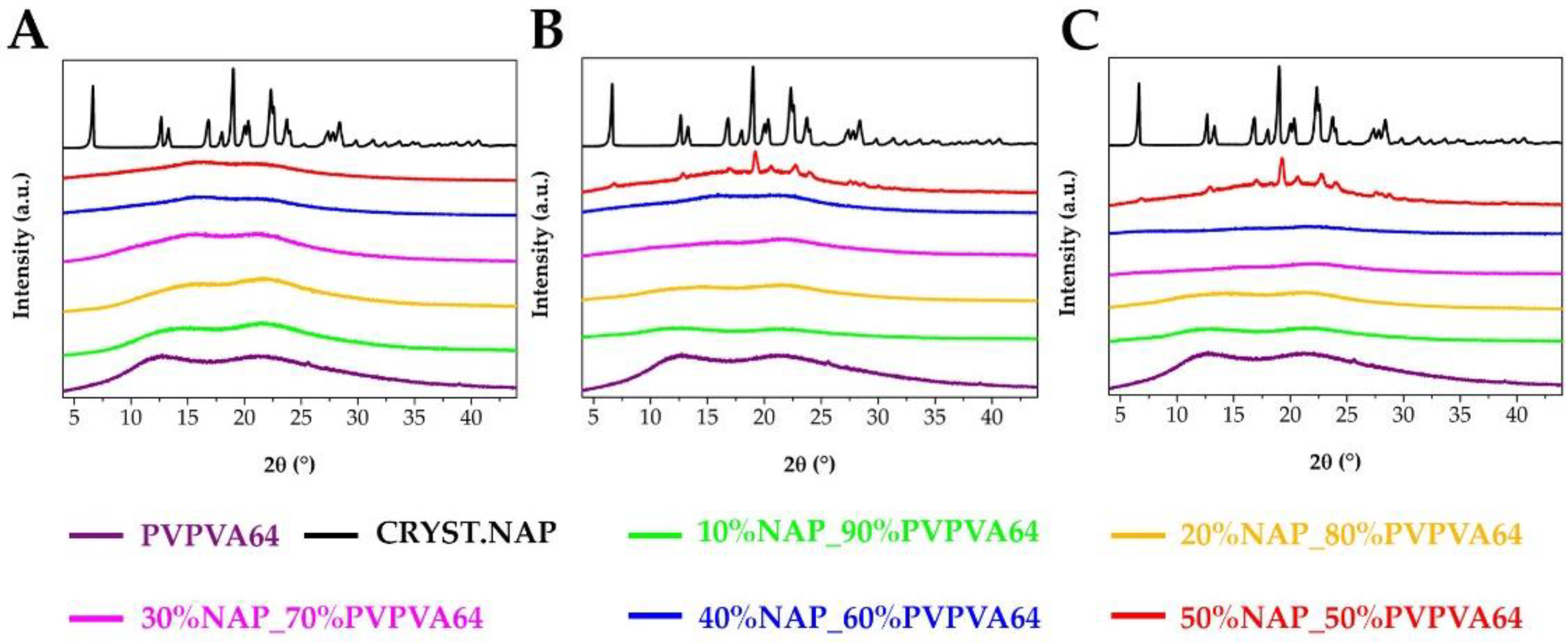
2.4. X-ray Powder Diffraction (XRPD)
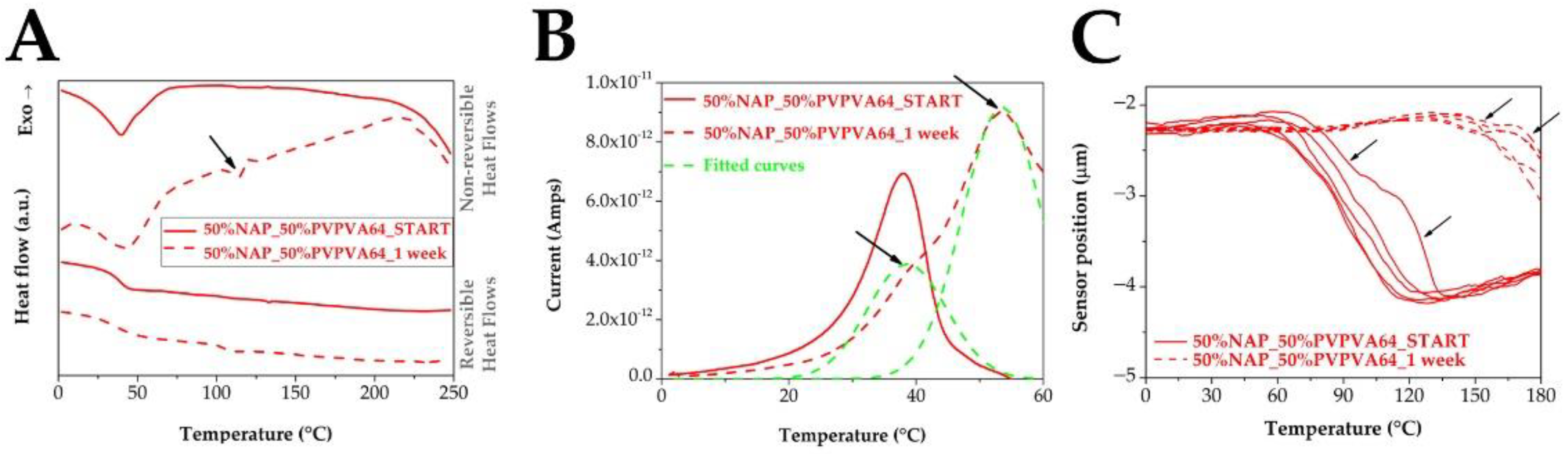
2.5. Modulated Differential Scanning Calorimetry (MDSC)
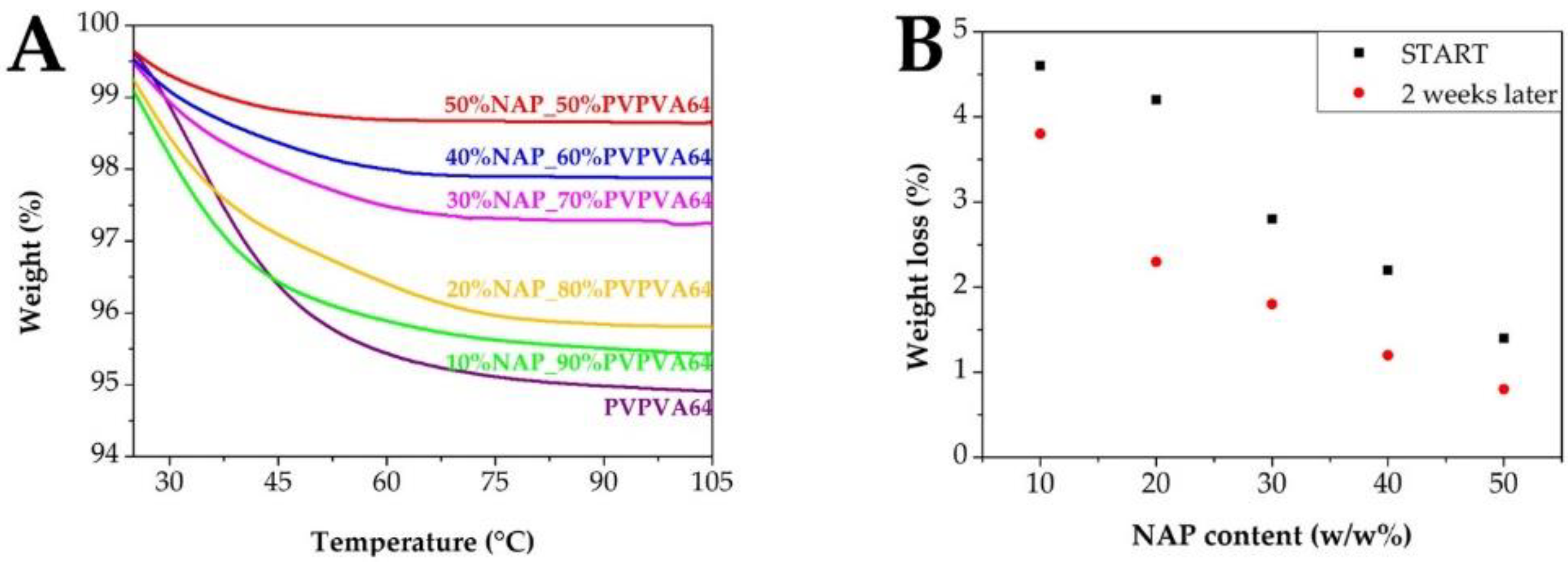
2.6. Thermogravimetric Analysis (TGA)
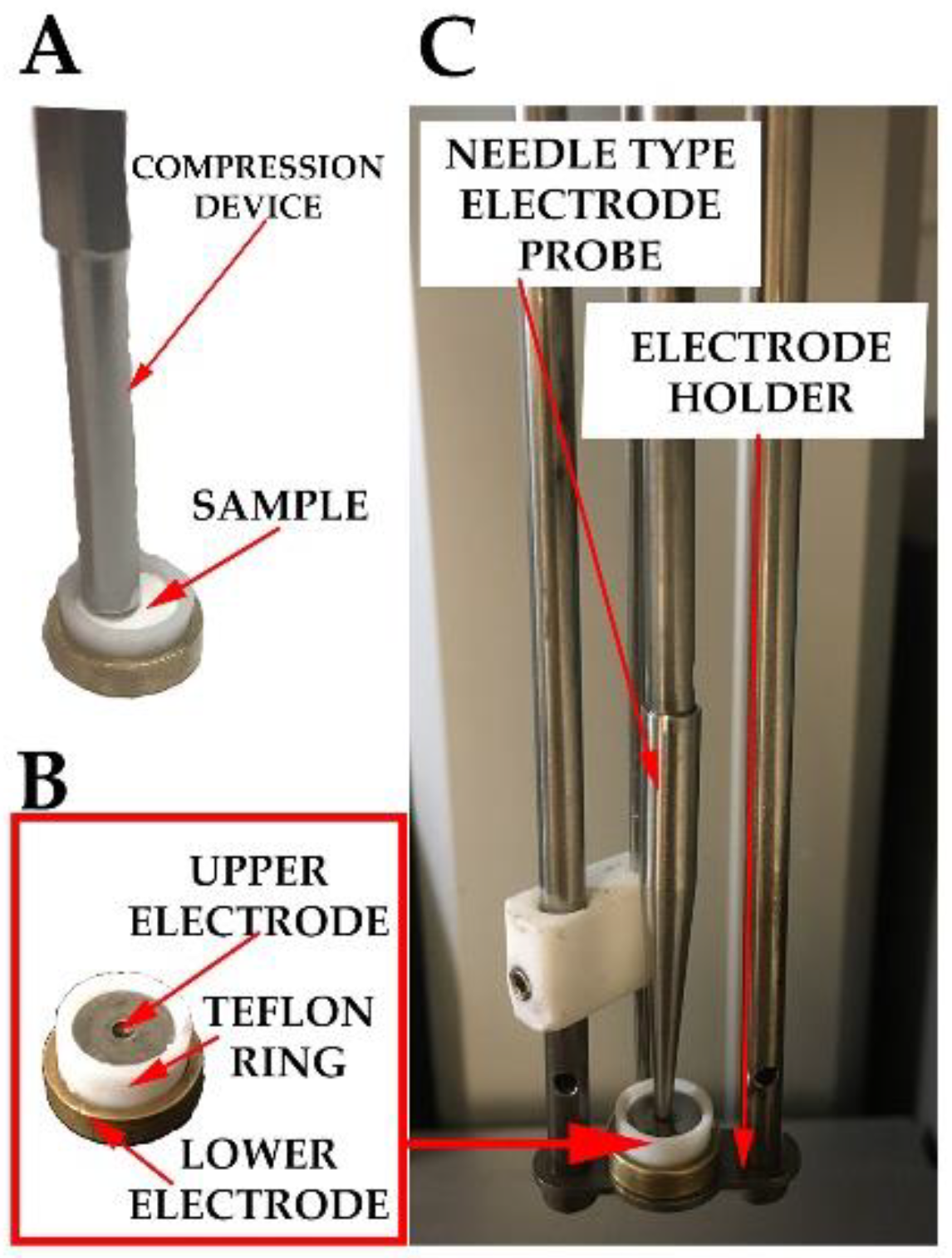
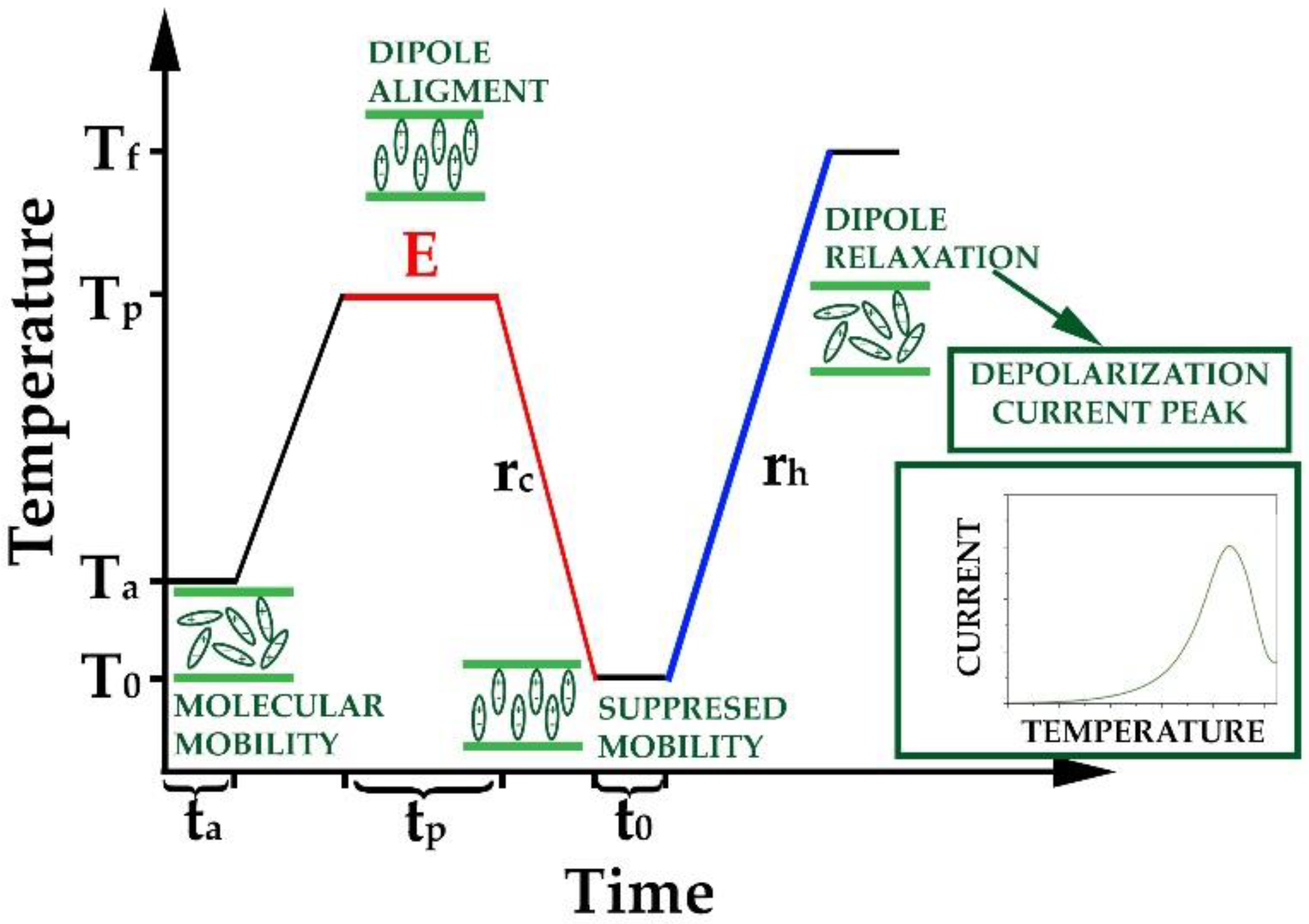
2.7. Thermally Stimulated Depolarization Current (TSDC)
2.8. Micro-Thermal Analysis (µTA)
2.9. Stress Stability Test
2.10. Polarized Light Microscopy (PLM)
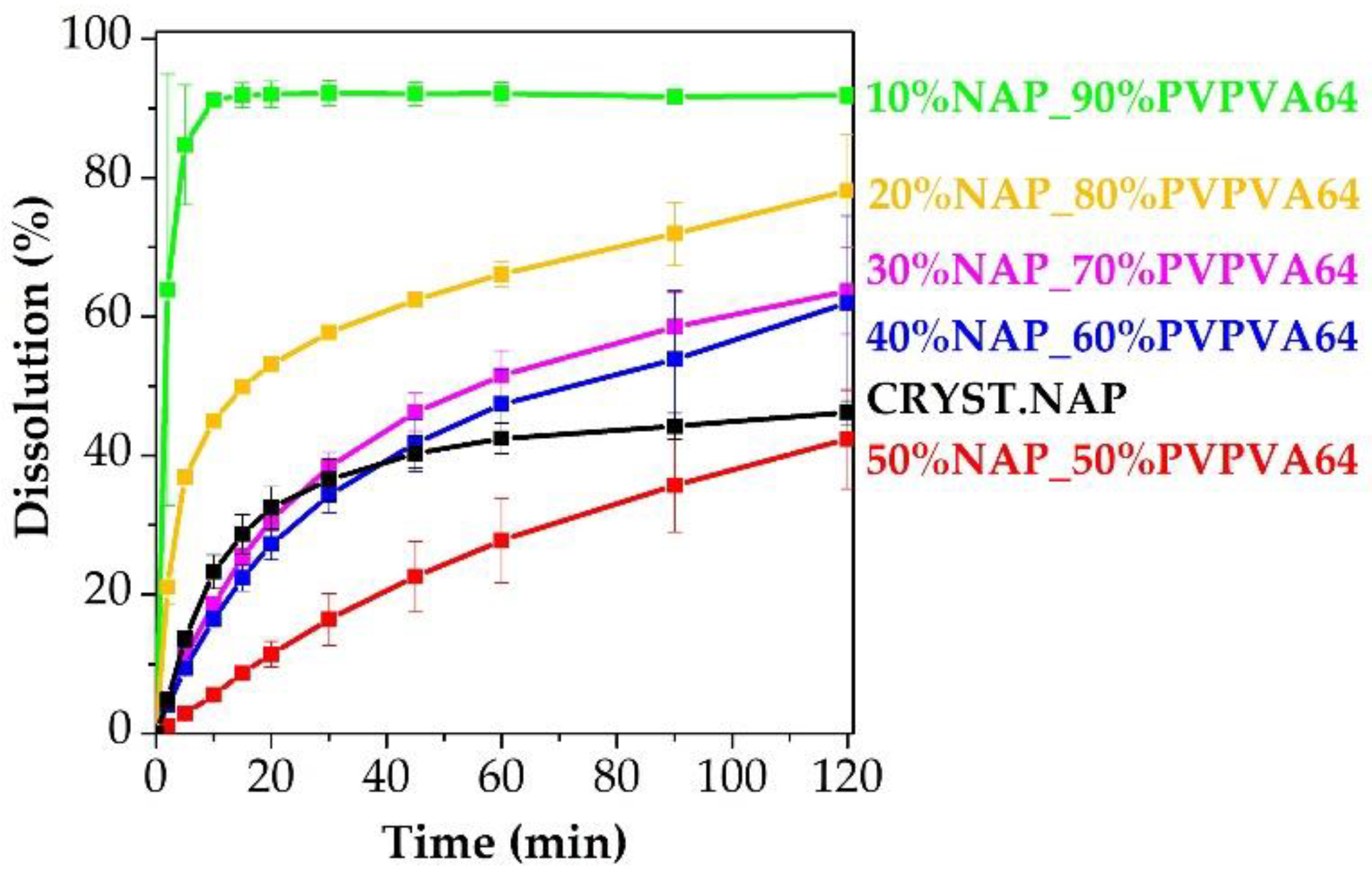
2.11. In Vitro Dissolution Testing
3. Results and Discussion
3.1. Preparation and Basic Characterization of the Samples
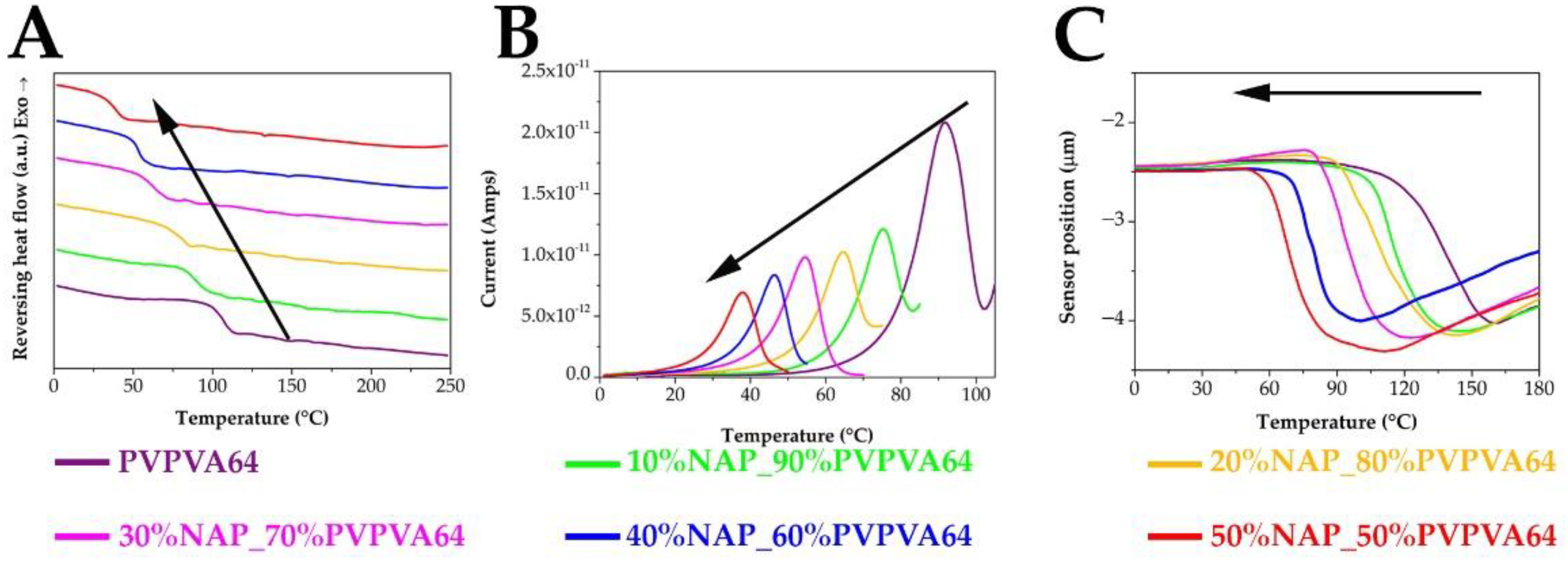
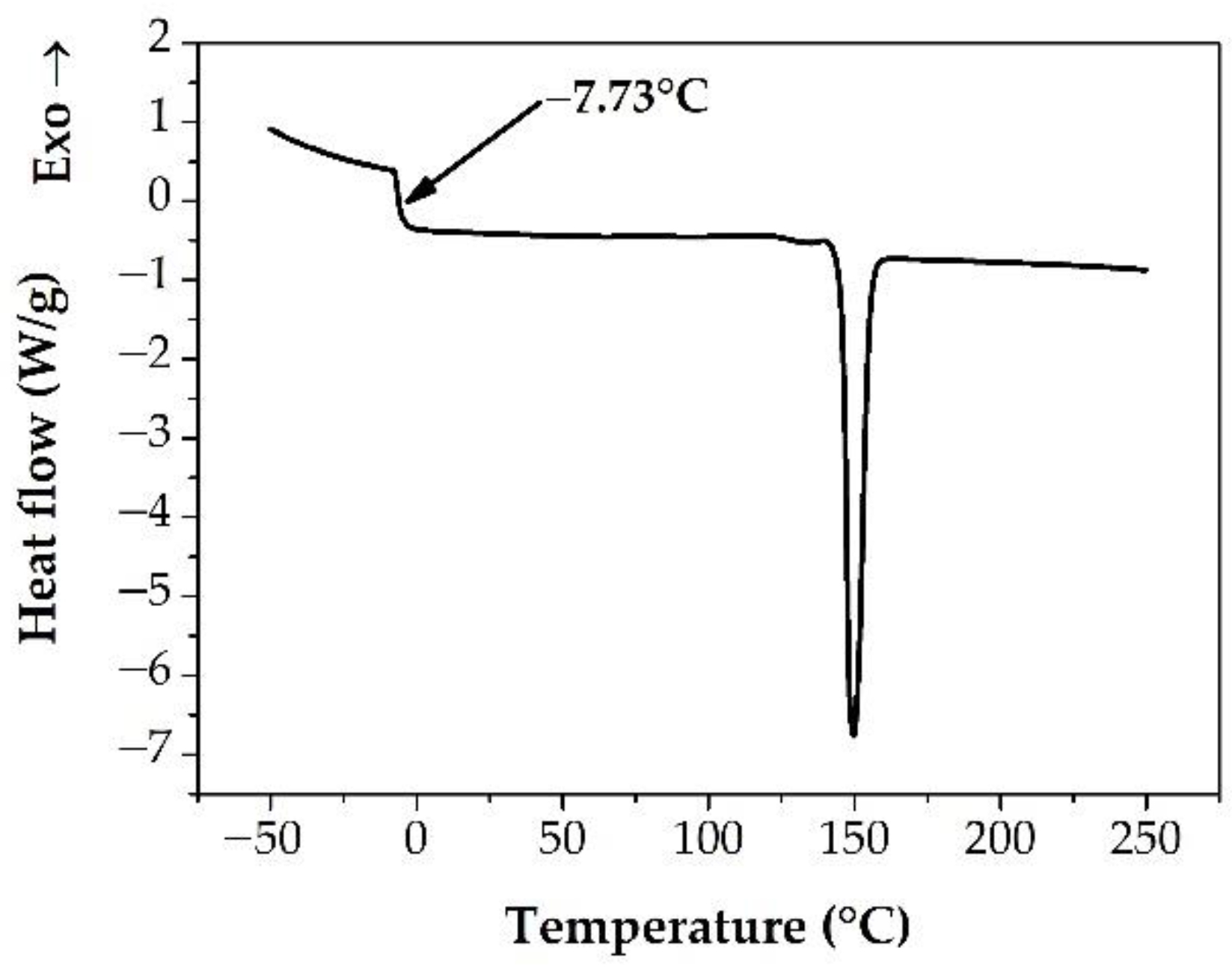
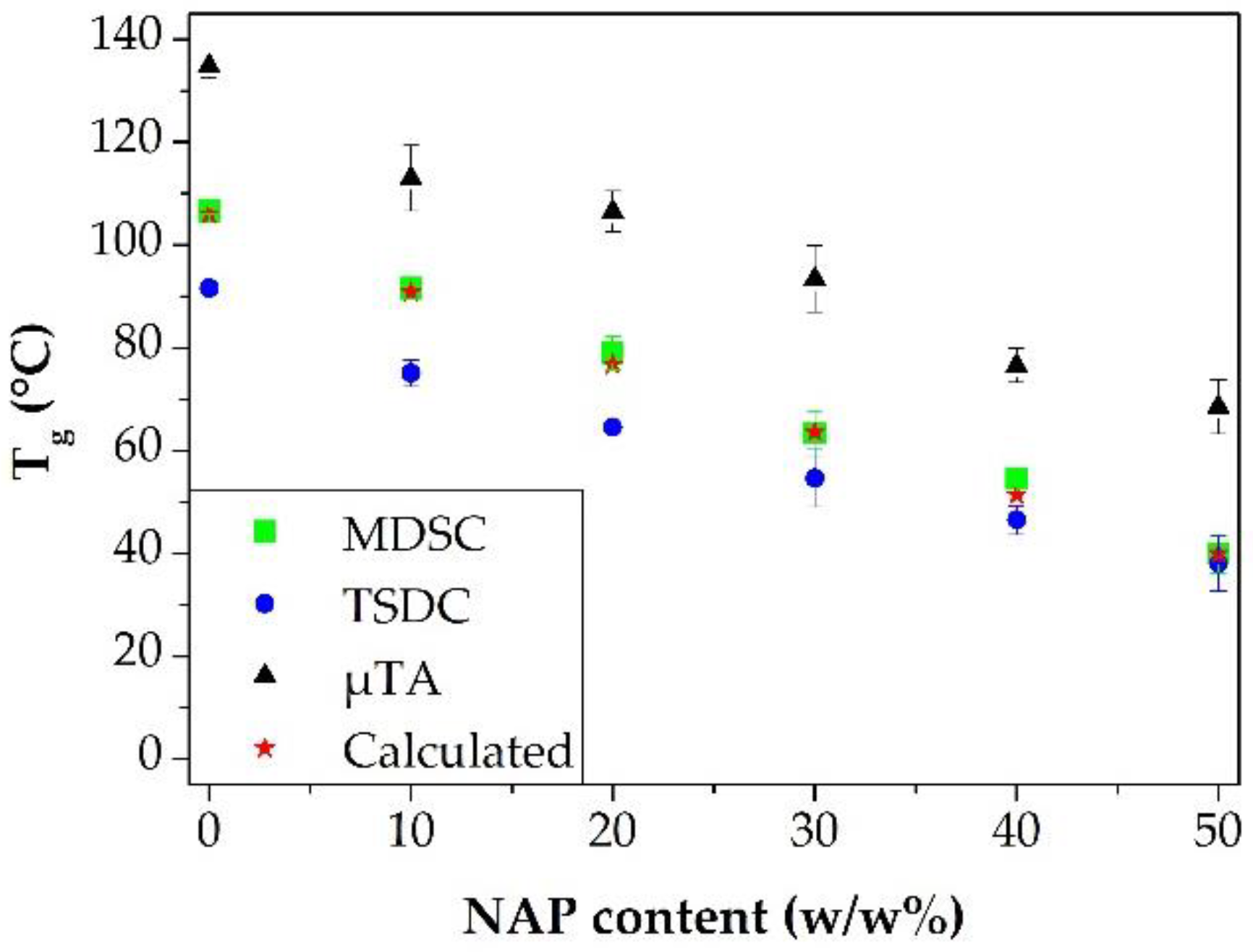
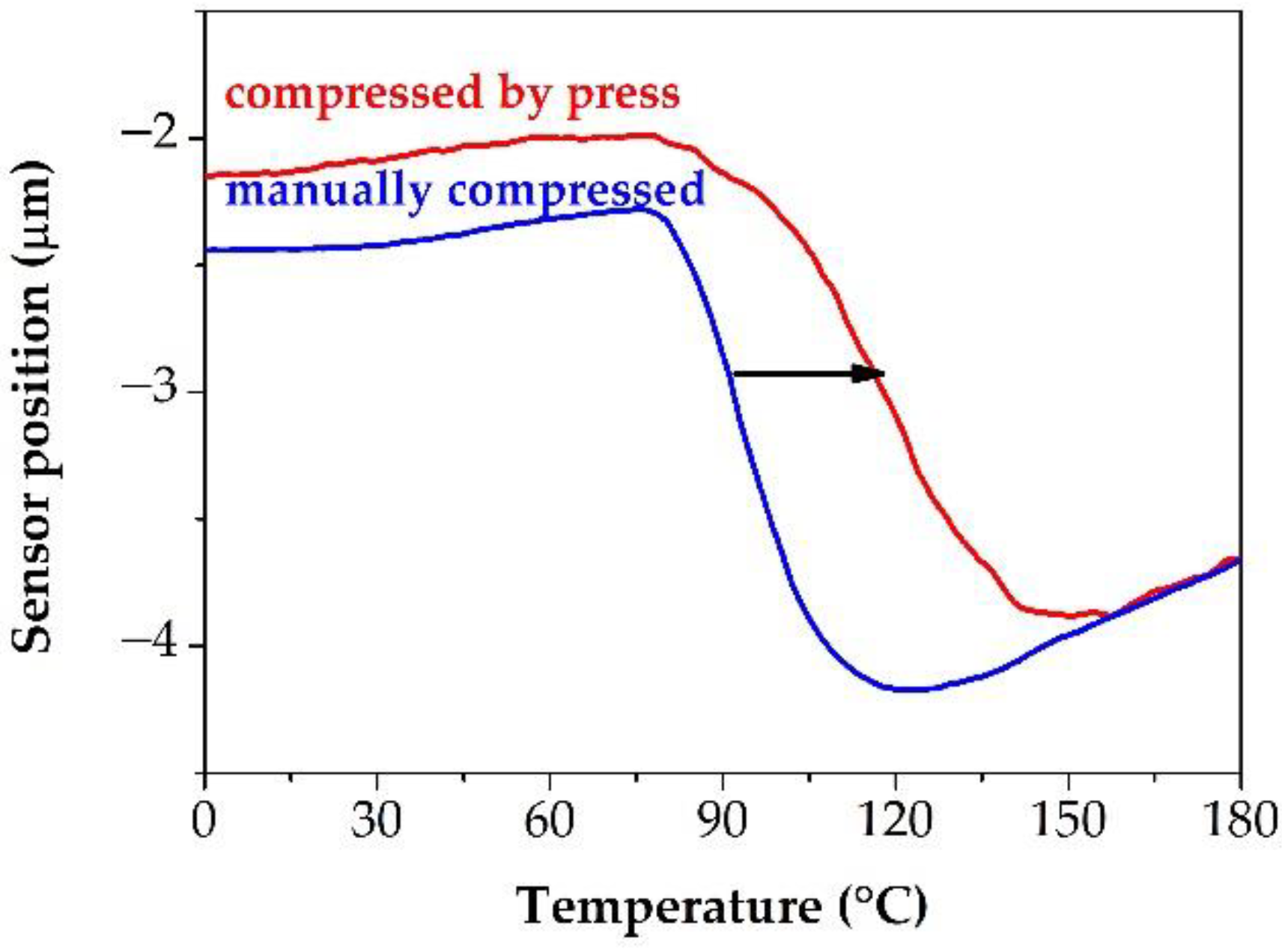
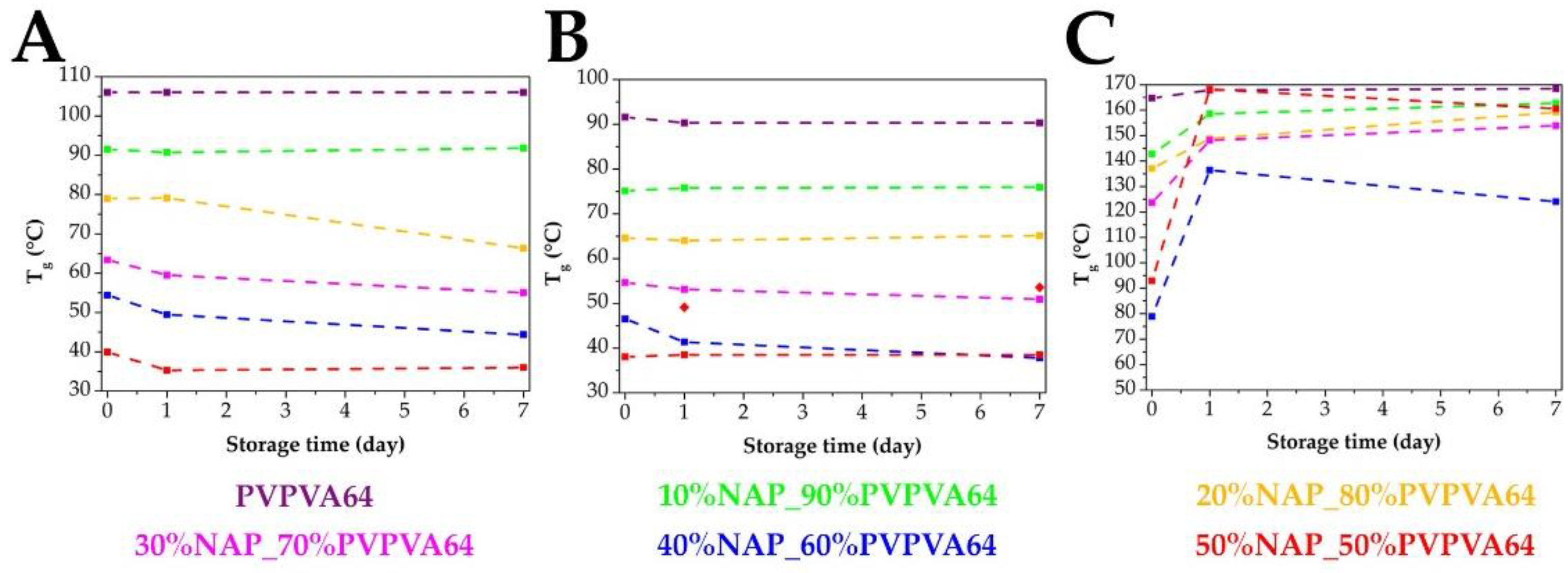
3.2. Measuring the Glass Transition Temperature with Different Methods
3.3. Effect of the Stress Stability Tests
3.4. Correlations between the Measured Tg Values and the Stability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Göke, K.; Lorenz, T.; Repanas, A.; Schneider, F.; Steiner, D.; Baumann, K.; Bunjes, H.; Dietzel, A.; Finke, J.H.; Glasmacher, B. Novel strategies for the formulation and processing of poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2018, 126, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Fahr, A.; Liu, X. Drug delivery strategies for poorly water-soluble drugs. Expert Opin. Drug Deliv. 2007, 4, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Babu, N.J.; Nangia, A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Skrdla, P.J.; Floyd, P.D.; Dell’Orco, P.C. Modeling recrystallization kinetics following the dissolution of amorphous drugs. Mol. Pharm. 2019, 17, 219–228. [Google Scholar] [CrossRef]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef]
- Graeser, K.A.; Patterson, J.E.; Zeitler, J.A.; Gordon, K.C.; Rades, T. Correlating thermodynamic and kinetic parameters with amorphous stability. Eur. J. Pharm. Sci. 2009, 37, 492–498. [Google Scholar] [CrossRef]
- Guo, Y.; Shalaev, E.; Smith, S. Physical stability of pharmaceutical formulations: Solid-state characterization of amorphous dispersions. Trends Anal. Chem. 2013, 49, 137–144. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [Green Version]
- Szafraniec-Szczęsny, J.; Antosik-Rogóż, A.; Kurek, M.; Gawlak, K.; Górska, A.; Peralta, S.; Knapik-Kowalczuk, J.; Kramarczyk, D.; Paluch, M.; Jachowicz, R. How does the addition of Kollidon® VA64 inhibit the recrystallization and improve ezetimibe dissolution from amorphous solid dispersions? Pharmaceutics 2021, 13, 147. [Google Scholar] [CrossRef]
- Uttaro, E.; Pudipeddi, M.; Schweighardt, A.; Zhao, F. To crush or not to crush: A brief review of novel tablets and capsules prepared from nanocrystal and amorphous solid dispersion technologies. Am. J. Health Syst. Pharm. 2021, 78, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Szabó, E.; Záhonyi, P.; Brecska, D.; Galata, D.n.L.; Mészáros, L.A.; Madarász, L.; Csorba, K.; Vass, P.; Hirsch, E.; Szafraniec-Szczęsny, J. Comparison of amorphous solid dispersions of spironolactone prepared by spray drying and electrospinning: The influence of the preparation method on the dissolution properties. Mol. Pharm. 2020, 18, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Impact of polymer type and relative humidity on the long-term physical stability of amorphous solid dispersions. Mol. Pharm. 2017, 14, 4374–4386. [Google Scholar] [CrossRef] [PubMed]
- Lehmkemper, K.; Kyeremateng, S.O.; Bartels, M.; Degenhardt, M.; Sadowski, G. Physical stability of API/polymer-blend amorphous solid dispersions. Eur. J. Pharm. Biopharm. 2018, 124, 147–157. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Alzghoul, A.; Mahlin, D.; Bergström, C.A. Physical stability of drugs after storage above and below the glass transition temperature: Relationship to glass-forming ability. Int. J. Pharm. 2015, 495, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Chen, H.; Wang, Y.; Wang, R.; Xu, J.; Zhang, C. Amorphous Solid Dispersions: Role of the Polymer and Its Importance in Physical Stability and In Vitro Performance. Pharmaceutics 2022, 14, 1747. [Google Scholar] [CrossRef]
- Kou, X.; Zhou, L. Stability of Amorphous Solid Dispersion. In Amorphous Solid Dispersions: Theory and Practice; Shah, N., Sandhu, H., Choi, D.S., Chokshi, H., Malick, A.W., Eds.; Springer: New York, NY, USA, 2014; pp. 515–544. [Google Scholar]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous solid dispersions: An update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef]
- DeBoyace, K.; Wildfong, P.L. The application of modeling and prediction to the formation and stability of amorphous solid dispersions. J. Pharm. Sci. 2018, 107, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.; Authelin, J.-R.; Leveder, C.; Segalini, A. A practical method to predict physical stability of amorphous solid dispersions. Pharm. Res. 2012, 29, 2792–2805. [Google Scholar] [CrossRef]
- Blaabjerg, L.I.; Bulduk, B.; Lindenberg, E.; Löbmann, K.; Rades, T.; Grohganz, H. Influence of glass forming ability on the physical stability of supersaturated amorphous solid dispersions. J. Pharm. Sci. 2019, 108, 2561–2569. [Google Scholar] [CrossRef]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, M.; Hancock, B.C.; Zografi, G. Crystallization of indomethacin from the amorphous state below and above its glass transition temperature. J. Pharm. Sci. 1994, 83, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Sibik, J.; Löbmann, K.; Rades, T.; Zeitler, J.A. Predicting crystallization of amorphous drugs with terahertz spectroscopy. Mol. Pharm. 2015, 12, 3062–3068. [Google Scholar] [CrossRef] [PubMed]
- Kissi, E.O.; Grohganz, H.; Löbmann, K.; Ruggiero, M.T.; Zeitler, J.A.; Rades, T. Glass-transition temperature of the β-relaxation as the major predictive parameter for recrystallization of neat amorphous drugs. J. Phys. Chem. B 2018, 122, 2803–2808. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.; Zografi, G. Commentary: Considerations in the measurement of glass transition temperatures of pharmaceutical amorphous solids. AAPS PharmSciTech. 2020, 21, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, J.A.; Taylor, L.S. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv. Drug Deliv. Rev. 2012, 64, 396–421. [Google Scholar] [CrossRef]
- Thomas, L.C. Modulated dsc® paper# 1 why modulated dsc®?; an overview and summary of advantages and disadvantages relative to traditional dsc. TA Instrum. Tech. Pap. TP 2005, 6, TP006. [Google Scholar]
- Reading, M.; Hahn, B.K.; Crowe, B.S. Method and Apparatus for Modulated Differential Analysis. U.S. Patent 5,224,775A, 6 July 1993. [Google Scholar]
- Royall, P.G.; Craig, D.Q.; Doherty, C. Characterisation of the glass transition of an amorphous drug using modulated DSC. Pharm. Res. 1998, 15, 1117–1121. [Google Scholar] [CrossRef]
- Dong, Y.; Ruan, Y.; Wang, H.; Zhao, Y.; Bi, D. Studies on glass transition temperature of chitosan with four techniques. J. Appl. Polym. Sci. 2004, 93, 1553–1558. [Google Scholar] [CrossRef]
- Mahlin, D.; Wood, J.; Hawkins, N.; Mahey, J.; Royall, P.G. A novel powder sample holder for the determination of glass transition temperatures by DMA. Int. J. Pharm. 2009, 371, 120–125. [Google Scholar] [CrossRef]
- Diogo, H.P.; Ramos, J.J.M. TSDC and DSC Investigation on the Molecular Mobility in the Amorphous Solid State and in the Glass Transformation Region of Two Benzodiazepine Derivatives: Diazepam and Nordazepam. J. Pharm. Sci. 2022, 111, 2239–2248. [Google Scholar] [CrossRef] [PubMed]
- Knapik-Kowalczuk, J.; Rams-Baron, M.; Paluch, M. Current research trends in dielectric relaxation studies of amorphous pharmaceuticals: Physical stability, tautomerism, and the role of hydrogen bonding. Trends Anal. Chem. 2021, 134, 116097. [Google Scholar] [CrossRef]
- Ramos, J.J.M.; Diogo, H.P. The determination of the glass transition temperature by thermally stimulated depolarization currents. Comparison with the performance of other techniques. Phase Transit. 2017, 90, 1061–1078. [Google Scholar] [CrossRef]
- Huang, Y.; Dai, W.-G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm. Sin. B 2014, 4, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Shmeis, R.A.; Wang, Z.; Krill, S.L. A mechanistic investigation of an amorphous pharmaceutical and its solid dispersions, part I: A comparative analysis by thermally stimulated depolarization current and differential scanning calorimetry. Pharm. Res. 2004, 21, 2025–2030. [Google Scholar] [CrossRef]
- Zhang, J.; Bunker, M.; Parker, A.; Madden-Smith, C.E.; Patel, N.; Roberts, C.J. The stability of solid dispersions of felodipine in polyvinylpyrrolidone characterized by nanothermal analysis. Int. J. Pharm. 2011, 414, 210–217. [Google Scholar] [CrossRef]
- Galop, M. Study of pharmaceutical solid dispersions by microthermal analysis. Pharm. Res. 2005, 22, 293–302. [Google Scholar] [CrossRef]
- Thayumanasundaram, S.; Venkatesan, T.R.; Ousset, A.; Van Hollebeke, K.; Aerts, L.; Wübbenhorst, M.; Van den Mooter, G. Complementarity of mDSC, DMA, and DRS Techniques in the Study of T g and Sub-T g Transitions in Amorphous Solids: PVPVA, Indomethacin, and Amorphous Solid Dispersions Based on Indomethacin/PVPVA. Mol. Pharm. 2022, 19, 2299–2315. [Google Scholar] [CrossRef]
- Ramos, J.J.M.; Piedade, M.F.M.; Diogo, H.P.; Viciosa, M.T. Thermal behavior and slow relaxation dynamics in amorphous efavirenz: A study by DSC, XRPD, TSDC, and DRS. J. Pharm. Sci. 2019, 108, 1254–1263. [Google Scholar] [CrossRef]
- Blaabjerg, L.I.; Lindenberg, E.; Löbmann, K.; Grohganz, H.; Rades, T. Glass forming ability of amorphous drugs investigated by continuous cooling and isothermal transformation. Mol. Pharm. 2016, 13, 3318–3325. [Google Scholar] [CrossRef]
- Lakshman, J.P.; Cao, Y.; Kowalski, J.; Serajuddin, A.T. Application of melt extrusion in the development of a physically and chemically stable high-energy amorphous solid dispersion of a poorly water-soluble drug. Mol. Pharm. 2008, 5, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Worku, Z.A.; Aarts, J.; Singh, A.; Van den Mooter, G. Drug–polymer miscibility across a spray dryer: A case study of naproxen and miconazole solid dispersions. Mol. Pharm. 2014, 11, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Balogh, A.; Farkas, B.; Faragó, K.; Farkas, A.; Wagner, I.; Verreck, G.; Nagy, Z.K.; Marosi, G. Melt-blown and electrospun drug-loaded polymer fiber mats for dissolution enhancement: A comparative study. J. Pharm. Sci. 2015, 104, 1767–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Instruments, T. µTA™ 2990 Micro-Thermal Analyzer. Available online: https://www.tainstruments.com/pdf/literature/TA_TA-243.pdf (accessed on 15 October 2022).
- Nagy, Z.K.; Balogh, A.; Démuth, B.; Pataki, H.; Vigh, T.; Szabó, B.; Molnár, K.; Schmidt, B.T.; Horák, P.; Marosi, G. High speed electrospinning for scaled-up production of amorphous solid dispersion of itraconazole. Int. J. Pharm. 2015, 480, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Kazsoki, A.; Szabó, P.; Süvegh, K.; Vörös, T.; Zelkó, R. Macro-and microstructural tracking of ageing-related changes of papaverine hydrochloride-loaded electrospun nanofibrous buccal sheets. J. Pharm. Biomed. Anal. 2017, 143, 62–67. [Google Scholar] [CrossRef]
- Li, X.-Y.; Wang, X.; Yu, D.-G.; Ye, S.; Kuang, Q.-K.; Yi, Q.-W.; Yao, X.-Z. Electrospun borneol-PVP nanocomposites. J. Nanomater. 2012, 2012, 731382. [Google Scholar] [CrossRef] [Green Version]
- Rumondor, A.C.; Taylor, L.S. Effect of polymer hygroscopicity on the phase behavior of amorphous solid dispersions in the presence of moisture. Mol. Pharm. 2010, 7, 477–490. [Google Scholar] [CrossRef]
- Prudic, A.; Kleetz, T.; Korf, M.; Ji, Y.; Sadowski, G. Influence of copolymer composition on the phase behavior of solid dispersions. Mol. Pharm. 2014, 11, 4189–4198. [Google Scholar] [CrossRef]
- Löbmann, K.; Laitinen, R.; Grohganz, H.; Gordon, K.C.; Strachan, C.; Rades, T. Coamorphous drug systems: Enhanced physical stability and dissolution rate of indomethacin and naproxen. Mol. Pharm. 2011, 8, 1919–1928. [Google Scholar] [CrossRef]
- Paudel, A.; Nies, E.; Van den Mooter, G. Relating hydrogen-bonding interactions with the phase behavior of naproxen/PVP K 25 solid dispersions: Evaluation of solution-cast and quench-cooled films. Mol. Pharm. 2012, 9, 3301–3317. [Google Scholar] [CrossRef]
- Gordon, M.; Taylor, J.S. Ideal copolymers and the second-order transitions of synthetic rubbers. I. Non-crystalline copolymers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Moura Ramos, J.J.; Correia, N.T.; Taveira-Marques, R.; Collins, G. The activation energy at T g and the fragility index of indomethacin, predicted from the influence of the heating rate on the temperature position and on the intensity of thermally stimulated depolarization current peak. Pharm. Res. 2002, 19, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Marossy, K. Practical approach to thermally stimulated discharge (TSD) method on polymers. J. Therm. Anal. Calorim. 2017, 129, 161–170. [Google Scholar] [CrossRef]
- Tobyn, M.; Brown, J.; Dennis, A.B.; Fakes, M.; Gamble, J.; Khimyak, Y.Z.; Mcgeorge, G.; Patel, C.; Sinclair, W.; Timmins, P. Amorphous drug-PVP dispersions: Application of theoretical, thermal and spectroscopic analytical techniques to the study of a molecule with intermolecular bonds in both the crystalline and pure amorphous state. J. Pharm. Sci. 2009, 98, 3456–3468. [Google Scholar] [CrossRef]
- Li, N.; Cape, J.L.; Mankani, B.R.; Zemlyanov, D.Y.; Shepard, K.B.; Morgen, M.M.; Taylor, L.S. Water-induced phase separation of spray-dried amorphous solid dispersions. Mol. Pharm. 2020, 17, 4004–4017. [Google Scholar] [CrossRef] [PubMed]
- Ayenew, Z.; Paudel, A.; Van den Mooter, G. Can compression induce demixing in amorphous solid dispersions? A case study of naproxen–PVP K25. Eur. J. Pharm. Biopharm. 2012, 81, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Saboo, S.; Kestur, U.S.; Flaherty, D.P.; Taylor, L.S. Congruent release of drug and polymer from amorphous solid dispersions: Insights into the role of drug-polymer hydrogen bonding, surface crystallization, and glass transition. Mol. Pharm. 2020, 17, 1261–1275. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | NAP (mg) | PVPVA64 (mg) | Ratio of the Solvents (v/v) | Amount of the Solvents |
|---|---|---|---|---|
| PVPVA64 | - | 2000.0 | DCM:EtOH 1:1 | 8 mL |
| 10%NAP_90%PVPVA64 | 222.2 | |||
| 20%NAP_80%PVPVA64 | 500.0 | |||
| 30%NAP_70%PVPVA64 | 857.1 | |||
| 40%NAP_60%PVPVA64 | 1333.3 | |||
| 50%NAP_50%PVPVA64 | 2000.0 | |||
| CRYST. NAP | Crystalline NAP was used as a reference as received, without any modification. | |||
| Sample | MDSC (°C) | TSDC (°C) |
|---|---|---|
| PVPVA64 | 0 | 14 |
| 10%NAP_90%PVPVA64 | 0 | 16 |
| 20%NAP_80%PVPVA64 | −5 | 12 |
| 30%NAP_70%PVPVA64 | −1 | 9 |
| 40%NAP_60%PVPVA64 | −2 | 5 |
| 50%NAP_50%PVPVA64 | 1 | 2 |
| START | 1 Day | 1 Week | |
|---|---|---|---|
| PVPVA64 |  |  |  |
| 10%NAP_ 90%PVPVA64 |  |  |  |
| 20%NAP_ 80%PVPVA64 |  |  |  |
| 30%NAP_ 70%PVPVA64 |  |  |  |
| 40%NAP_ 60%PVPVA64 |  |  |  |
| 50%NAP_ 50%PVPVA64 |  |  |  |
| START | 1 Day | 1 Week | |
|---|---|---|---|
| 10%NAP_ 90%PVPVA64 |  |  |  |
| 20%NAP_ 80%PVPVA64 |  |  |  |
| 30%NAP_ 70%PVPVA64 |  |  |  |
| 40%NAP_ 60%PVPVA64 |  |  |  |
| 50%NAP_ 50%PVPVA64 |  |  |  |
| CRYST. NAP |  | PVPVA64 |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabó, E.; Haraszti, A.; Záhonyi, P.; Vadas, D.; Csontos, I.; Nagy, Z.K.; Van den Mooter, G.; Marosi, G. Evaluation of Different Thermoanalytical Methods for the Analysis of the Stability of Naproxen-Loaded Amorphous Solid Dispersions. Pharmaceutics 2022, 14, 2508. https://doi.org/10.3390/pharmaceutics14112508
Szabó E, Haraszti A, Záhonyi P, Vadas D, Csontos I, Nagy ZK, Van den Mooter G, Marosi G. Evaluation of Different Thermoanalytical Methods for the Analysis of the Stability of Naproxen-Loaded Amorphous Solid Dispersions. Pharmaceutics. 2022; 14(11):2508. https://doi.org/10.3390/pharmaceutics14112508
Chicago/Turabian StyleSzabó, Edina, Anna Haraszti, Petra Záhonyi, Dániel Vadas, István Csontos, Zsombor Kristóf Nagy, Guy Van den Mooter, and György Marosi. 2022. "Evaluation of Different Thermoanalytical Methods for the Analysis of the Stability of Naproxen-Loaded Amorphous Solid Dispersions" Pharmaceutics 14, no. 11: 2508. https://doi.org/10.3390/pharmaceutics14112508