

Oral Delivery of Niclosamide as an Amorphous Solid Dispersion That Generates Amorphous Nanoparticles during Dissolution
 , ,
, ,  ,
, 
Abstract
:
1. Introduction
2. Methods
2.1. Preparation of the Niclosamide ASD Granules
2.2. Preparation of the Enteric Capsules Containing Niclosamide ASD Granules
2.3. Preparation of the Enteric-Coated Tablets of Niclosamide ASD
2.4. HPLC Analysis
2.5. Dissolution Testing of the Granules, Capsules, and Tablets
2.6. Acid Uptake Test
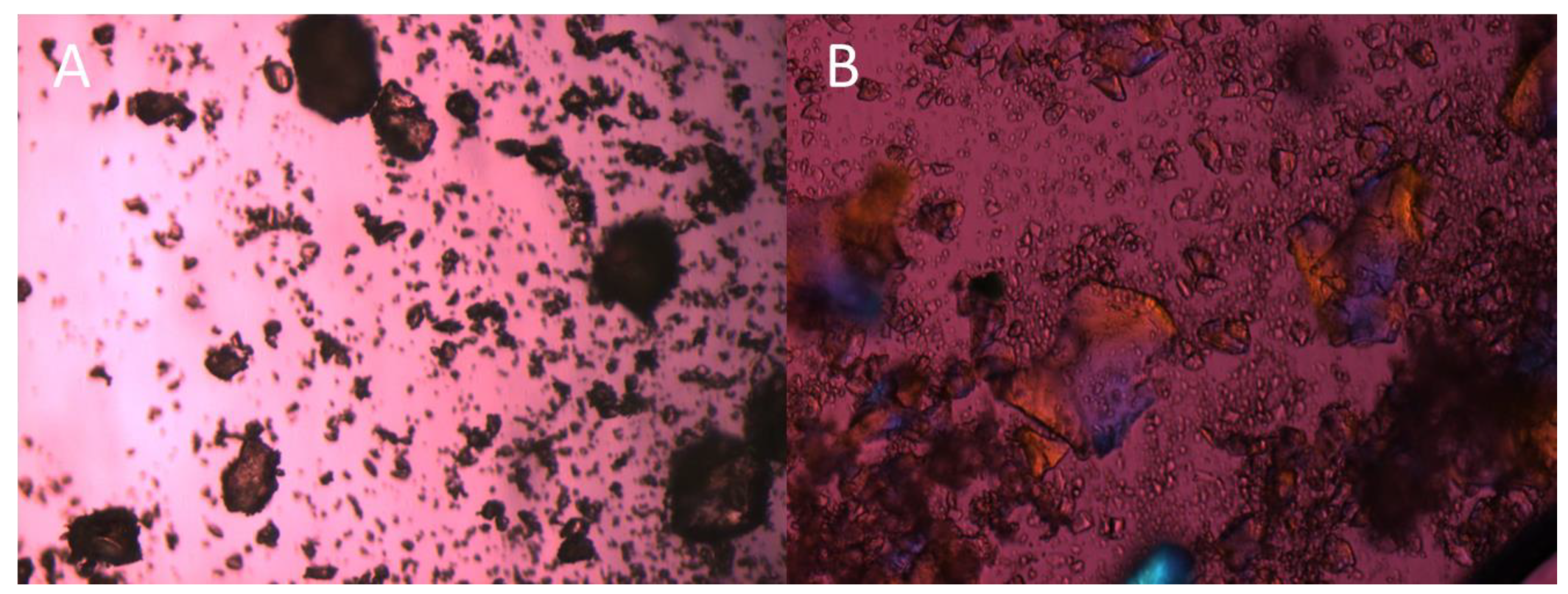
2.7. Polarized Light Microscopy (PLM)
2.8. Wide-Angle X-ray Scattering (WAXS)
2.9. Cryogenic Transmission Electron Microscopy (Cryo-TEM)
2.10. Administration of Niclosamide ASD to Beagle Dogs
3. Results and Discussion
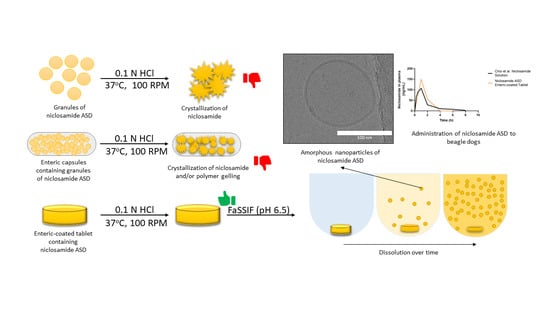
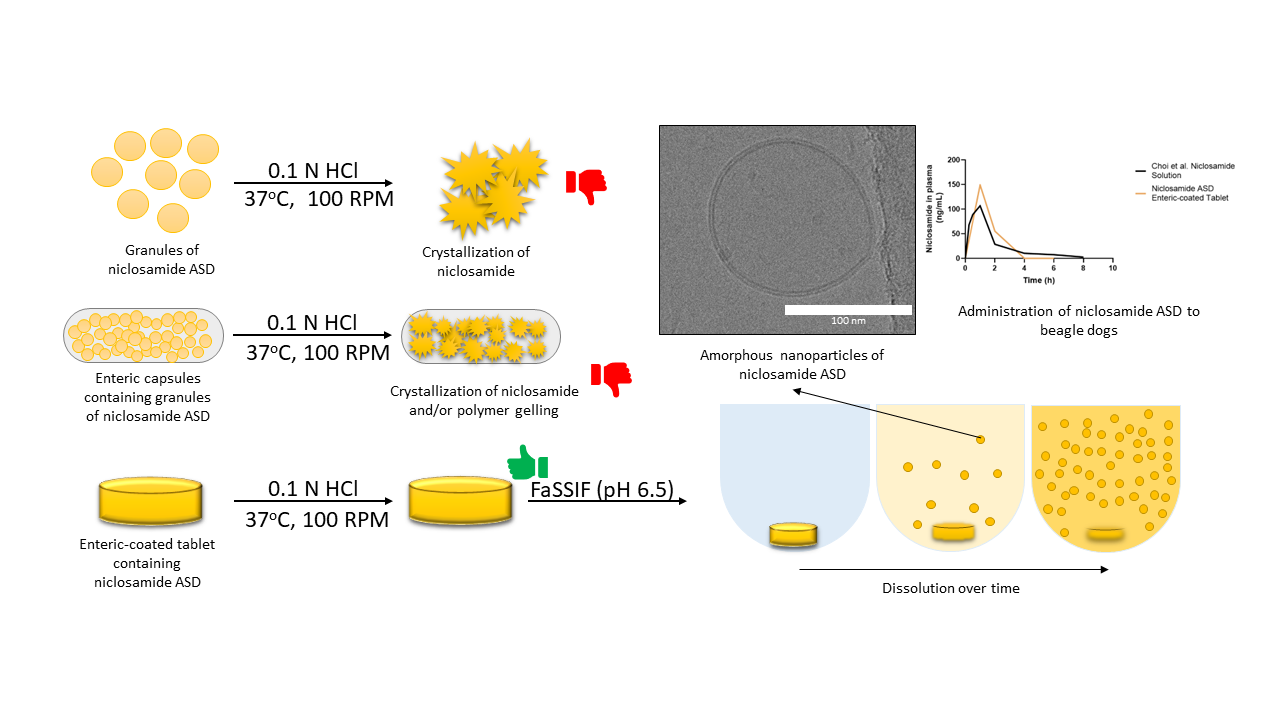
3.1. Niclosamide ASD Generated Amorphous Nanoparticles in Biorelevant Media
3.2. Niclosamide ASD Dissolution Performance and Stability Were Impaired When Formulated as an Enteric Capsule Formulation
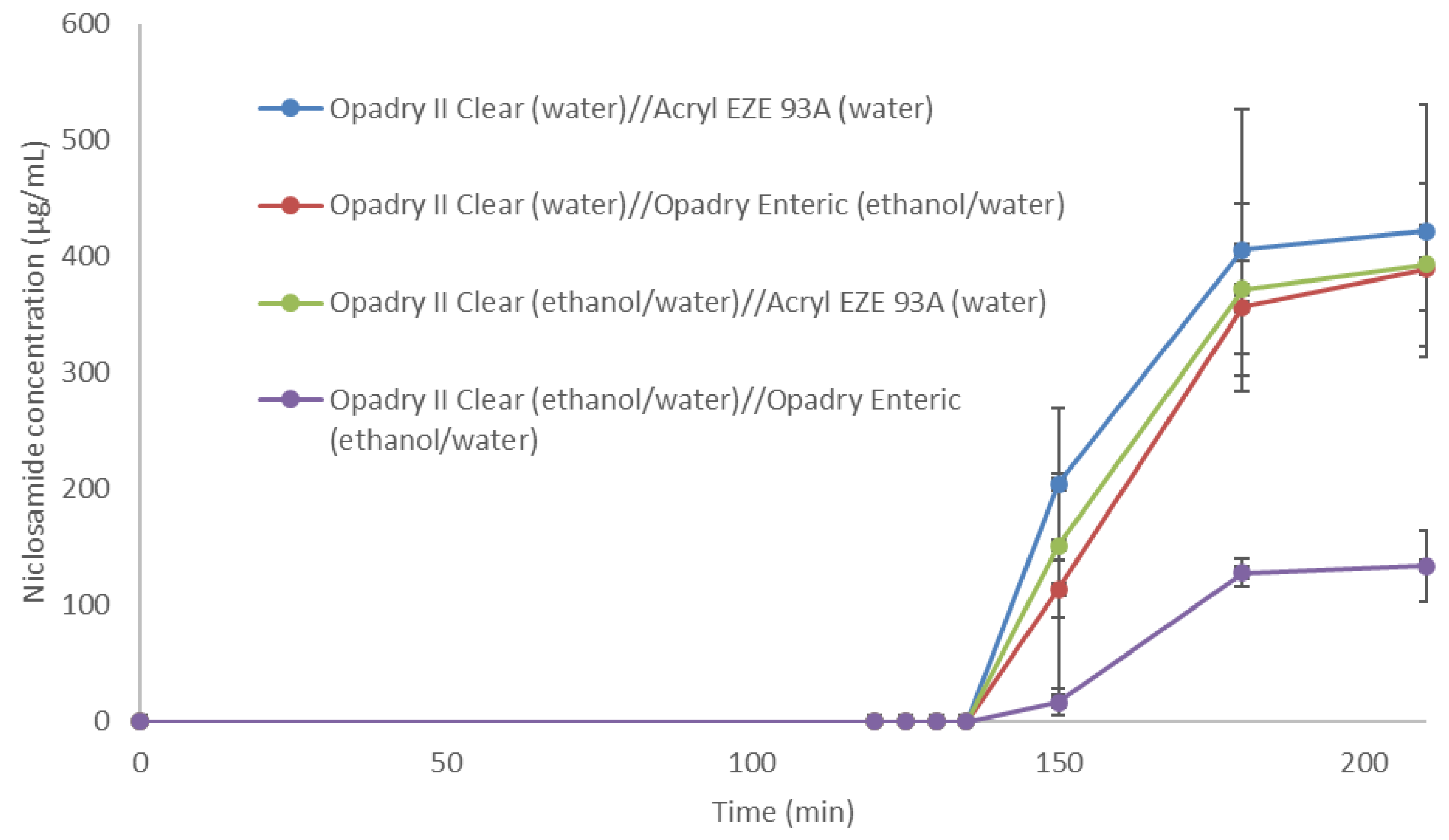
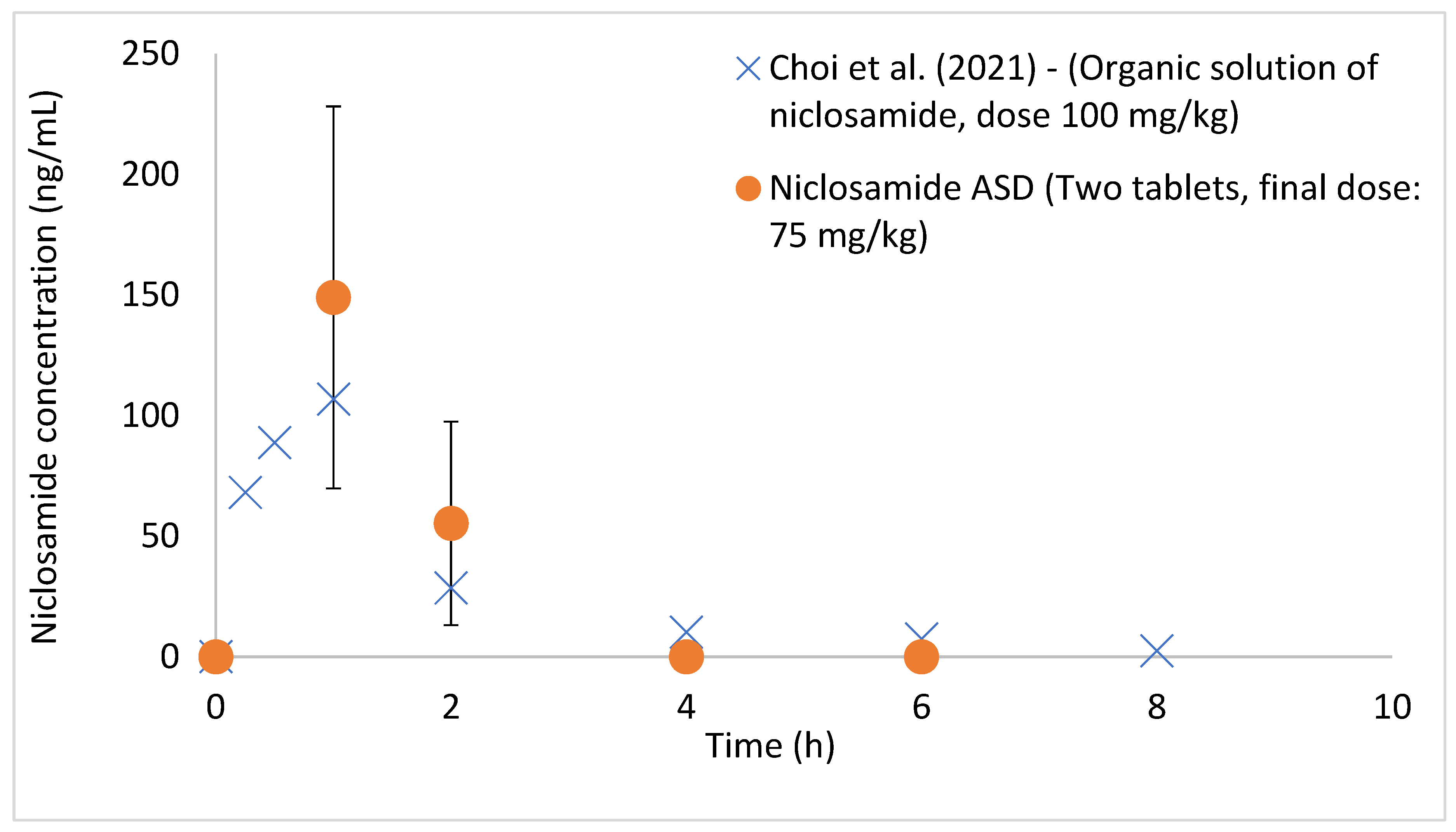
3.3. Niclosamide ASD Was Protected by Enteric-Coated Tablet Formulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aungst, B.J. Optimizing Oral Bioavailability in Drug Discovery: An Overview of Design and Testing Strategies and Formulation Options. J. Pharm. Sci. 2017, 106, 921–929. [Google Scholar] [CrossRef]
- Bayliss, M.K.; Butler, J.; Feldman, P.L.; Green, D.V.S.; Leeson, P.D.; Palovich, M.R.; Taylor, A.J. Quality Guidelines for Oral Drug Candidates: Dose, Solubility and Lipophilicity. Drug Discov. Today 2016, 21, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Loftsson, T.; Brewster, M.E. Pharmaceutical Applications of Cyclodextrins: Basic Science and Product Development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef]
- Jermain, S.V.; Brough, C.; Williams, R.O. Amorphous Solid Dispersions and Nanocrystal Technologies for Poorly Water-Soluble Drug Delivery—An Update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef]
- Formulating Poorly Water Soluble Drugs; Williams, R.O.; Watts, A.B., III; Miller, D.A. (Eds.) AAPS Advances in the Pharmaceutical Sciences Series; Springer International Publishing: Cham, Switzerland, 2016; Volume 22, ISBN 978-3-319-42607-5. [Google Scholar]
- Ashwathy, P.; Anto, A.T.; Sudheesh, M.S. A Mechanistic Review on the Dissolution Phase Behavior and Supersaturation Stabilization of Amorphous Solid Dispersions. Drug Dev. Ind. Pharm. 2021, 47, 1–11. [Google Scholar] [CrossRef]
- Qian, F.; Huang, J.; Hussain, M.A. Drug–Polymer Solubility and Miscibility: Stability Consideration and Practical Challenges in Amorphous Solid Dispersion Development. J. Pharm. Sci. 2010, 99, 2941–2947. [Google Scholar] [CrossRef] [PubMed]
- Van den Mooter, G. The Use of Amorphous Solid Dispersions: A Formulation Strategy to Overcome Poor Solubility and Dissolution Rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef] [PubMed]
- Saboo, S.; Mugheirbi, N.A.; Zemlyanov, D.Y.; Kestur, U.S.; Taylor, L.S. Congruent Release of Drug and Polymer: A “Sweet Spot” in the Dissolution of Amorphous Solid Dispersions. J. Control. Release 2019, 298, 68–82. [Google Scholar] [CrossRef]
- Harmon, P.; Galipeau, K.; Xu, W.; Brown, C.; Wuelfing, W.P. Mechanism of Dissolution-Induced Nanoparticle Formation from a Copovidone-Based Amorphous Solid Dispersion. Mol. Pharm. 2016, 13, 1467–1481. [Google Scholar] [CrossRef]
- Ricarte, R.G.; Li, Z.; Johnson, L.M.; Ting, J.M.; Reineke, T.M.; Bates, F.S.; Hillmyer, M.A.; Lodge, T.P. Direct Observation of Nanostructures during Aqueous Dissolution of Polymer/Drug Particles. Macromolecules 2017, 50, 3143–3152. [Google Scholar] [CrossRef]
- Tres, F.; Posada, M.M.; Hall, S.D.; Mohutsky, M.A.; Taylor, L.S. Mechanistic Understanding of the Phase Behavior of Supersaturated Solutions of Poorly Water-Soluble Drugs. Int. J. Pharm. 2018, 543, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Mann, A.K.P.; Van Duong, T.; Ormes, J.D.; Okoh, G.A.; Hermans, A.; Taylor, L.S. Drug Release and Nanodroplet Formation from Amorphous Solid Dispersions: Insight into the Roles of Drug Physicochemical Properties and Polymer Selection. Mol. Pharm. 2021, 18, 2066–2081. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.-W.; Kuhn, R.; Hu, C.; Zhang, W.; Chiang, P.-C.; Chen, J.Z.; Hau, J.; Estevez, A.; Nagapudi, K.; Leung, D.H. Impact of Surfactant Selection and Incorporation on in Situ Nanoparticle Formation from Amorphous Solid Dispersions. Int. J. Pharm. 2021, 607, 120980. [Google Scholar] [CrossRef] [PubMed]
- Correa Soto, C.E.; Gao, Y.; Indulkar, A.S.; Ueda, K.; Zhang, G.G.Z.; Taylor, L.S. Impact of Surfactants on the Performance of Clopidogrel-Copovidone Amorphous Solid Dispersions: Increased Drug Loading and Stabilization of Nanodroplets. Pharm. Res. 2022, 39, 167–188. [Google Scholar] [CrossRef]
- Liu, L.; Chen, L.; Müllers, W.; Serno, P.; Qian, F. Water-Resistant Drug–Polymer Interaction Contributes to the Formation of Nano-Species during the Dissolution of Felodipine Amorphous Solid Dispersions. Mol. Pharm. 2022, 19, 2888–2899. [Google Scholar] [CrossRef]
- Démuth, B.; Nagy, Z.K.; Balogh, A.; Vigh, T.; Marosi, G.; Verreck, G.; Van Assche, I.; Brewster, M.E. Downstream Processing of Polymer-Based Amorphous Solid Dispersions to Generate Tablet Formulations. Int. J. Pharm. 2015, 486, 268–286. [Google Scholar] [CrossRef]
- Lugtu-Pe, J.A.; Ghaffari, A.; Chen, K.; Kane, A.; Wu, X.Y. Development of Controlled Release Amorphous Solid Dispersions (CRASD) Using Polyvinyl Acetate-Based Release Retarding Materials: Effect of Dosage Form Design. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2018, 124, 319–327. [Google Scholar] [CrossRef]
- Puri, V.; Dantuluri, A.K.; Bansal, A.K. Investigation of Atypical Dissolution Behavior of an Encapsulated Amorphous Solid Dispersion. J. Pharm. Sci. 2011, 100, 2460–2468. [Google Scholar] [CrossRef]
- Agrawal, A.; Dudhedia, M.; Deng, W.; Shepard, K.; Zhong, L.; Povilaitis, E.; Zimny, E. Development of Tablet Formulation of Amorphous Solid Dispersions Prepared by Hot Melt Extrusion Using Quality by Design Approach. AAPS PharmSciTech 2016, 17, 214–232. [Google Scholar] [CrossRef]
- Akbuga, J.; Gürsoy, A.; Yetimoĝlu, F. Preparation and Properties of Tablets Prepared from Furosemide-PVP Solid Dispersion Systems. Drug Dev. Ind. Pharm. 1988, 14, 2091–2108. [Google Scholar] [CrossRef]
- Xi, H.; Ren, J.; Novak, J.M.; Kemp, E.; Johnson, G.; Klinzing, G.; Johnson, M.A.; Xu, W. The Effect of Inorganic Salt on Disintegration of Tablets with High Loading of Amorphous Solid Dispersion Containing Copovidone. Pharm. Res. 2020, 37, 70. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A.; Premont, R.T.; Wang, J. Niclosamide: Beyond an Antihelminthic Drug. Cell. Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef]
- Xu, J.; Shi, P.-Y.; Li, H.; Zhou, J. Broad Spectrum Antiviral Agent Niclosamide and Its Therapeutic Potential. ACS Infect. Dis. 2020, 6, 909–915. [Google Scholar] [CrossRef]
- Jara, M.O.; Warnken, Z.N.; Williams, R.O. Amorphous Solid Dispersions and the Contribution of Nanoparticles to In Vitro Dissolution and In Vivo Testing: Niclosamide as a Case Study. Pharmaceutics 2021, 13, 97. [Google Scholar] [CrossRef]
- Elkhabaz, A.; Sarkar, S.; Simpson, G.J.; Taylor, L.S. Characterization of Phase Transformations for Amorphous Solid Dispersions of a Weakly Basic Drug upon Dissolution in Biorelevant Media. Pharm. Res. 2019, 36, 174. [Google Scholar] [CrossRef]
- de Villiers, M.M.; Mahlatji, M.D.; van Tonder, E.C.; Malan, S.F.; Lötter, A.P.; Liebenberg, W. Comparison of the Physical and Chemical Stability of Niclosamide Crystal Forms in Aqueous Versus Nonaqueous Suspensions. Drug Dev. Ind. Pharm. 2004, 30, 581–592. [Google Scholar] [CrossRef]
- Hövelmann, J.; Stawski, T.M.; Besselink, R.; Freeman, H.M.; Dietmann, K.M.; Mayanna, S.; Pauw, B.R.; Benning, L.G. A Template-Free and Low Temperature Method for the Synthesis of Mesoporous Magnesium Phosphate with Uniform Pore Structure and High Surface Area. Nanoscale 2019, 11, 6939–6951. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Katai, H.; Higashi, K.; Ueda, K.; Kawakami, K.; Moribe, K. Cryo-TEM and AFM Observation of the Time-Dependent Evolution of Amorphous Probucol Nanoparticles Formed by the Aqueous Dispersion of Ternary Solid Dispersions. Mol. Pharm. 2019, 16, 2184–2198. [Google Scholar] [CrossRef]
- Clulow, A.J.; Parrow, A.; Hawley, A.; Khan, J.; Pham, A.C.; Larsson, P.; Bergström, C.A.S.; Boyd, B.J. Characterization of Solubilizing Nanoaggregates Present in Different Versions of Simulated Intestinal Fluid. J. Phys. Chem. B 2017, 121, 10869–10881. [Google Scholar] [CrossRef]
- Kabedev, A.; Hossain, S.; Hubert, M.; Larsson, P.; Bergström, C.A.S. Molecular Dynamics Simulations Reveal Membrane Interactions for Poorly Water-Soluble Drugs: Impact of Bile Solubilization and Drug Aggregation. J. Pharm. Sci. 2021, 110, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Khoshakhlagh, P.; Johnson, R.; Langguth, P.; Nawroth, T.; Schmueser, L.; Hellmann, N.; Decker, H.; Szekely, N.K. Fasted-State Simulated Intestinal Fluid “FaSSIF-C”, a Cholesterol Containing Intestinal Model Medium for in Vitro Drug Delivery Development. J. Pharm. Sci. 2015, 104, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Kloefer, B.; Hoogevest, P.; Moloney, R.; Kuentz, M.; Leigh, M.; Dressma, J. Study of a Standardized Taurocholate-Lecithin Powder for Preparing the Biorelevant Media FeSSIF and FaSSIF. Dissolution Technol. 2010, 8, 6–13. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Alzghoul, A.; Kaialy, W.; Mahlin, D.; Bergström, C.A.S. Computational Predictions of Glass-Forming Ability and Crystallization Tendency of Drug Molecules. Mol. Pharm. 2014, 11, 3123–3132. [Google Scholar] [CrossRef] [PubMed]
- Vuai, S.A.; Sahini, M.G.; Onoka, I.; Kiruri, L.W.; Shadrack, D.M. Cation–π Interactions Drive Hydrophobic Self-Assembly and Aggregation of Niclosamide in Water. RSC Adv. 2021, 11, 33136–33147. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Piao, H.; Rejinold, N.S.; Jin, G.; Choi, G.; Choy, J.-H. Niclosamide–Clay Intercalate Coated with Nonionic Polymer for Enhanced Bioavailability toward COVID-19 Treatment. Polymers 2021, 13, 1044. [Google Scholar] [CrossRef]
- Barbosa, E.J.; Löbenberg, R.; de Araujo, G.L.B.; Bou-Chacra, N.A. Niclosamide Repositioning for Treating Cancer: Challenges and Nano-Based Drug Delivery Opportunities. Eur. J. Pharm. Biopharm. 2019, 141, 58–69. [Google Scholar] [CrossRef]
- Mook, R.A.; Wang, J.; Ren, X.-R.; Chen, M.; Spasojevic, I.; Barak, L.S.; Lyerly, H.K.; Chen, W. Structure-Activity Studies of Wnt/β-Catenin Inhibition in the Niclosamide Chemotype: Identification of Derivatives with Improved Drug Exposure. Bioorg. Med. Chem. 2015, 23, 5829–5838. [Google Scholar] [CrossRef] [Green Version]
- Natera, J.; Gatica, E.; Challier, C.; Possetto, D.; Massad, W.; Miskoski, S.; Pajares, A.; García, N.A. On the Photooxidation of the Multifunctional Drug Niclosamide. A Kinetic Study in the Presence of Vitamin B2 and Visible Light. Redox Rep. 2015, 20, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Needham, D. The PH Dependence of Niclosamide Solubility, Dissolution, and Morphology: Motivation for Potentially Universal Mucin-Penetrating Nasal and Throat Sprays for COVID19, Its Variants and Other Viral Infections. Pharm. Res. 2022, 39, 115–141. [Google Scholar] [CrossRef]
- Devarakonda, B.; Hill, R.A.; Liebenberg, W.; Brits, M.; de Villiers, M.M. Comparison of the Aqueous Solubilization of Practically Insoluble Niclosamide by Polyamidoamine (PAMAM) Dendrimers and Cyclodextrins. Int. J. Pharm. 2005, 304, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Paghadar, C.; Vadia, N.H. Development and Validation of Stability Indicating RP-HPLC and HPTLC for Determination of Niclosamide in Bulk and in Synthetic Mixture. Arab. J. Chem. 2019, 12, 1803–1814. [Google Scholar] [CrossRef] [Green Version]
- Shantier, S.; Elobaid, E.; Gadkariem, E.; Wagiealla, S. Stability Studies on Niclosamide Using Derivative Spectroscopic and Chromatographic Methods. World J. Pharm. Res. 2015, 4, 122–132. [Google Scholar]
- Zaazaa, H.E.; Abdelrahman, M.M.; Ali, N.W.; Magdy, M.A.; Abdelkawy, M. Kinetic Study and Mechanism of Niclosamide Degradation. Spectrochim. Acta A. Mol. Biomol. Spectrosc. 2014, 132, 655–662. [Google Scholar] [CrossRef]
- Johnsirani, P.; Ch, V.; Lingesh, A.; Naidu, V.G.M.; Ch, N.; Satheeshkumar, N. Isolation, Characterization Using LC-ESI-QTOF, NMR and in Vitro Cytotoxicity Assay of Niclosamide Forced Degradation Products. J. Pharm. Biomed. Anal. 2017, 136, 148–155. [Google Scholar] [CrossRef]
- Smith, A.M.; Ingham, A.; Grover, L.M.; Perrie, Y. Polymer Film Formulations for the Preparation of Enteric Pharmaceutical Capsules. J. Pharm. Pharmacol. 2010, 62, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Badawy, S.I.F.; Hussain, M.A. Microenvironmental PH Modulation in Solid Dosage Forms. J. Pharm. Sci. 2007, 96, 948–959. [Google Scholar] [CrossRef]
- Hughey, J.R.; Keen, J.M.; Miller, D.A.; Kolter, K.; Langley, N.; McGinity, J.W. The Use of Inorganic Salts to Improve the Dissolution Characteristics of Tablets Containing Soluplus®-Based Solid Dispersions. Eur. J. Pharm. Sci. 2013, 48, 758–766. [Google Scholar] [CrossRef]
- Takano, R.; Maurer, R.; Jacob, L.; Stowasser, F.; Stillhart, C.; Page, S. Formulating Amorphous Solid Dispersions: Impact of Inorganic Salts on Drug Release from Tablets Containing Itraconazole-HPMC Extrudate. Mol. Pharm. 2020, 17, 2768–2778. [Google Scholar] [CrossRef]
- Taniguchi, C.; Kawabata, Y.; Wada, K.; Yamada, S.; Onoue, S. Microenvironmental PH-Modification to Improve Dissolution Behavior and Oral Absorption for Drugs with PH-Dependent Solubility. Expert Opin. Drug Deliv. 2014, 11, 505–516. [Google Scholar] [CrossRef]
- Al-Tabakha, M.M.; Arida, A.I.; Fahelelbom, K.M.S.; Sadek, B.; Abu Jarad, R.A. Performances of New Generation of Delayed Release Capsules. J. Young Pharm. 2014, 7, 36–44. [Google Scholar] [CrossRef]
- Fu, M.; Al-Gousous, J.; Blechar, J.A.; Langguth, P. Enteric Hard Capsules for Targeting the Small Intestine: Positive Correlation between In Vitro Disintegration and Dissolution Times. Pharmaceutics 2020, 12, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, S.D.; Kundu, I.; Swamy, N.P.; Crull, G.B.; Pan, D.; Zhao, J.; Shah, R.P.; Venkatesh, C.; Vig, B.; Varia, S.A.; et al. Cross-Linking of Poly (Vinyl Alcohol) Films under Acidic and Thermal Stress. Eur. J. Pharm. Sci. 2020, 152, 105429. [Google Scholar] [CrossRef]
- Khan, A.; Majeedullah; Khan, S.A. Tablets and Capsules. In Essentials of Industrial Pharmacy; Khan, S.A., Ed.; AAPS Advances in the Pharmaceutical Sciences Series; Springer International Publishing: Cham, Switzerland, 2022; pp. 95–122. ISBN 978-3-030-84977-1. [Google Scholar]
- Missaghi, S.; Young, C.; Fegely, K.; Rajabi-Siahboomi, A.R. Delayed Release Film Coating Applications on Oral Solid Dosage Forms of Proton Pump Inhibitors: Case Studies. Drug Dev. Ind. Pharm. 2010, 36, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Hansell, J.; Nuneviller, F., III; Rajabi-Siahboomi, A.R. Evaluation of Recent Advances in Continuous Film Coating Processes. Drug Dev. Ind. Pharm. 2010, 36, 227–233. [Google Scholar] [CrossRef]
- Choi, H.-I.; Kim, T.; Lee, S.-W.; Woo Kim, J.; Ju Noh, Y.; Kim, G.-Y.; Jin Park, H.; Chae, Y.-J.; Lee, K.-R.; Kim, S.-J.; et al. Bioanalysis of Niclosamide in Plasma Using Liquid Chromatography-Tandem Mass and Application to Pharmacokinetics in Rats and Dogs. J. Chromatogr. B 2021, 1179, 122862. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Seal Coating | Enteric Coating | %Weight Gain after 2 h in 0.1 N HCl |
|---|---|---|
| Opadry® II Clear (water) | Acryl-EZE® 93A (water) | 2.87 ± 0.53 |
| Opadry® II Clear (water) | Opadry® Enteric (ethanol/water) | 1.28 ± 0.58 |
| Opadry® II Clear (ethanol/water) | Acryl-EZE® 93A (water) | 3.08 ± 0.46 |
| Opadry® II Clear (ethanol/water) | Opadry® Enteric (ethanol/water) | 0.94 ± 0.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jara, M.O.; Warnken, Z.N.; Sahakijpijarn, S.; Thakkar, R.; Kulkarni, V.R.; Christensen, D.J.; Koleng, J.J.; Williams, R.O., III. Oral Delivery of Niclosamide as an Amorphous Solid Dispersion That Generates Amorphous Nanoparticles during Dissolution. Pharmaceutics 2022, 14, 2568. https://doi.org/10.3390/pharmaceutics14122568
Jara MO, Warnken ZN, Sahakijpijarn S, Thakkar R, Kulkarni VR, Christensen DJ, Koleng JJ, Williams RO III. Oral Delivery of Niclosamide as an Amorphous Solid Dispersion That Generates Amorphous Nanoparticles during Dissolution. Pharmaceutics. 2022; 14(12):2568. https://doi.org/10.3390/pharmaceutics14122568
Chicago/Turabian StyleJara, Miguel O., Zachary N. Warnken, Sawittree Sahakijpijarn, Rishi Thakkar, Vineet R. Kulkarni, Dale J. Christensen, John J. Koleng, and Robert O. Williams, III. 2022. "Oral Delivery of Niclosamide as an Amorphous Solid Dispersion That Generates Amorphous Nanoparticles during Dissolution" Pharmaceutics 14, no. 12: 2568. https://doi.org/10.3390/pharmaceutics14122568