

Arene Variation of Highly Cytotoxic Tridentate Naphthoquinone-Based Ruthenium(II) Complexes and In-Depth In Vitro Studies
 , , , , , , , , and
, , , , , , , , and
Abstract
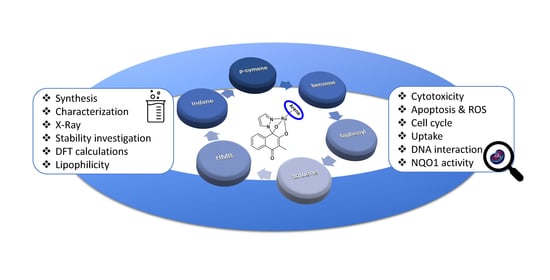
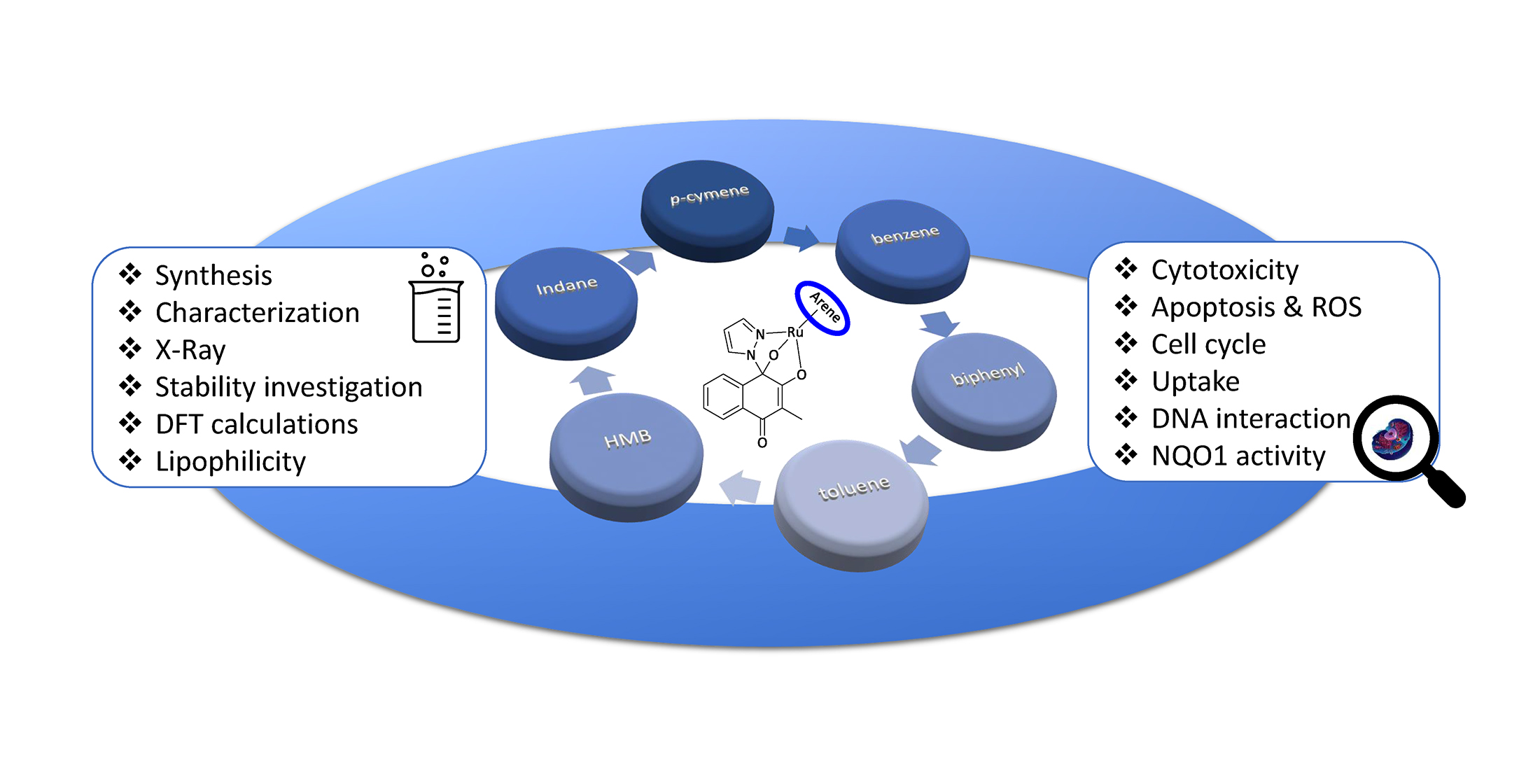
:
1. Introduction
2. Materials and Methods
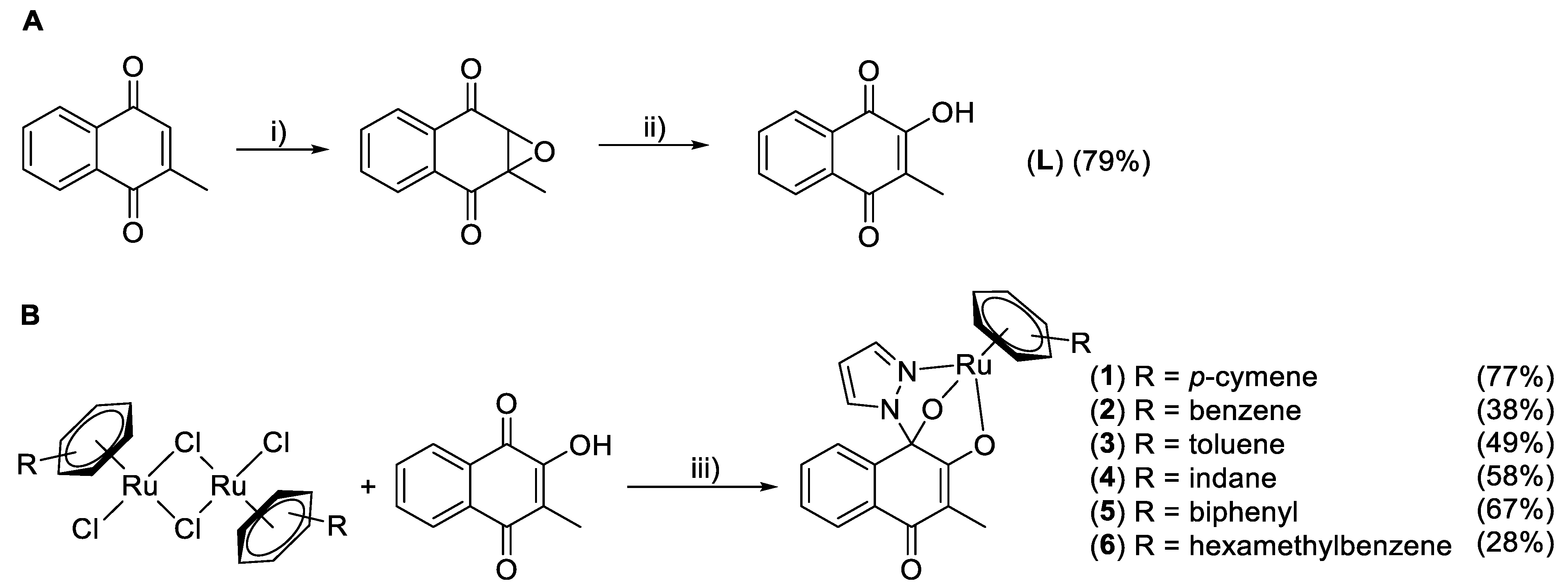
2.1. Synthesis
General Procedure for Complex Syntheses (2–6) [17]
2.2. HPLC-MS Stability Measurements
2.3. Theoretical Simulations
2.4. Cell Culture
2.5. MTT Assay
2.6. Colony Formation Assay
2.7. Cellular Ruthenium Accumulation
2.8. Cell Cycle Studies
2.9. Apoptosis Assay (Annexin V/PI)
2.10. DCFH/DA Assay
2.11. DNA Interaction Assay
2.12. NQO1 Activity Assay
3. Results and Discussion
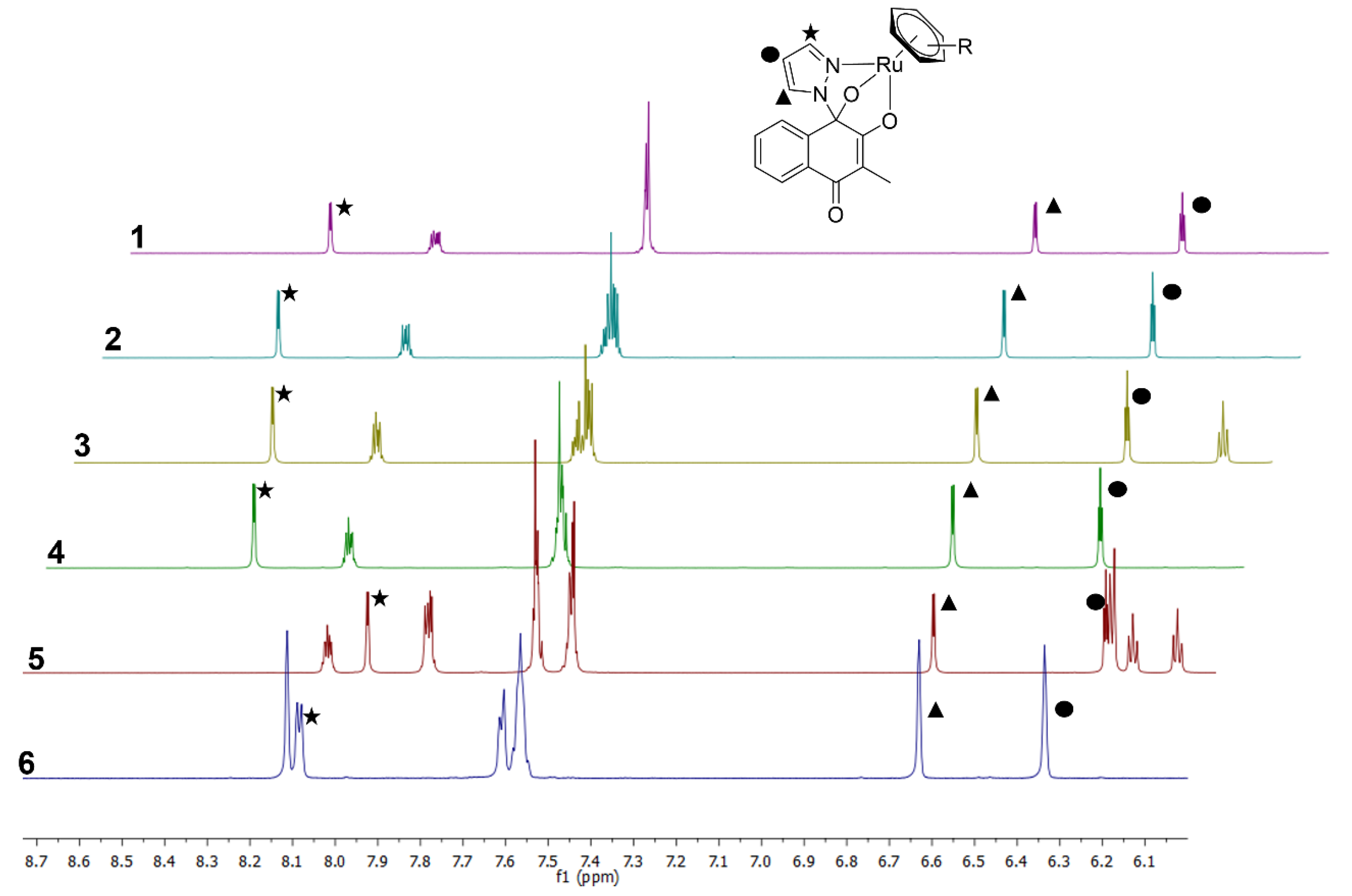
3.1. Syntheses and Characterization
3.2. X-ray Crystallographic Studies
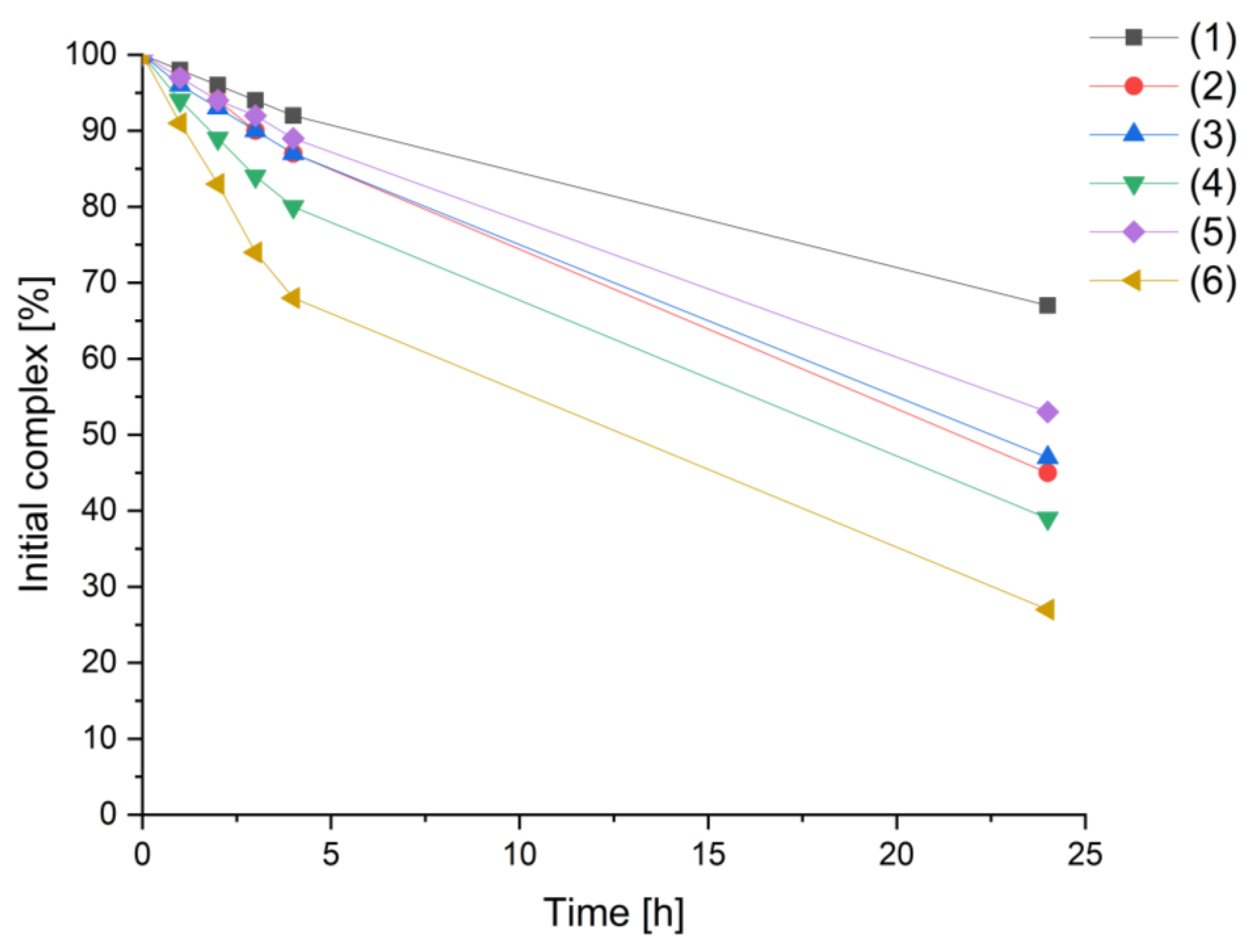
3.3. Stability Studies
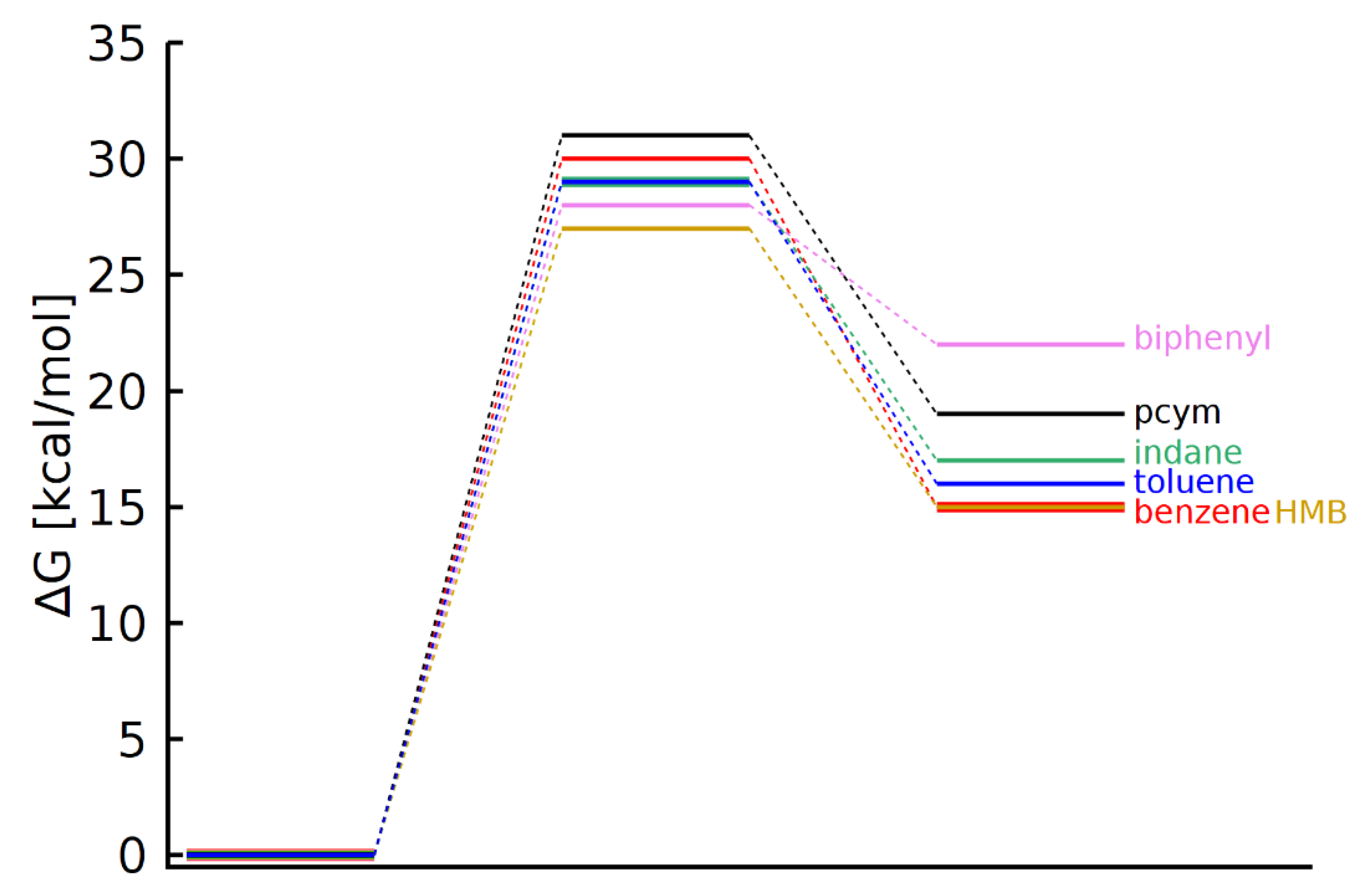
3.4. DFT Calculations
3.5. Biological Studies
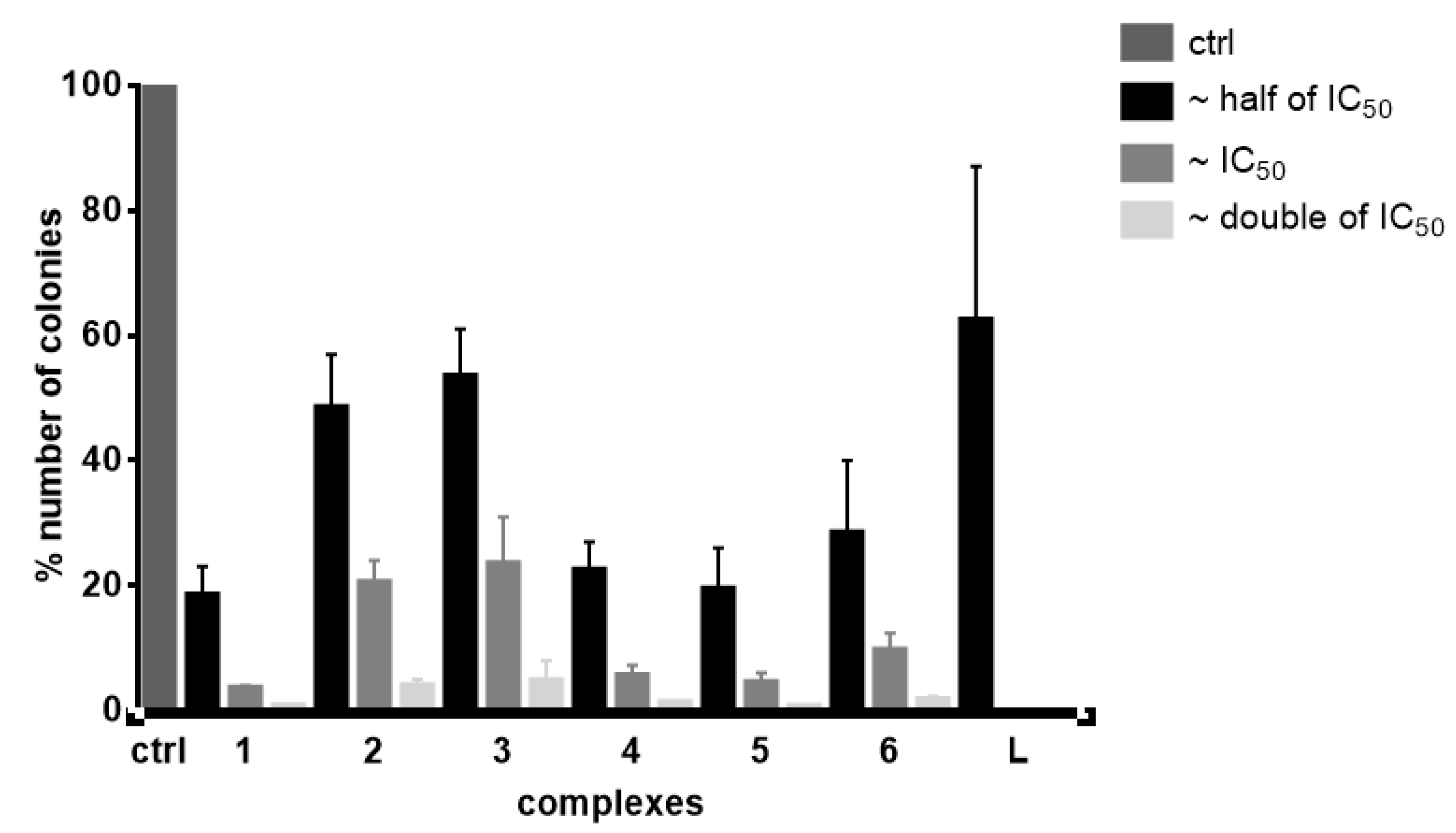
3.5.1. Effects on Cell Proliferation and Cytotoxicity
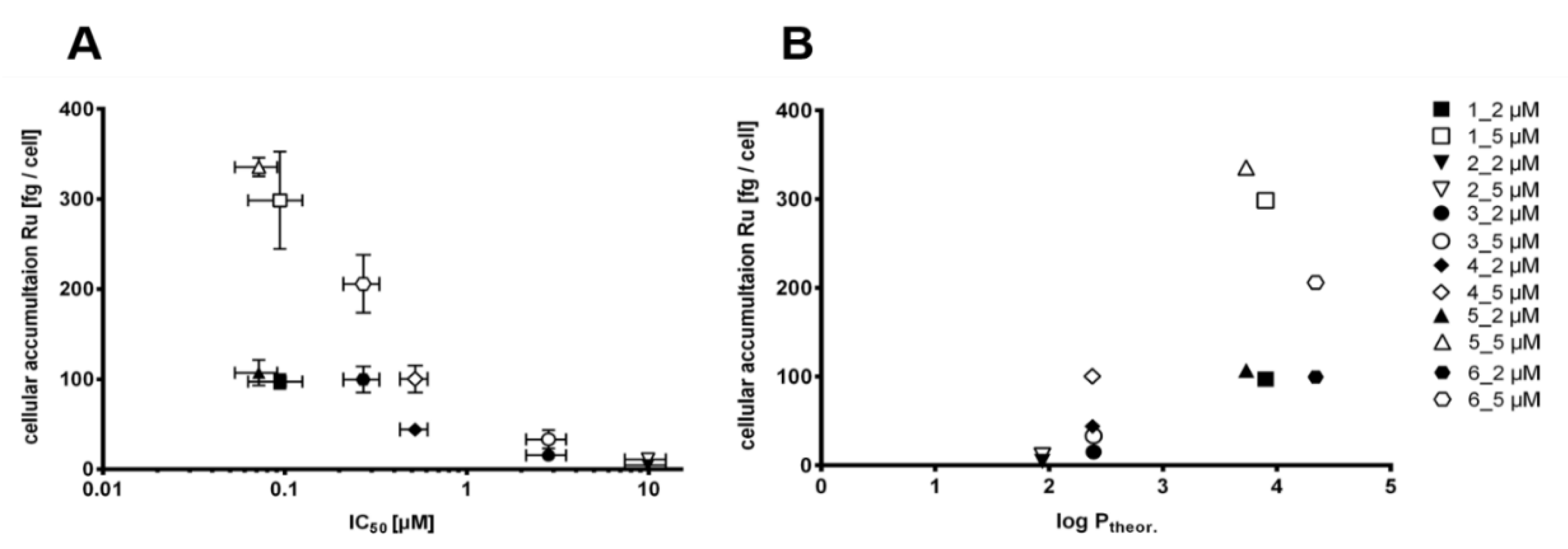
3.5.2. Cellular Accumulation
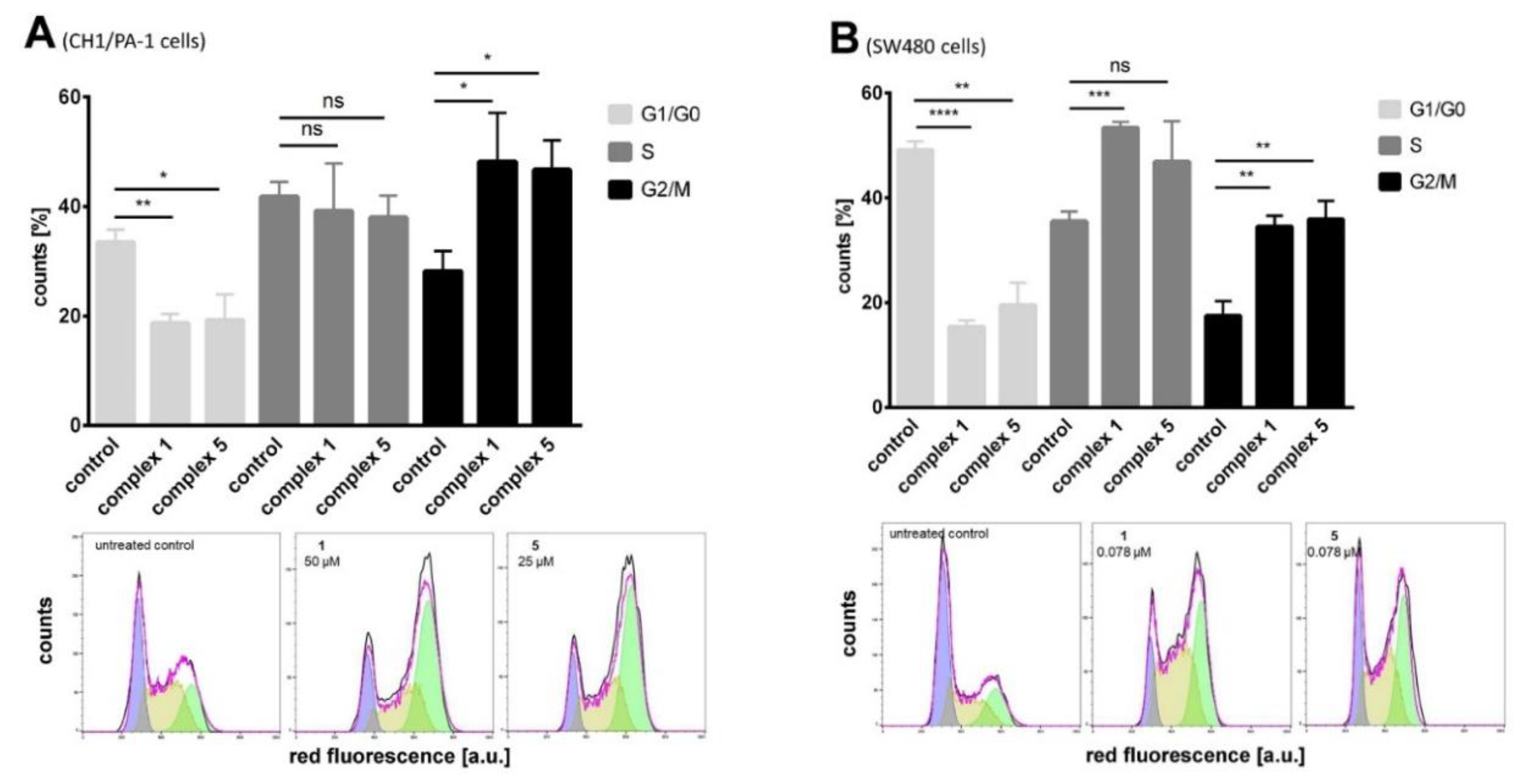
3.5.3. Analysis of Cell Cycle Distribution
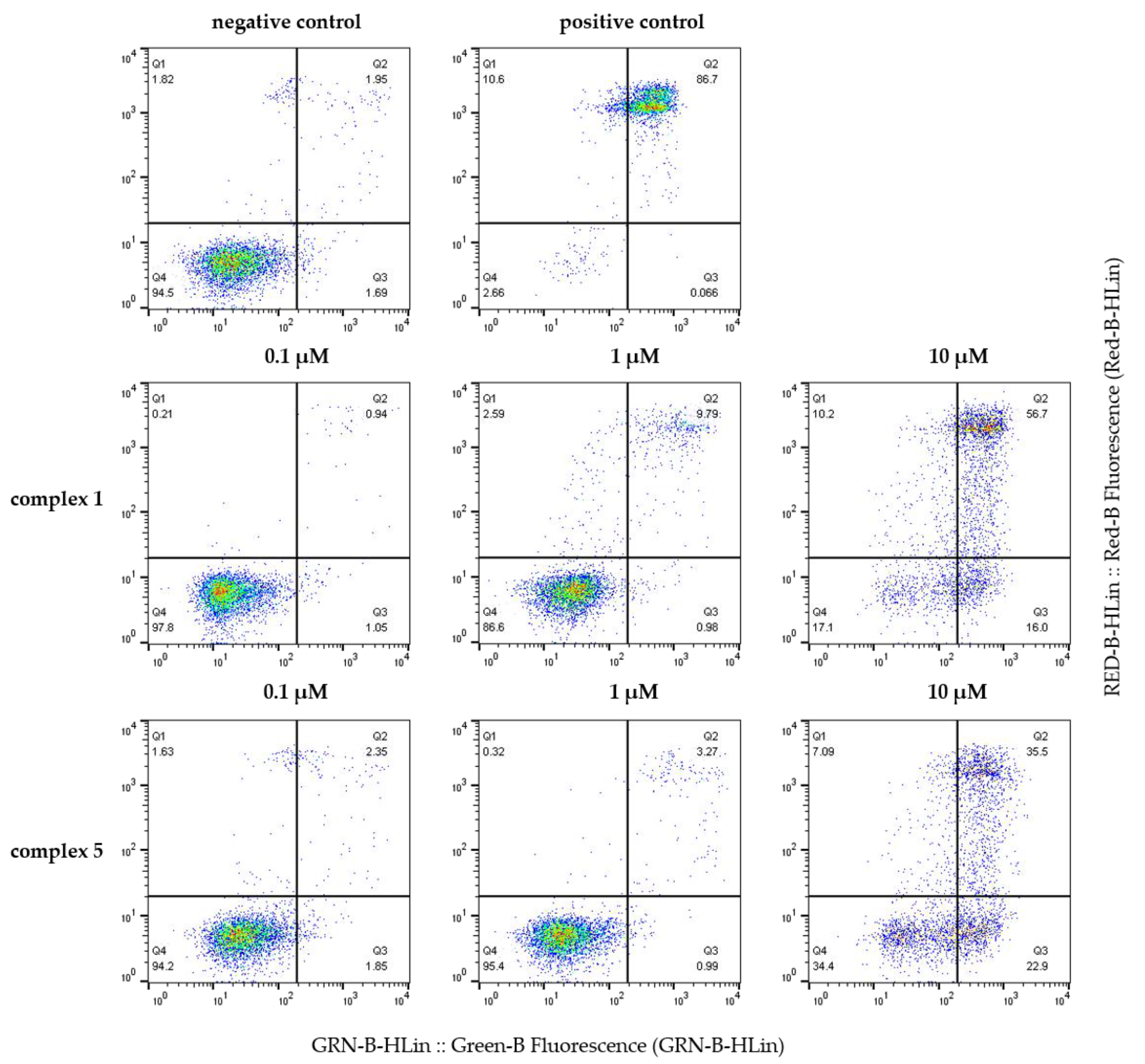
3.5.4. Apoptosis Induction
3.5.5. ROS Assay
3.5.6. DNA Interaction Studies
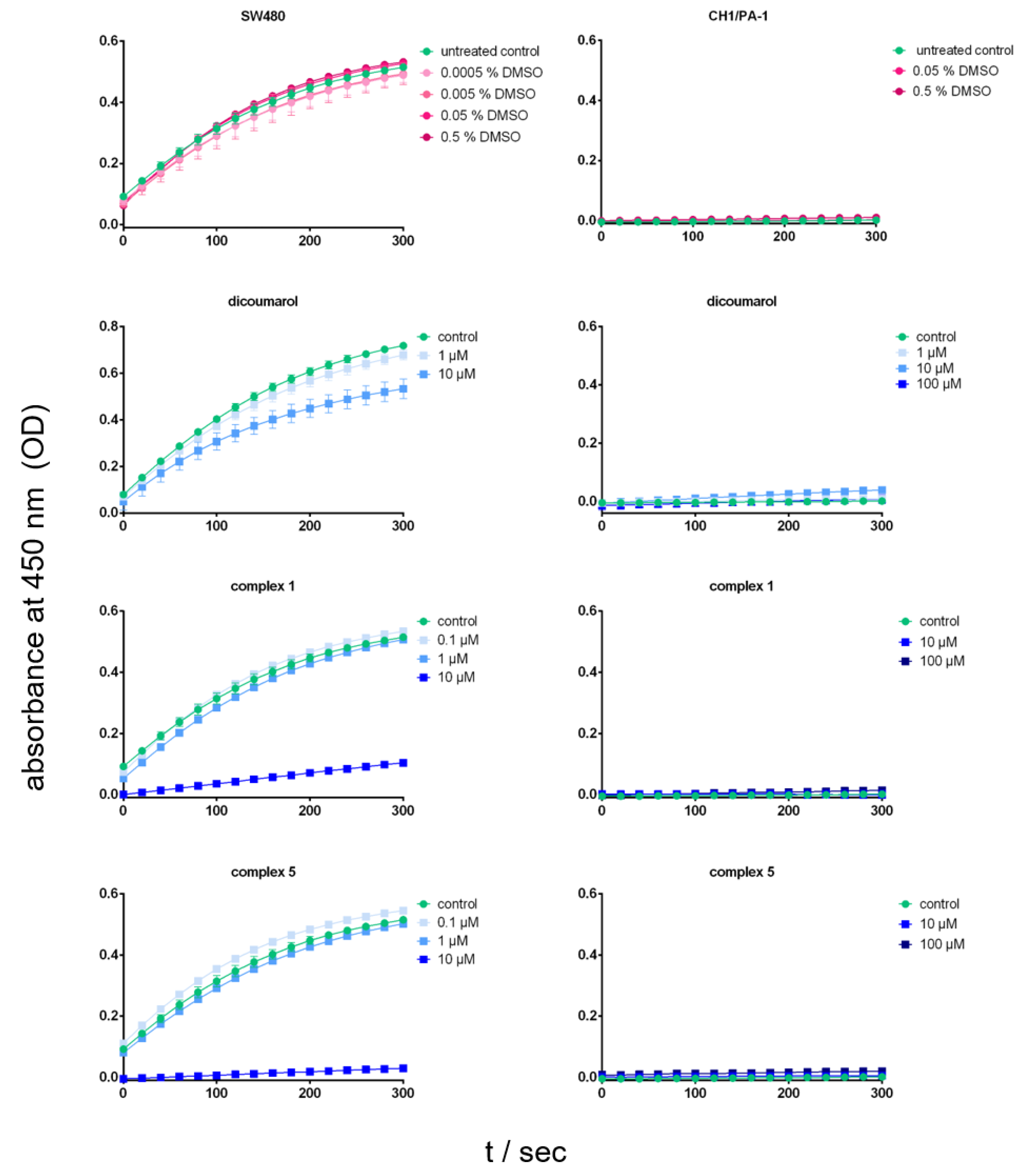
3.5.7. NQO1 Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Cheff, D.M.; Hall, M.D. A drug of such damned nature. 1 challenges and opportunities in translational platinum drug research. J. Med. Chem. 2017, 60, 4517–4532. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment. Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Köpf, H.; Köpf-Maier, P. Titanocene Dichloride—The first metallocene with cancerostatic activity. Angew. Chem. Int. Ed. Engl. 1979, 18, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Guo, Z.; Sadler, P. Titanium(IV) targets phosphoesters on nucleotides: Implications for the mechanism of action of the anticancer drug titanocene dichloride. J. Biol. Inorg. Chem. 2001, 6, 698–707. [Google Scholar] [CrossRef]
- Vatansever, H.S.; Kabadayı, H.; Korkmaz, M.; Özdal-Kurt, F.; Kavukcu, S.B.; Türkmen, H. Apoptotic properties of rutheinum complexes on different type of cancer cell lines. Proceedings 2018, 2, 1593. [Google Scholar] [CrossRef] [Green Version]
- Intravesical Photodynamic Therapy (PDT) in BCG Refractory/Intolerant Non-Muscle Invasive Bladder Cancer (NMIBC) Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03945162 (accessed on 13 October 2022).
- BOLD-100 in Combination With FOLFOX for the Treatment of Advanced Solid Tumours. Available online: https://clinicaltrials.gov/ct2/show/NCT04421820 (accessed on 13 October 2022).
- Adhireksan, Z.; Davey, G.E.; Campomanes, P.; Groessl, M.; Clavel, C.M.; Yu, H.; Nazarov, A.A.; Yeo, C.H.F.; Ang, W.H.; Dröge, P.; et al. Ligand substitutions between ruthenium-cymene compounds can control protein versus DNA targeting and anticancer activity. Nat. Commun. 2014, 5, 3462. [Google Scholar] [CrossRef] [Green Version]
- Pizarro, A.M.; Habtemariam, A.; Sadler, P.J. Activation mechanisms for organometallic anticancer complexes. In Medicinal Organometallic Chemistry; Jaouen, G., Metzler-Nolte, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 21–56. [Google Scholar]
- Peacock, A.F.A.; Habtemariam, A.; Fernández, R.; Walland, V.; Fabbiani, F.P.A.; Parsons, S.; Aird, R.E.; Jodrell, D.I.; Sadler, P.J. Tuning the reactivity of Osmium(II) and Ruthenium(II) arene complexes under physiological conditions. J. Am. Chem. Soc. 2006, 128, 1739–1748. [Google Scholar] [CrossRef]
- Ang, W.H.; Daldini, E.; Scolaro, C.; Scopelliti, R.; Juillerat-Jeannerat, L.; Dyson, P.J. Development of organometallic ruthenium−arene anticancer drugs that resist hydrolysis. Inorg. Chem. 2006, 45, 9006–9013. [Google Scholar] [CrossRef]
- Pereyra, C.E.; Dantas, R.F.; Ferreira, S.B.; Gomes, L.P.; Silva-Jr, F.P. The diverse mechanisms and anticancer potential of naphthoquinones. Cancer Cell Int. 2019, 19, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackl, C.M.; Schoenhacker-Alte, B.; Klose, M.H.M.; Henke, H.; Legina, M.S.; Jakupec, M.A.; Berger, W.; Keppler, B.K.; Brüggemann, O.; Teasdale, I.; et al. Synthesis and in vivo anticancer evaluation of poly(organo)phosphazene-based metallodrug conjugates. Dalton Trans. 2017, 46, 12114–12124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandioller, W.; Balsano, E.; Meier, S.M.; Jungwirth, U.; Göschl, S.; Roller, A.; Jakupec, M.A.; Berger, W.; Keppler, B.K.; Hartinger, C.G. Organometallic anticancer complexes of lapachol: Metal centre-dependent formation of reactive oxygen species and correlation with cytotoxicity. Chem. Commun. 2013, 49, 3348–3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, H.; Wernitznig, D.; Hejl, M.; Gajic, N.; Jakupec, M.A.; Kandioller, W.; Keppler, B.K. Novel phthiocol-based organometallics with tridentate coordination motif and their unexpected cytotoxic behaviour. Dalton Trans. 2020, 49, 1393–1397. [Google Scholar] [CrossRef]
- Babak, M.V.; Meier, S.M.; Legin, A.A.; Adib Razavi, M.S.; Roller, A.; Jakupec, M.A.; Keppler, B.K.; Hartinger, C.G. Am(m)ines make the difference: Organoruthenium Am(m)ine complexes and their chemistry in anticancer drug development. Chem. A Eur. J. 2013, 19, 4308–4318. [Google Scholar] [CrossRef]
- Geisler, H.; Westermayr, J.; Cseh, K.; Wenisch, D.; Fuchs, V.; Harringer, S.; Plutzar, S.; Gajic, N.; Hejl, M.; Jakupec, M.A.; et al. Tridentate 3-substituted naphthoquinone ruthenium arene complexes: Synthesis, characterization, aqueous behavior, and theoretical and biological studies. Inorg. Chem. 2021, 60, 9805–9819. [Google Scholar] [CrossRef]
- Martínez-De-León, C.G.; del Carmen Flores Vallejo, R.; Rodríguez-Álvarez, A.; Villareal, M.L.; Grévy, J.-M. Synthesis, characterization and cytotoxic activity of cationic half-sandwich Ru(ii) complexes stabilized by iminophosphorane N,N,S and N,N,Se tridentate ligands. New J. Chem. 2020, 44, 20676–20687. [Google Scholar] [CrossRef]
- Bennett, M.A.; Huang, T.N.; Matheson, T.W.; Smith, A.K.; Ittel, S.; Nickerson, W. 16. (η6-Hexamethylbenzene)Ruthenium Complexes. In Inorganic Syntheses; John and Wiley and Sons: Hoboken, NJ, USA, 1982; pp. 74–78. [Google Scholar]
- Ma, L.; Ma, R.; Wang, Z.; Yiu, S.-M.; Zhu, G. Heterodinuclear Pt(iv)–Ru(ii) anticancer prodrugs to combat both drug resistance and tumor metastasis. Chem. Commun. 2016, 52, 10735–10738. [Google Scholar] [CrossRef] [Green Version]
- Queyriaux, N.; Giannoudis, E.; Lefebvre, J.F.; Artero, V.; Chavarot-Kerlidou, M. Synthesis of Ruthenium Tris-Diimine photosensitizers substituted by four methylphosphonate anchoring groups for dye-sensitized photoelectrochemical cell applications. Eur. J. Inorg. Chem. 2019, 2019, 2154–2161. [Google Scholar] [CrossRef]
- Kathawate, L.; Gejji, S.P.; Yeole, S.D.; Verma, P.L.; Puranik, V.G.; Salunke-Gawali, S. The first naphthosemiquinone complex of K+ with vitamin K3 analog: Experiment and density functional theory. J. Mol. Struct. 2015, 1088, 56–63. [Google Scholar] [CrossRef]
- Zhu, R.; Xing, L.; Wang, X.; Cheng, C.; Liu, B.; Hu, Y. Practical Preparation of Diosphenols by Ring Opening of α,β-Epoxyketones Catalyzed by Silica Gel Supported Acids. Synlett 2007, 2007, 2267–2271. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 054107. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- York, D.M.; Karplus, M. A smooth solvation potential based on the conductor-like screening model. J. Phys. Chem. A 1999, 103, 11060–11079. [Google Scholar] [CrossRef]
- Available online: https://www.abcam.com/nqo1-activity-assay-kit-ab184867.html# (accessed on 13 October 2022).
- Condon, S.E.; Buron, C.; Tippmann, E.M.; Tinner, C.; Platz, M.S. Generation and characterization of phenylsulfanylcarbene. Org. Lett. 2004, 6, 815–818. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, M.J.; More O’Ferrall, R.A.; Boyd, D.R.; Lam, C.M.; O’Donoghue, A.C. Substituent effects on the dehydration of arene hydrates in aqueous solution. J. Phys. Org. Chem. 2013, 26, 989–996. [Google Scholar] [CrossRef] [Green Version]
- Di, L.; Kerns, E.H. Stability challenges in drug discovery. Chem. Biodivers. 2009, 6, 1875–1886. [Google Scholar] [CrossRef]
- Geisler, H.; Harringer, S.; Wenisch, D.; Urban, R.; Jakupec, M.A.; Kandioller, W.; Keppler, B.K. Systematic study on the cytotoxic potency of commonly used dimeric metal precursors in human cancer cell lines. ChemistryOpen 2022, 11, e202200019. [Google Scholar] [CrossRef]
- Harringer, S.; Hejl, M.; Enyedi, É.A.; Jakupec, M.A.; Galanski, M.S.; Keppler, B.K.; Dyson, P.J.; Varbanov, H.P. Multifunctional Pt(IV) prodrug candidates featuring the carboplatin core and deferoxamine. Dalton Trans. 2021, 50, 8167–8178. [Google Scholar] [CrossRef]
- Varbanov, H.P.; Goschl, S.; Heffeter, P.; Theiner, S.; Roller, A.; Jensen, F.; Jakupec, M.A.; Berger, W.; Galanski, M.S.; Keppler, B.K. A novel class of bis- and tris-chelate diam(m)inebis(dicarboxylato)platinum(IV) complexes as potential anticancer prodrugs. J. Med. Chem. 2014, 57, 6751–6764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, B.; Zheng, L.; Yang, K.; Li, Y.; Hu, M.; He, D. Target ROS to induce apoptosis and cell cycle arrest by 5,7-dimethoxy-1,4-naphthoquinone derivative. Bioorg. Med. Chem. Lett. 2018, 28, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, Y.H.; Shen, G.N.; Piao, X.J.; Xu, W.T.; Zhang, Y.; Wang, J.R.; Feng, Y.C.; Li, J.Q.; Zhang, Y.; et al. Two novel 1,4-naphthoquinone derivatives induce human gastric cancer cell apoptosis and cell cycle arrest by regulating reactive oxygen speciesmediated MAPK/Akt/STAT3 signaling pathways. Mol. Med. Rep. 2019, 20, 2571–2582. [Google Scholar] [CrossRef] [Green Version]
- Jan, R.; Chaudhry, G.E. Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Ma, T.; Yang, P.; Dammann, I.; Lin, Z.; Mougharbel, A.S.; Li, M.X.; Adascalitei, F.; Mitea, R.; Silvestru, C.; Thorstenson, C.; et al. Tetra-(p-tolyl)antimony(III)-Containing Heteropolytungstates, [{(p-tolyl)Sb(III)}4(A-alpha-XW9O34)2](n-) (X = P, As, or Ge): Synthesis, structure, and study of antibacterial and antitumor activity. Inorg. Chem. 2020, 59, 2978–2987. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside—Preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef]
- Goschl, S.; Schreiber-Brynzak, E.; Pichler, V.; Cseh, K.; Heffeter, P.; Jungwirth, U.; Jakupec, M.A.; Berger, W.; Keppler, B.K. Comparative studies of oxaliplatin-based platinum(iv) complexes in different in vitro and in vivo tumor models. Met. Integr. Biometal Sci. 2017, 9, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Bey, E.A.; Bentle, M.S.; Reinicke, K.E.; Dong, Y.; Yang, C.R.; Girard, L.; Minna, J.D.; Bornmann, W.G.; Gao, J.; Boothman, D.A. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc. Natl. Acad. Sci. USA 2007, 104, 11832–11837. [Google Scholar] [CrossRef]
- Srijiwangsa, P.; Na Bangchang, K. Roles of NAD (P) H-Quinone Oxidoreductase 1 (NQO1) on cancer progression and chemoresistance. J. Clin. Exp. Oncol. 2017, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Pidugu, L.S.; Mbimba, J.C.; Ahmad, M.; Pozharski, E.; Sausville, E.A.; Emadi, A.; Toth, E.A. A direct interaction between NQO1 and a chemotherapeutic dimeric naphthoquinone. BMC Struct. Biol. 2016, 16, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The crystal structure of NAD(P)H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef] [PubMed]
- Bruker AXS. Bruker SAINT v.838B Copyright(C); Bruker AXS: Billerica, MA, USA, 2005–2019. [Google Scholar]
- Sheldrick, G.M. Sadabs; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Hubschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G. SHELXS v 2016/4. 2015. [Google Scholar]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Spek, A. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ru-O1 | Ru-O2 | Ru-N2 | C1-N1 | M-Arene | |
|---|---|---|---|---|---|
| 1(a) | 2.059(2) | 2.121(2) | 2.096(3) | 1.528(4) | 1.651 |
| 2 | 2.077(2) | 2.086(2) | 2.088(2) | 1.514(3) | 1.650 |
| 3 | 2.057(2) | 2.104(2) | 2.111(3) | 1.507(4) | 1.655 |
| 4 | 2.049(1) | 2.116(1) | 2.086(2) | 1.516(2) | 1.649 |
| 6 | 2.062(1) | 2.117(1) | 2.094(2) | 1.523(2) | 1.659 |
| Arene | Reactant [a.u.] | Transition State [a.u.] | Aqua-Complex [a.u.] |
|---|---|---|---|
| p-Cymene (1) | −1356.045552 | −1432.265348 | −1432.285092 |
| Benzene (2) | −1199.219445 | −1275.437208 | −1275.459690 |
| Toluene (3) | −1238.431142 | −1314.648960 | −1314.670467 |
| Indane (4) | −1315.656248 | −1391.875753 | −1391.894567 |
| Biphenyl (5) | −1429.689526 | −1505.909387 | −1505.919352 |
| HMB (6) | −1434.464410 | −1510.686025 | −1510.704607 |
| IC50 Values [µM] | ||||||
|---|---|---|---|---|---|---|
| A549 | SW480 | CH1/PA-1 | ||||
| Compound | 24 h | 96 h | 24 h | 96 h | 24 h | 96 h |
| 1 | 3.8 ± 2.6 | 1.2 ± 0.2 (a) | 0.46 ± 0.14 | 0.094 ± 0.031 (a) | 59 ± 18 | >50 (a) |
| 2 | >200 | 141 ± 38 | 31 ± 3 | 9.9 ± 2.5 | 170 ± 23 | 157 ± 8 |
| 3 | >200 | 72 ± 12 | 11 ± 2 | 2.8 ± 0.7 | 121 ± 18 | 138 ± 10 |
| 4 | 119 ± 21 | 9.1 ± 1.5 | 3.5 ± 1.6 | 0.52 ± 0.09 | 146 ± 18 | 88 ± 10 |
| 5 | 4.3 ± 1.6 | 1.2 ± 0.2 | 0.77 ± 0.24 | 0.072 ± 0.019 | 42 ± 4 | 30 ± 3 |
| 6 | 38 ± 21 | 3.4 ± 0.6 | 2.8 ± 1.6 | 0.27 ± 0.06 | 57 ± 16 | 35 ± 4 |
| L | >400 | 210 ± 32 (b) | >400 | 116 ± 37 (b) | >400 | 129 ± 29 (b) |
| [Ru(p-cym)Cl2]2 (c) | n.d. | >100 | n.d. | >100 | n.d. | >100 |
| [Ru(benzene)Cl2]2 (c) | n.d. | >100 | n.d. | >100 | n.d. | >100 |
| [Ru(toluene)Cl2]2 (c) | n.d. | >100 | n.d. | >100 | n.d. | >100 |
| [Ru(indane)Cl2]2 (c) | n.d. | 257 ± 66 | n.d. | 166 ± 35 | n.d. | 51 ± 13 |
| Cisplatin | n.d. | 3.8 ± 1.0 | n.d. | 2.3 ± 0.2 | n.d. | 0.073 ± 0.001 |
| Carboplatin (d) | n.d. | 38 ± 3 | n.d. | 42 ± 10 | n.d. | 0.79 ± 0.11 |
| Oxaliplatin (e) | n.d. | 0.98 ± 0.21 | n.d. | 0.29 ± 0.05 | n.d. | 0.18 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cseh, K.; Geisler, H.; Stanojkovska, K.; Westermayr, J.; Brunmayr, P.; Wenisch, D.; Gajic, N.; Hejl, M.; Schaier, M.; Koellensperger, G.; et al. Arene Variation of Highly Cytotoxic Tridentate Naphthoquinone-Based Ruthenium(II) Complexes and In-Depth In Vitro Studies. Pharmaceutics 2022, 14, 2466. https://doi.org/10.3390/pharmaceutics14112466
Cseh K, Geisler H, Stanojkovska K, Westermayr J, Brunmayr P, Wenisch D, Gajic N, Hejl M, Schaier M, Koellensperger G, et al. Arene Variation of Highly Cytotoxic Tridentate Naphthoquinone-Based Ruthenium(II) Complexes and In-Depth In Vitro Studies. Pharmaceutics. 2022; 14(11):2466. https://doi.org/10.3390/pharmaceutics14112466
Chicago/Turabian StyleCseh, Klaudia, Heiko Geisler, Kristina Stanojkovska, Julia Westermayr, Philipp Brunmayr, Dominik Wenisch, Natalie Gajic, Michaela Hejl, Martin Schaier, Gunda Koellensperger, and et al. 2022. "Arene Variation of Highly Cytotoxic Tridentate Naphthoquinone-Based Ruthenium(II) Complexes and In-Depth In Vitro Studies" Pharmaceutics 14, no. 11: 2466. https://doi.org/10.3390/pharmaceutics14112466