
Extracellular Vesicles as Delivery Vehicles for Therapeutic Nucleic Acids in Cancer Gene Therapy: Progress and Challenges

Abstract
:1. Introduction
2. EVs as Nucleic Acid Drug Delivery Vehicles
3. Improvements in EV Drug Delivery Systems
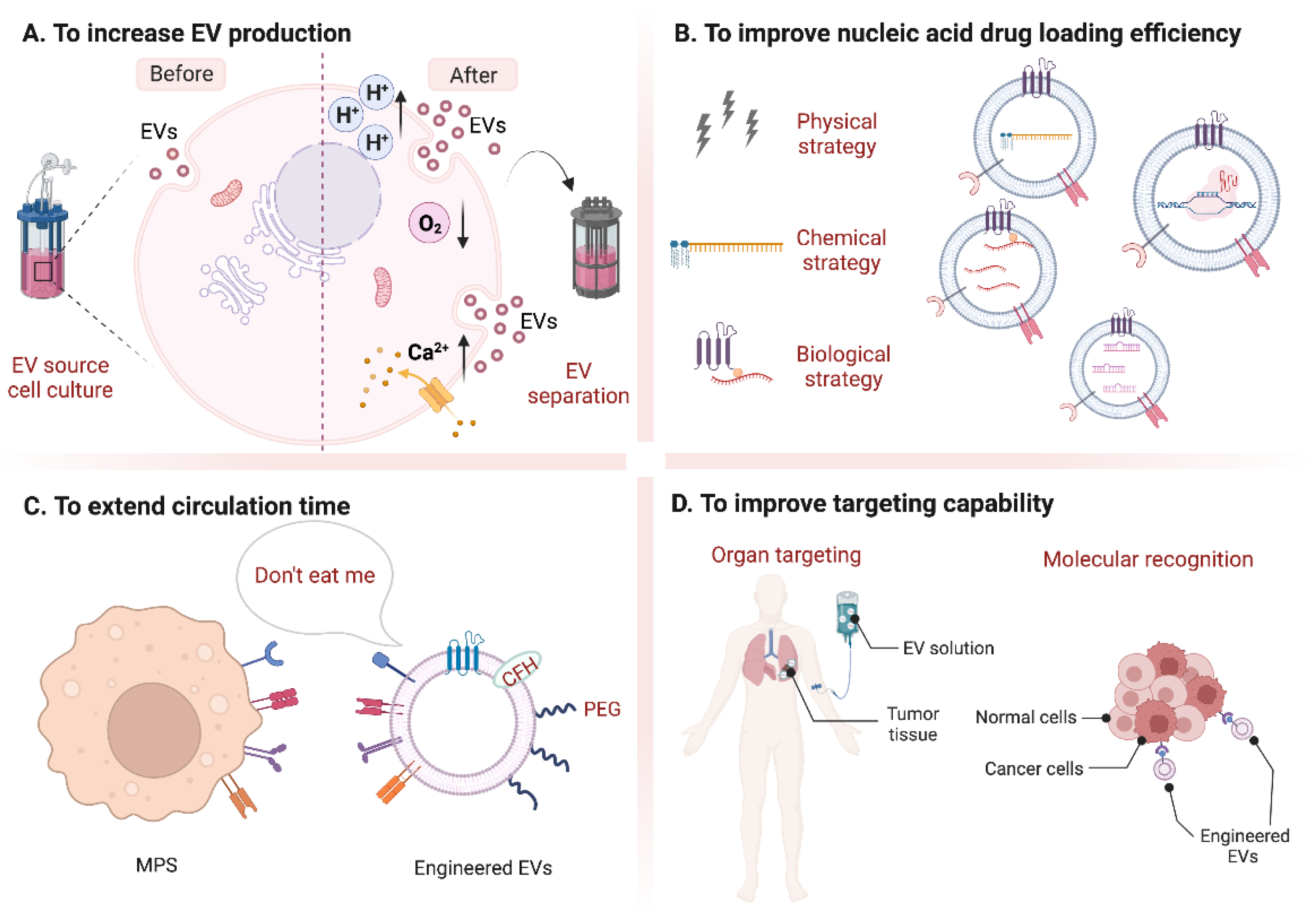
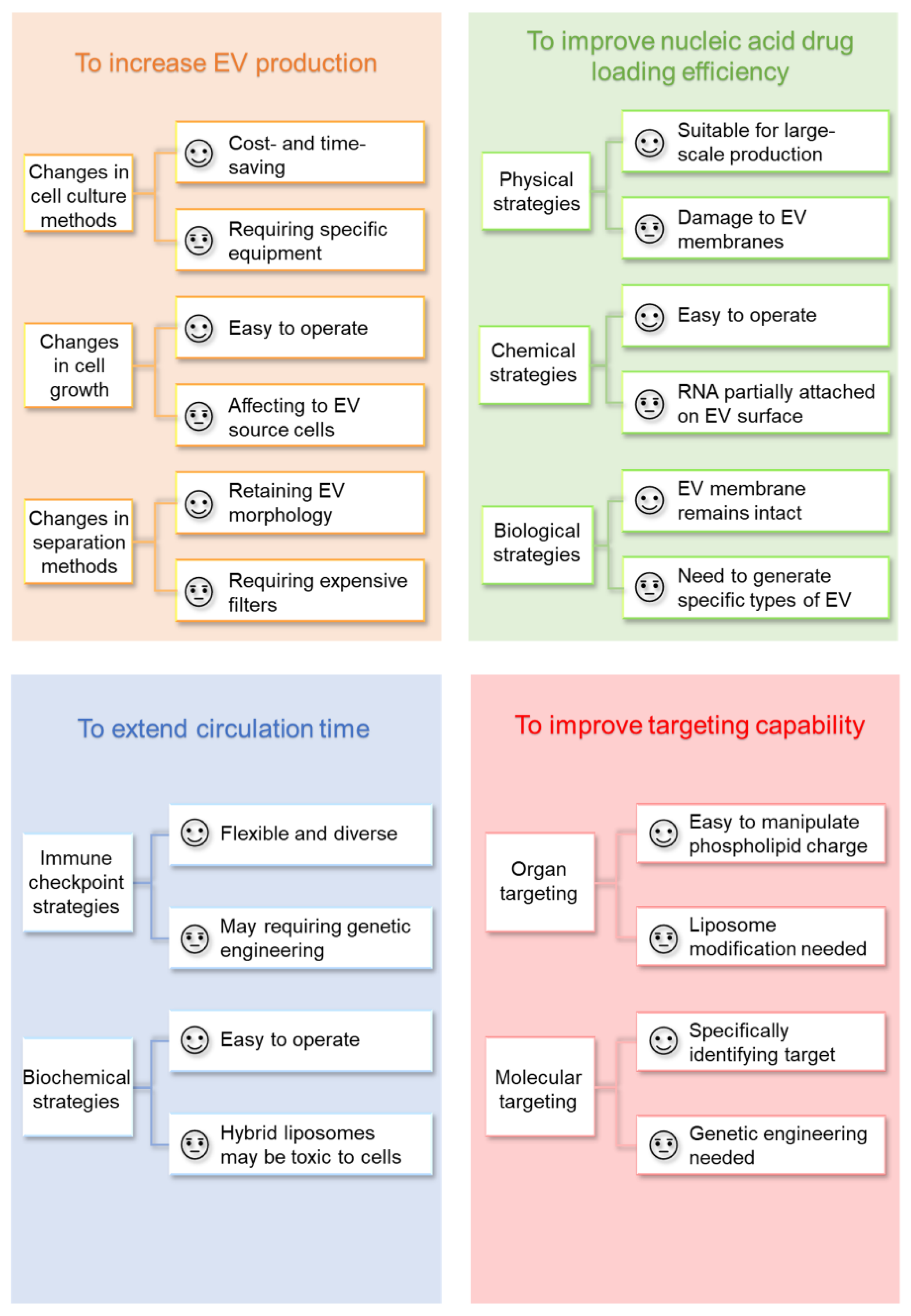
3.1. To Increase EV Production
3.1.1. Changing Cell Culture Methods
3.1.2. Changing EV Separation Methods
3.1.3. Inducing EV Secretion by Ca2+-Dependent Regulation
3.1.4. Inducing EV Secretion by Stressed Culture Conditions
3.2. To Improve Nucleic Acid Drug Loading Efficiency
3.2.1. Physical Strategies
3.2.2. Chemical Strategies
3.2.3. Biological Strategies
3.3. To Extend Circulation Time
3.3.1. Immune Checkpoint Strategies
3.3.2. Biochemical Strategies
3.4. To Improve Targeting Capability
3.4.1. Organ Targeting
3.4.2. Molecular Targeting
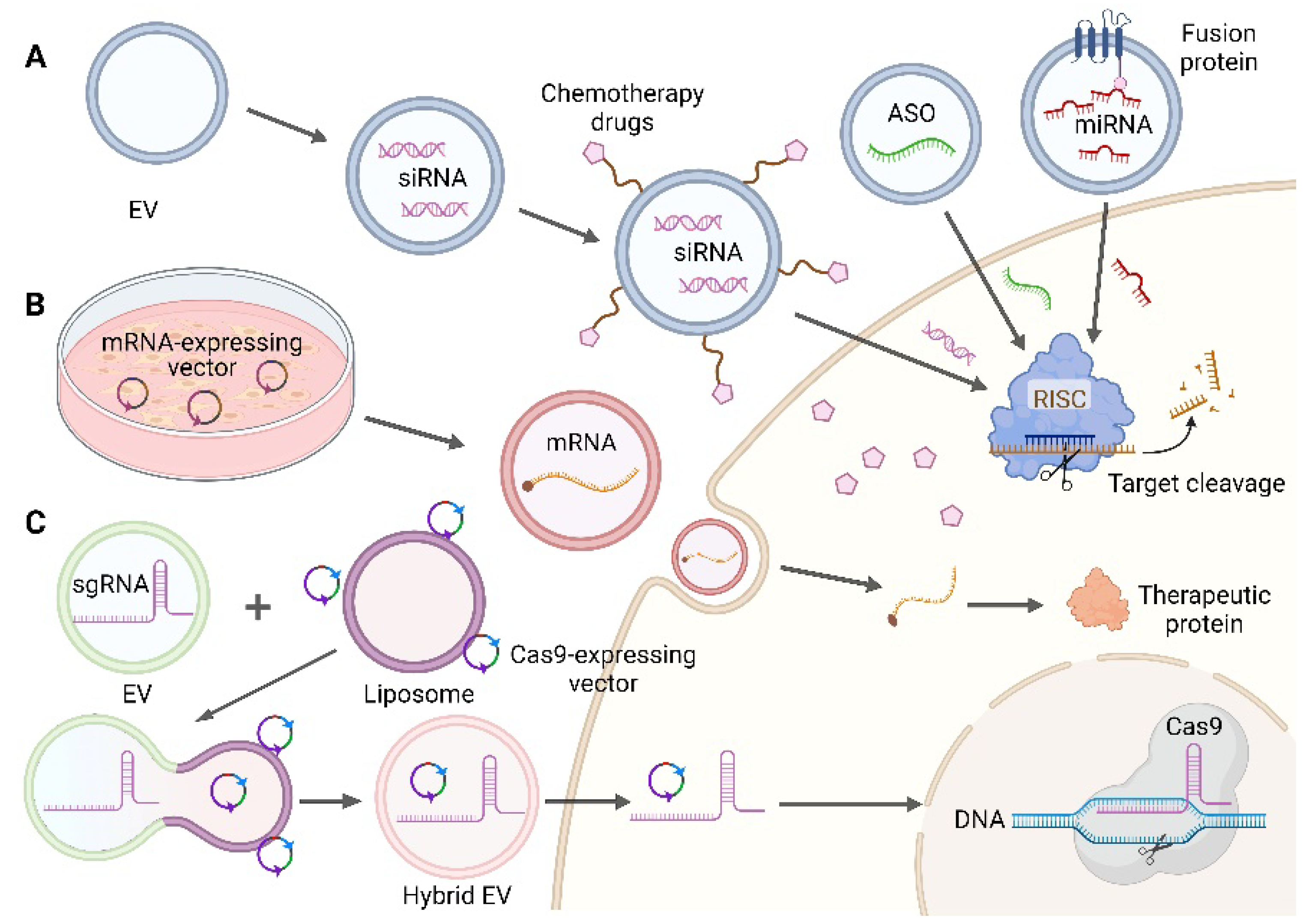
4. Nucleic Acid Drugs Delivered by EVs for Cancer Therapy
4.1. EVs for Delivery of Small Nucleic Acid Drugs
4.1.1. ASO
4.1.2. Single-Stranded miRNA
4.1.3. Double-Stranded siRNA
4.1.4. Combination Therapy
4.2. EVs for Delivery of mRNA
4.3. EVs for Delivery of CRISPR/Cas9 System
4.3.1. Plasmids
4.3.2. sgRNA and Cas9 mRNA
4.3.3. RNP
4.4. Clinical Status of EV-Based Nucleic Acid Drug Delivery Systems
5. Conclusions and Research Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EV | Extracellular vesicle |
| DDS | Drug delivery system |
| LNP | Lipid nanoparticle |
| MSC | Mesenchymal stromal cell |
| TFF | Tangential flow filtration |
| MVB | Multivesicular body |
| CNP | Cellular nanoporation |
| HSP | Heat shock protein |
| DC | Dendritic cell |
| EE | Encapsulation efficiency |
| SIRPα | Signal regulatory protein α |
| MPS | Mononuclear phagocyte system |
| Siglec-10 | Sialic acid-binding Ig-like lectin 10 |
| EPR | Enhanced permeability and retention |
| ASO | Antisense oligonucleotide |
| siRNA | Small interfering RNA |
| miRNA | Microrna |
| hsiRNA | Hydrophobically modified sirna |
| CRISPR/Cas | Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein |
| sgRNA | Single guide RNA |
| OMV | Outer membrane vesicle |
| PEG | Polyethylenglycol |
| PEI | Polyethylenimine |
| RVG | Rabies virus glycoprotein |
| TME | Tumor microenvironment |
| RNP | Ribonucleoprotein |
| dCas9 | Dead or deactivated Cas9 |
References
- Global Cancer Facts Reported from the WHO. Available online: https://www.who.int/en/news-room/fact-sheets/detail/cancer (accessed on 25 August 2022).
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. Opinion—How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Rajadas, J.; Seifalian, A.M. Exosomes as nano-theranostic delivery platforms for gene therapy. Adv. Drug Deliv. Rev. 2013, 65, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.W.; Yuan, H.B.; Sun, D.X. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Nordmeier, S.; Ke, W.; Afonin, K.A.; Portnoy, V. Exosome mediated delivery of functional nucleic acid nanoparticles (NANPs). Nanomed. Nanotechnol. Biol. Med. 2020, 30, 102285. [Google Scholar] [CrossRef]
- Xue, V.W.; Wong, S.C.C.; Song, G.; Cho, W.C.S. Promising RNA-based cancer gene therapy using extracellular vesicles for drug delivery. Expert Opin. Biol. Ther. 2020, 20, 767–777. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Dammes, N.; Peer, D. Paving the Road for RNA Therapeutics. Trends Pharm. Sci. 2020, 41, 755–775. [Google Scholar] [CrossRef]
- Ando, H.; Ishida, T. An RNAi therapeutic, DFP-10825, for intraperitoneal and intrapleural malignant cancers. Adv. Drug Deliv. Rev. 2020, 154–155, 27–36. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.Q.; Yin, H.F.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Tarasov, V.V.; Svistunov, A.A.; Chubarev, V.N.; Dostdar, S.A.; Sokolov, A.V.; Brzecka, A.; Sukocheva, O.; Neganova, M.E.; Klochkov, S.G.; Somasundaram, S.G.; et al. Extracellular vesicles in cancer nanomedicine. Semin. Cancer Biol. 2021, 69, 212–225. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, B.; Ocansey, D.K.W.; Xu, W.; Qian, H. Extracellular vesicles: A bright star of nanomedicine. Biomaterials 2021, 269, 120467. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Rezaie, J.; Ahmadi, M.; Ravanbakhsh, R.; Mojarad, B.; Mahbubfam, S.; Shaban, S.A.; Shadi, K.; Berenjabad, N.J.; Etemadi, T. Tumor-derived extracellular vesicles: The metastatic organotropism drivers. Life Sci. 2022, 289, 120216. [Google Scholar] [CrossRef]
- Baek, G.; Choi, H.; Kim, Y.; Lee, H.C.; Choi, C. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Therapeutics and as a Drug Delivery Platform. Stem Cells Transl. Med. 2019, 8, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Xing, H.N.; Xun, Z.; Yang, T.Z.; Zhao, X.Y.; Cai, C.F.; Wang, D.K.; Ding, P.T. Functionalized extracellular vesicles as advanced therapeutic nanodelivery systems. Eur. J. Pharm. Sci. 2018, 121, 34–46. [Google Scholar] [CrossRef]
- Guo, J.F.; Bourre, L.; Soden, D.M.; O’Sullivan, G.C.; O’Driscoll, C. Can non-viral technologies knockdown the barriers to siRNA delivery and achieve the next generation of cancer therapeutics? Biotechnol. Adv. 2011, 29, 402–417. [Google Scholar] [CrossRef] [Green Version]
- Walther, W.; Stein, U. Viral vectors for gene transfer—A review of their use in the treatment of human diseases. Drugs 2000, 60, 249–271. [Google Scholar] [CrossRef]
- Van den Boorn, J.G.; Schlee, M.; Coch, C.; Hartmann, G. SiRNA delivery with exosome nanoparticles. Nat. Biotechnol. 2011, 29, 325–326. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.X.; Li, M.; Lee, C.M.; Chakraborty, S.; Kim, H.W.; Bao, G.; Leong, K.W. CRISPR/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem. Rev. 2017, 117, 9874–9906. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, C.; Wang, C.; Jankovic, K.E.; Dong, Y. Lipids and Lipid Derivatives for RNA Delivery. Chem. Rev. 2021, 121, 12181–12277. [Google Scholar] [CrossRef]
- Murphy, D.E.; de Jong, O.G.; Evers, M.J.W.; Nurazizah, M.; Schiffelers, R.M.; Vader, P. Natural or Synthetic RNA Delivery: A Stoichiometric Comparison of Extracellular Vesicles and Synthetic Nanoparticles. Nano Lett. 2021, 21, 1888–1895. [Google Scholar] [CrossRef]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 148–156. [Google Scholar] [CrossRef]
- Yeo, R.W.; Lai, R.C.; Zhang, B.; Tan, S.S.; Yin, Y.; Teh, B.J.; Lim, S.K. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 336–341. [Google Scholar] [CrossRef]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Azad, A.; et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef] [Green Version]
- Pirisinu, M.; Tin Chanh, P.; Zhang, D.X.; Tran Nguyen, H.; Lap Thi, N.; Le, M.T.N. Extracellular vesicles as natural therapeutic agents and innate drug delivery systems for cancer treatment: Recent advances, current obstacles, and challenges for clinical translation. Semin. Cancer Biol. 2022, 80, 340–355. [Google Scholar] [CrossRef]
- Koh, E.; Lee, E.J.; Nam, G.H.; Hong, Y.; Cho, E.; Yang, Y.; Kim, I.S. Exosome-SIRP alpha, a CD47 blockade increases cancer cell phagocytosis. Biomaterials 2017, 121, 121–129. [Google Scholar] [CrossRef]
- Lara, P.; Palma-Florez, S.; Salas-Huenuleo, E.; Polakovicova, I.; Guerrero, S.; Lobos-Gonzalez, L.; Campos, A.; Munoz, L.; Jorquera-Cordero, C.; Varas-Godoy, M.; et al. Gold nanoparticle based double-labeling of melanoma extracellular vesicles to determine the specificity of uptake by cells and preferential accumulation in small metastatic lung tumors. J. Nanobiotechnol. 2020, 18, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.J.; Wu, J.Y.; Wang, J.M.; Hu, X.B.; Cai, J.X.; Xiang, D.X. Gemcitabine loaded autologous exosomes for effective and safe chemotherapy of pancreatic cancer. Acta Biomater. 2020, 101, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Yong, T.Y.; Zhang, X.Q.; Bie, N.N.; Zhang, H.B.; Zhang, X.T.; Li, F.Y.; Hakeem, A.; Hu, J.; Gan, L.; Santos, H.A.; et al. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat. Commun. 2019, 10, 3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, M.; van de Wakker, S.; de Groot, E.; de Jong, O.; Gitz-François, J.; Seinen, C.; Sluijter, J.; Schiffelers, R.; Vader, P. Functional siRNA Delivery by Extracellular Vesicle-Liposome Hybrid Nanoparticles. Adv. Healthc. Mater. 2022, 11, e2101202. [Google Scholar] [CrossRef]
- Massaro, C.; Sgueglia, G.; Frattolillo, V.; Baglio, S.; Altucci, L.; Dell’Aversana, C. Advances and Future Perspectives inExtracellular Vesicle-Based Nucleic Acid Delivery: Current Cancer Therapeutic Strategies. Pharmaceutics 2020, 12, 980. [Google Scholar] [CrossRef]
- Van den Boorn, J.G.; Dassler, J.; Coch, C.; Schlee, M.; Hartmann, G. Exosomes as nucleic acid nanocarriers. Adv. Drug Deliv. Rev. 2013, 65, 331–335. [Google Scholar] [CrossRef]
- Yang, B.; Chen, Y.; Shi, J. Exosome Biochemistry and Advanced Nanotechnology for Next-Generation Theranostic Platforms. Adv. Mater 2019, 31, e1802896. [Google Scholar] [CrossRef]
- Richter, M.; Vader, P.; Fuhrmann, G. Approaches to surface engineering of extracellular vesicles. Adv. Drug Deliv. Rev. 2021, 173, 416–426. [Google Scholar] [CrossRef]
- Parada, N.; Romero-Trujillo, A.; Georges, N.; Alcayaga-Miranda, F. Camouflage strategies for therapeutic exosomes evasion from phagocytosis. J. Adv. Res. 2021, 31, 61–74. [Google Scholar] [CrossRef]
- Jhan, Y.Y.; Prasca-Chamorro, D.; Zuniga, G.P.; Moore, D.M.; Kumar, S.A.; Gaharwar, A.K.; Bishop, C.J. Engineered extracellular vesicles with synthetic lipids via membrane fusion to establish efficient gene delivery. Int. J. Pharm. 2020, 573, 118802. [Google Scholar] [CrossRef]
- Xu, L.; Faruqu, F.N.; Lim, Y.M.; Lim, K.Y.; Liam-Or, R.; Walters, A.A.; Lavender, P.; Fear, D.; Wells, C.M.; Wang, J.T.-W.; et al. Exosome-mediated RNAi of PAK4 prolongs survival of pancreatic cancer mouse model after loco-regional treatment. Biomaterials 2021, 264, 120369. [Google Scholar] [CrossRef]
- Cao, J.; Wang, B.; Tang, T.; Lv, L.; Ding, Z.; Li, Z.; Hu, R.; Wei, Q.; Shen, A.; Fu, Y.; et al. Three-dimensional culture of MSCs produces exosomes with improved yield and enhanced therapeutic efficacy for cisplatin-induced acute kidney injury. Stem Cell Res. Ther. 2020, 11, 206. [Google Scholar] [CrossRef]
- Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z.; Clayton, A. Increased exosome production from tumour cell cultures using the Integra CELLine Culture System. J. Immunol. Methods 2008, 335, 98–105. [Google Scholar] [CrossRef]
- De Almeida Fuzeta, M.; Bernardes, N.; Oliveira, F.D.; Costa, A.C.; Fernandes-Platzgummer, A.; Farinha, J.P.; Rodrigues, C.A.V.; Jung, S.; Tseng, R.J.; Milligan, W.; et al. Scalable Production of Human Mesenchymal Stromal Cell-Derived Extracellular Vesicles Under Serum-/Xeno-Free Conditions in a Microcarrier-Based Bioreactor Culture System. Front Cell Dev. Biol. 2020, 8, 553444. [Google Scholar] [CrossRef]
- Watson, D.C.; Bayik, D.; Srivatsan, A.; Bergamaschi, C.; Valentin, A.; Niu, G.; Bear, J.; Monninger, M.; Sun, M.; Morales-Kastresana, A.; et al. Efficient production and enhanced tumor delivery of engineered extracellular vesicles. Biomaterials 2016, 105, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Phan, J.; Kumar, P.; Hao, D.; Gao, K.; Farmer, D.; Wang, A. Engineering mesenchymal stem cells to improve their exosome efficacy and yield for cell-free therapy. J. Extracell. Vesicles 2018, 7, 1522236. [Google Scholar] [CrossRef]
- Savina, A.; Furlan, M.; Vidal, M.; Colombo, M.I. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J. Biol. Chem. 2003, 278, 20083–20090. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Messenger, S.W.; Woo, S.S.; Sun, Z.; Martin, T.F.J. A Ca2+-stimulated exosome release pathway in cancer cells is regulated by Munc13-4. J. Cell Biol. 2018, 217, 2877–2890. [Google Scholar] [CrossRef] [Green Version]
- Ekstrom, E.J.; Bergenfelz, C.; von Bulow, V.; Serifler, F.; Carlemalm, E.; Jonsson, G.; Andersson, T.; Leandersson, K. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol. Cancer 2014, 13, 88. [Google Scholar] [CrossRef]
- Yang, Z.G.; Shi, J.F.; Xie, J.; Wang, Y.F.; Sun, J.Y.; Liu, T.Z.; Zhao, Y.R.; Zhao, X.T.; Wang, X.M.; Ma, Y.F.; et al. Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation. Nat. Biomed. Eng. 2020, 4, 69–83. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, X.; Yao, Q.; Liu, Y.; Zhang, H.; Dong, Z. HIF-1-mediated production of exosomes during hypoxia is protective in renal tubular cells. Am J. Physiol. Ren. Physiol. 2017, 313, F906–F913. [Google Scholar] [CrossRef] [Green Version]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef] [Green Version]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [Green Version]
- Campbell, C.R.; Berman, A.E.; Weintraub, N.L.; Tang, Y.L. Electrical stimulation to optimize cardioprotective exosomes from cardiac stem cells. Med. Hypotheses 2016, 88, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Kim, J.H. Platinum Nanoparticles Enhance Exosome Release in Human Lung Epithelial Adenocarcinoma Cancer Cells (A549): Oxidative Stress and the Ceramide Pathway are Key Players. Int. J. Nanomed. 2021, 16, 515–538. [Google Scholar] [CrossRef]
- Haraszti, R.A.; Miller, R.; Stoppato, M.; Sere, Y.Y.; Coles, A.; Didiot, M.C.; Wollacott, R.; Sapp, E.; Dubuke, M.L.; Li, X.; et al. Exosomes Produced from 3D Cultures of MSCs by Tangential Flow Filtration Show Higher Yield and Improved Activity. Mol. Ther. 2018, 26, 2838–2847. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Rhim, W.K.; Yoo, Y.I.; Kim, D.S.; Ko, K.W.; Heo, Y.; Park, C.G.; Han, D.K. Defined MSC exosome with high yield and purity to improve regenerative activity. J. Tissue Eng. 2021, 12, 20417314211008626. [Google Scholar] [CrossRef]
- Chen, J.; Li, P.; Zhang, T.; Xu, Z.; Huang, X.; Wang, R.; Du, L. Review on Strategies and Technologies for Exosome Isolation and Purification. Front. Bioeng. Biotechnol. 2021, 9, 811971. [Google Scholar] [CrossRef]
- Jeyaram, A.; Lamichhane, T.N.; Wang, S.; Zou, L.; Dahal, E.; Kronstadt, S.M.; Levy, D.; Parajuli, B.; Knudsen, D.R.; Chao, W.; et al. Enhanced Loading of Functional miRNA Cargo via pH Gradient Modification of Extracellular Vesicles. Mol. Ther. 2020, 28, 975–985. [Google Scholar] [CrossRef]
- Zhao, L.; Gu, C.; Gan, Y.; Shao, L.; Chen, H.; Zhu, H. Exosome-mediated siRNA delivery to suppress postoperative breast cancer metastasis. J. Control Release 2020, 318, 1–15. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, A.J.; Mager, I.; de Jong, O.G.; Varela, M.A.; Schiffelers, R.M.; El Andaloussi, S.; Wood, M.J.A.; Vader, P. Functional Delivery of Lipid-Conjugated siRNA by Extracellular Vesicles. Mol. Ther. 2017, 25, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraszti, R.A.; Miller, R.; Didiot, M.C.; Biscans, A.; Alterman, J.F.; Hassler, M.R.; Roux, L.; Echeverria, D.; Sapp, E.; DiFiglia, M.; et al. Optimized Cholesterol-siRNA Chemistry Improves Productive Loading onto Extracellular Vesicles. Mol. Ther. 2018, 26, 1973–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didiot, M.C.; Hall, L.M.; Coles, A.H.; Haraszti, R.A.; Godinho, B.M.; Chase, K.; Sapp, E.; Ly, S.; Alterman, J.F.; Hassler, M.R.; et al. Exosome-mediated Delivery of Hydrophobically Modified siRNA for Huntingtin mRNA Silencing. Mol. Ther. 2016, 24, 1836–1847. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Wu, J.H.; Gu, W.H.; Huang, Y.L.; Tong, Z.C.; Huang, L.J.; Tan, J.L. Exosome-Liposome Hybrid Nanoparticles Deliver CRISPR/Cas9 System in MSCs. Adv. Sci. 2018, 5, 1700611. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.L.; Zhou, X.Y.; Wei, M.Y.; Gao, X.T.; Zhao, L.B.; Shi, R.J.; Sun, W.Q.; Duan, Y.Y.; Yang, G.D.; Yuan, L.J. In Vitro and in Vivo RNA Inhibition by CD9-HuR Functionalized Exosomes Encapsulated with miRNA or CRISPR/dCas9. Nano Lett. 2019, 19, 19–28. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Wan, Z.; Wang, C.; Lu, F.; Wei, M.; Wang, D.; Hao, Q. Designer exosomes for targeted and efficient ferroptosis induction in cancer via chemo-photodynamic therapy. Theranostics 2021, 11, 8185–8196. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, X.; Tang, J.; Lv, Q.; Liu, J. Gene-engineered exosomes-thermosensitive liposomes hybrid nanovesicles by the blockade of CD47 signal for combined photothermal therapy and cancer immunotherapy. Biomaterials 2021, 275, 120964. [Google Scholar] [CrossRef]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Barkal, A.A.; Weiskopf, K.; Kao, K.S.; Gordon, S.R.; Rosental, B.; Yiu, Y.Y.; George, B.M.; Markovic, M.; Ring, N.G.; Tsai, J.M.; et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 2018, 19, 76–84. [Google Scholar] [CrossRef]
- Gordon, S.R.; Aute, R.L.M.; Dulken, B.W.; Hutter, G.; George, B.M.; Ccracken, M.N.M.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [Green Version]
- Daassi, D.; Mahoney, K.M.; Freeman, G.J. The importance of exosomal PDL1 in tumour immune evasion. Nat. Rev. Immunol. 2020, 20, 209–215. [Google Scholar] [CrossRef]
- Piffoux, M.; Silva, A.K.A.; Wilhelm, C.; Gazeau, F.; Tareste, D. Modification of Extracellular Vesicles by Fusion with Liposomes for the Design of Personalized Biogenic Drug Delivery Systems. ACS Nano 2018, 12, 6830–6842. [Google Scholar] [CrossRef]
- Bushey, R.T.; Gottlin, E.B.; Campa, M.J.; Patz, E.F., Jr. Complement factor H protects tumor cell-derived exosomes from complement-dependent lysis and phagocytosis. PLoS ONE 2021, 16, e0252577. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Gomari, H.; Moghadam, M.F.; Soleimani, M. Targeted cancer therapy using engineered exosome as a natura drug delivery vehicle. Oncotargets Ther. 2018, 11, 5753–5762. [Google Scholar] [CrossRef] [Green Version]
- Limoni, S.K.; Moghadam, M.F.; Moazzeni, S.M.; Gomari, H.; Salimi, F. Engineered Exosomes for Targeted Transfer of siRNA to HER2 Positive Breast Cancer Cells. Appl. Biochem. Biotechnol. 2019, 187, 352–364. [Google Scholar] [CrossRef]
- Liang, G.F.; Zhu, Y.L.; Ali, D.J.; Tian, T.; Xu, H.T.; Si, K.; Sun, B.; Chen, B.A.; Xiao, Z.D. Engineered exosomes for targeted co-delivery of miR-21 inhibitor and chemotherapeutics to reverse drug resistance in colon cancer. J. Nanobiotechnol. 2020, 18, 10. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Aleza, C.G.; Roffler, S.R.; van Solinge, W.W.; Vader, P.; Schiffelers, R.M. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J. Extracell. Vesicles 2016, 5, 31053. [Google Scholar] [CrossRef]
- Li, L.; He, D.; Guo, Q.; Zhang, Z.; Ru, D.; Wang, L.; Gong, K.; Liu, F.; Duan, Y.; Li, H. Exosome-liposome hybrid nanoparticle codelivery of TP and miR497 conspicuously overcomes chemoresistant ovarian cancer. J. Nanobiotechnol. 2022, 20, 50. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Gong, C.N.; Wang, Z.; Xia, Q.M.; Gu, F.F.; Hu, C.L.; Zhang, L.J.; Guo, H.L.; Gao, S. A33 antibody-functionalized exosomes for targeted delivery of doxorubicin against colorectal cancer. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1973–1985. [Google Scholar] [CrossRef]
- Liu, J.; Ye, Z.L.; Xiang, M.X.; Chang, B.C.; Cui, J.Y.; Ji, T.T.; Zhao, L.; Li, Q.L.; Deng, Y.; Xu, L.M.; et al. Functional extracellular vesicles engineered with lipid-grafted hyaluronic acid effectively reverse cancer drug resistance. Biomaterials 2019, 223, 119475. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Emam, S.E.; Ando, H.; Abu Lila, A.S.; Shimizu, T.; Ukawa, M.; Okuhira, K.; Ishima, Y.; Mahdy, M.A.; Ghazy, F.-e.S.; Ishida, T. A Novel Strategy to Increase the Yield of Exosomes (Extracellular Vesicles) for an Expansion of Basic Research. Biol. Pharm. Bull. 2018, 41, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Bellavia, D.; Raimondo, S.; Calabrese, G.; Forte, S.; Cristaldi, M.; Patinella, A.; Memeo, L.; Manno, M.; Raccosta, S.; Diana, P.; et al. Interleukin 3-receptor targeted exosomes inhibit in vitro and in vivo Chronic Myelogenous Leukemia cell growth. Theranostics 2017, 7, 1333–1345. [Google Scholar] [CrossRef]
- Lang, F.; Hossain, A.; Gumin, J.; Momin, E.; Shimizu, Y.; Ledbetter, D.; Shahar, T.; Yamashita, S.; Parker Kerrigan, B.; Fueyo, J.; et al. Mesenchymal stem cells as natural biofactories for exosomes carrying miR-124a in the treatment of gliomas. Neuro-Oncology 2018, 20, 380–390. [Google Scholar] [CrossRef] [Green Version]
- El-Andaloussi, S.; Lee, Y.; Lakhal-Littleton, S.; Li, J.; Seow, Y.; Gardiner, C.; Alvarez-Erviti, L.; Sargent, I.L.; Wood, M.J. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat. Protoc. 2012, 7, 2112–2126. [Google Scholar] [CrossRef]
- Stapulionis, R. Electric pulse-induced precipitation of biological macromolecules in electroporation. Bioelectrochem. Bioenerg. 1999, 48, 249–254. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.A.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control Release 2013, 172, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.K.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogene Knockdown via Active Loading of Small RNAs into Extracellular Vesicles by Sonication. Cell. Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Kandimalla, R.; Wallen, M.; Tyagi, N.; Wilcher, S.; Yan, J.; Schultz, D.J.; Spencer, W.; et al. Exosome-mediated delivery of RNA and DNA for gene therapy. Cancer Lett. 2021, 505, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Belhadj, Z.; He, B.; Deng, H.; Song, S.; Zhang, H.; Wang, X.; Dai, W.; Zhang, Q. A combined “eat me/don’t eat me” strategy based on extracellular vesicles for anticancer nanomedicine. J. Extracell. Vesicles 2020, 9, 1806444. [Google Scholar] [CrossRef]
- Matozaki, T.; Murata, Y.; Okazawa, H.; Ohnishi, H. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009, 19, 72–80. [Google Scholar] [CrossRef]
- Oldenborg, P.A. CD47: A Cell Surface Glycoprotein Which Regulates Multiple Functions of Hematopoietic Cells in Health and Disease. ISRN Hematol. 2013, 2013, 614619. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.W.; Li, F.X.Z.; Liu, Y.W.; Rao, S.S.; Yin, H.; Huang, J.; Chen, C.Y.; Hu, Y.; Zhang, Y.; Tan, Y.J.; et al. Aptamer-functionalized exosomes from bone marrow stromal cells target bone to promote bone regeneration. Nanoscale 2019, 11, 20884–20892. [Google Scholar] [CrossRef]
- Sun, D.; Zhuang, X.; Zhang, S.; Deng, Z.-B.; Grizzle, W.; Miller, D.; Zhang, H.-G. Exosomes are endogenous nanoparticles that can deliver biological information between cells. Adv. Drug Deliv. Rev. 2013, 65, 342–347. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef]
- Saias, L.; Gomes, A.; Cazales, M.; Ducommun, B.; Lobjois, V. Cell-Cell Adhesion and Cytoskeleton Tension Oppose Each Other in Regulating Tumor Cell Aggregation. Cancer Res. 2015, 75, 2426–2433. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Yang, Y.; Oh, S.J.; Hong, Y.; Seo, M.; Jang, M. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J. Control Release 2017, 266, 8–16. [Google Scholar] [CrossRef]
- Kim, G.; Kim, M.; Lee, Y.; Byun, J.W.; Hwang, D.W.; Lee, M. Systemic delivery of microRNA-21 antisense oligonucleotides to the brain using T7-peptide decorated exosomes. J. Control Release 2020, 317, 273–281. [Google Scholar] [CrossRef]
- Jia, G.; Han, Y.; An, Y.L.; Ding, Y.A.; He, C.; Wang, X.H.; Tang, Q.S. NRP-1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials 2018, 178, 302–316. [Google Scholar] [CrossRef]
- Smyth, T.; Petrova, K.; Payton, N.M.; Persaud, I.; Redzic, J.S.; Gruner, M.W.; Smith-Jones, P.; Anchordoquy, T.J. Surface Functionalization of Exosomes Using Click Chemistry. Bioconjug. Chem. 2014, 25, 1777–1784. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Gebeyehu, A.; Kommineni, N.; Meckes, D.G., Jr.; Sachdeva, M.S. Role of Exosomes for Delivery of Chemotherapeutic Drugs. Crit. Rev. Ther. Drug Carr. Syst. 2021, 38, 53–97. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, Y.; Chen, X.; Ning, T.; Chen, H.; Guo, Q.; Zhang, Y.; Liu, P.; Zhang, Y.; Li, C.; et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials 2021, 268, 120546. [Google Scholar] [CrossRef]
- Naseri, Z.; Oskuee, R.K.; Forouzandeh-moghadam, M.; Jaafari, M.R. Delivery of LNA-antimiR-142-3p by Mesenchymal Stem Cells-Derived Exosomes to Breast Cancer Stem Cells Reduces Tumorigenicity. Stem Cell Rev. Rep. 2020, 16, 541–556. [Google Scholar] [CrossRef]
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Esau, C.C.; Monia, B.P. Therapeutic potential for microRNAs. Adv. Drug Deliv. Rev. 2007, 59, 101–114. [Google Scholar] [CrossRef]
- Peng, B.; Nguyen, T.M.; Jayasinghe, M.K.; Gao, C.; Pham, T.T.; Vu, L.T.; Yeo, E.Y.M.; Yap, G.; Wang, L.; Goh, B.C.; et al. Robust delivery of RIG-I agonists using extracellular vesicles for anti-cancer immunotherapy. J. Extracell. Vesicles 2022, 11, e12187. [Google Scholar] [CrossRef]
- Kamerkar, S.; Leng, C.; Burenkova, O.; Jang, S.C.; McCoy, C.; Zhang, K.; Dooley, K.; Kasera, S.; Zi, T.; Siso, S.; et al. Exosome-mediated genetic reprogramming of tumor-associated macrophages by exoASO-STAT6 leads to potent monotherapy antitumor activity. Sci. Adv. 2022, 8, eabj7002. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Wang, H.; Zhang, X.; Sun, X.; Zhang, J.; Ma, J. Exosomal miR-200c suppresses chemoresistance of docetaxel in tongue squamous cell carcinoma by suppressing TUBB3 and PPP2R1B. Aging 2020, 12, 6756–6773. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sawada, K.; Miyamoto, M.; Shimizu, A.; Yamamoto, M.; Kinose, Y.; Nakamura, K.; Kawano, M.; Kodama, M.; Hashimoto, K.; et al. Exploring the potential of engineered exosomes as delivery systems for tumor-suppressor microRNA replacement therapy in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 527, 153–161. [Google Scholar] [CrossRef]
- Liu, X.X.; Wang, W.; Samarsky, D.; Liu, L.; Xu, Q.; Zhang, W.Q.; Zhu, G.Z.; Wu, P.; Zuo, X.L.; Deng, H.L.; et al. Tumor-targeted in vivo gene silencing via systemic delivery of cRGD-conjugated siRNA. Nucleic Acids Res. 2014, 42, 11805–11817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Liu, B.; Fan, J.; Xue, C.; Lu, Y.; Li, C.; Cui, D. Engineered mesenchymal stem cell-derived exosomes with high CXCR4 levels for targeted siRNA gene therapy against cancer. Nanoscale 2022, 14, 4098–4113. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Chen, Y.; Li, B.; Sugimoto, H.; Yang, S.; Yang, C.; LeBleu, V.S.; McAndrews, K.M.; Kalluri, R. Therapeutic targeting of STAT3 with small interference RNAs and antisense oligonucleotides embedded exosomes in liver fibrosis. FASEB J. 2021, 35, e21557. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.A.; Tian, J.; Wang, Z.; Gao, Y.; Wu, X.; Ding, X.Y.; Qiang, L.; Li, G.R.; Han, Z.M.; Yuan, Y.F.; et al. Functional exosome-mediated co-delivery of doxorubicin and hydrophobically modified microRNA 159 for triple-negative breast cancer therapy. J. Nanobiotechnol. 2019, 17, 93. [Google Scholar] [CrossRef]
- Jhan, Y.Y.; Palou Zuniga, G.; Singh, K.A.; Gaharwar, A.K.; Alge, D.L.; Bishop, C.J. Polymer-Coated Extracellular Vesicles for Selective Codelivery of Chemotherapeutics and siRNA to Cancer Cells. ACS Appl. Bio Mater. 2021, 4, 1294–1306. [Google Scholar] [CrossRef]
- Ahmadi, M.; Hassanpour, M.; Rezaie, J. Engineered extracellular vesicles: A novel platform for cancer combination therapy and cancer immunotherapy. Life Sci. 2022, 308, 120935. [Google Scholar] [CrossRef]
- Munoz, J.L.; Bliss, S.A.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Delivery of Functional Anti-miR-9 by Mesenchymal Stem Cell-derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol. Ther. Nucleic Acids 2013, 2, e126. [Google Scholar] [CrossRef]
- Huang, L.; Rong, Y.; Tang, X.; Yi, K.; Qi, P.; Hou, J.; Liu, W.; He, Y.; Gao, X.; Yuan, C.; et al. Engineered exosomes as an in situ DC-primed vaccine to boost antitumor immunity in breast cancer. Mol. Cancer 2022, 21, 45. [Google Scholar] [CrossRef]
- Paunovska, K.; Loughrey, D.; Dahlman, J. Drug delivery systems for RNA therapeutics. Nat. Reviews. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef]
- Youn, H.; Chune, J.-K. Modified mRNA as an alternative to plasmid DNA (pDNA) for transcript replacement and vaccination therapy. Expert Opin. Biol. Ther. 2015, 15, 1337–1348. [Google Scholar] [CrossRef] [Green Version]
- Altanerova, U.; Benejova, K.; Altanerova, V.; Tyciakova, S.; Rychly, B.; Szomolanyi, P.; Ciampor, F.; Cihova, M.; Repiska, V.; Ondicova, K.; et al. Dental pulp mesenchymal stem/stromal cells labeled with iron sucrose release exosomes and cells applied intra-nasally migrate to intracerebral glioblastoma. Neoplasma 2016, 63, 925–933. [Google Scholar] [CrossRef]
- Altanerova, U.; Jakubechova, J.; Benejova, K.; Priscakova, P.; Repiska, V.; Babelova, A.; Smolkova, B.; Altaner, C. Intracellular prodrug gene therapy for cancer mediated by tumor cell suicide gene exosomes. Int. J. Cancer 2021, 148, 128–139. [Google Scholar] [CrossRef]
- Wang, J.H.; Forterre, A.V.; Zhao, J.; Frimannsson, D.O.; Delcayre, A.; Antes, T.J.; Efron, B.; Jeffrey, S.S.; Pegram, M.D.; Matin, A.C. Anti-HER2 scFv-Directed Extracellular Vesicle-Mediated mRNA-Based Gene Delivery Inhibits Growth of HER2-Positive Human Breast Tumor Xenografts by Prodrug Activation. Mol. Cancer Ther. 2018, 17, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Forterre, A.V.; Wang, J.H.; Delcayre, A.; Kim, K.; Green, C.; Pegram, M.D.; Jeffrey, S.S.; Matin, A.C. Extracellular Vesicle-Mediated In Vitro Transcribed mRNA Delivery for Treatment of HER2+ Breast Cancer Xenografts in Mice by Prodrug CB1954 without General Toxicity. Mol. Cancer Ther. 2020, 19, 858–867. [Google Scholar] [CrossRef]
- Shapiro, L.; Losick, R. Delivering the message: How a novel technology enabled the rapid development of effective vaccines. Cell 2021, 184, 5271–5274. [Google Scholar] [CrossRef]
- Maugeri, M.; Nawaz, M.; Papadimitriou, A.; Angerfors, A.; Camponeschi, A.; Na, M.; Holtta, M.; Skantze, P.; Johansson, S.; Sundqvist, M.; et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 2019, 10, 4333. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ma, X.; Yue, Y.; Zhang, K.; Cheng, K.; Feng, Q.; Ma, N.; Liang, J.; Zhang, T.; Zhang, L.; et al. Rapid Surface Display of mRNA Antigens by Bacteria-Derived Outer Membrane Vesicles for a Personalized Tumor Vaccine. Adv. Mater. 2022, 34, e2109984. [Google Scholar] [CrossRef]
- Oppel, F.; Schuermann, M.; Goon, P.; Albers, A.E.; Sudhoff, H. Specific Targeting of Oncogenes Using CRISPR Technology. Cancer Res. 2018, 78, 5506–5512. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Huang, H.; Liu, H.; Xi, J.; Ning, J.; Zeng, W.; Shen, C.; Zhang, T.; Yu, G.; Xu, Q.; et al. Friend or Foe? Evidence Indicates Endogenous Exosomes Can Deliver Functional gRNA and Cas9 Protein. Small 2019, 15, 1902686. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, Z.; Zhao, L.; Qin, Y.; Cai, H.; Geng, Z.; Zhu, X.; Zhang, W.; Zhang, Y.; Tan, J.; et al. Tropism-facilitated delivery of CRISPR/Cas9 system with chimeric antigen receptor-extracellular vesicles against B-cell malignancies. J. Control Release 2020, 326, 455–467. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef]
- Han, Z.; Zhou, D.; Wang, J.; Jiang, B.; Liu, X. Reflections on drug resistance to KRAS(G12C) inhibitors and gene silencing/editing tools for targeting mutant KRAS in cancer treatment. Biochim. Biophys. Acta—Rev. Cancer 2022, 1877, 188677. [Google Scholar] [CrossRef]
- Murty, T.; Mackall, C.L. Gene editing to enhance the efficacy of cancer cell therapies. Mol. Ther. 2021, 29, 3153–3162. [Google Scholar] [CrossRef]
- Schulz-Siegmund, M.; Aigner, A. Nucleic acid delivery with extracellular vesicles. Adv. Drug Deliv. Rev. 2021, 173, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Gee, P.; Lung, M.S.Y.; Okuzaki, Y.; Sasakawa, N.; Iguchi, T.; Makita, Y.; Hozumi, H.; Miura, Y.; Yang, L.F.; Iwasaki, M.; et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat. Commun. 2020, 11, 1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Lyu, P.; Yoo, K.; Yadav, M.K.; Singh, R.; Atala, A.; Lu, B. Engineered extracellular vesicles as versatile ribonucleoprotein delivery vehicles for efficient and safe CRISPR genome editing. J. Extracell. Vesicles 2021, 10, e12076. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Tan, J.; Wu, C.; Zhang, J.; Liu, T.; Fan, C.; Li, J.; Zhang, Y. Extracellular vesicles engineered with valency-controlled DNA nanostructures deliver CRISPR/Cas9 system for gene therapy. Nucleic Acids Res. 2020, 48, 8870–8882. [Google Scholar] [CrossRef]
- Jiang, X.C.; Zhang, T.; Gao, J.Q. The in vivo fate and targeting engineering of crossover vesicle-based gene delivery system. Adv. Drug Deliv. Rev. 2022, 187, 114324. [Google Scholar] [CrossRef]
- Gunassekaran, G.R.; Vadevoo, S.M.P.; Baek, M.-C.; Lee, B. M1 macrophage exosomes engineered to foster M1 polarization and target the IL-4 receptor inhibit tumor growth by reprogramming tumor-associated macrophages into M1-like macrophages. Biomaterials 2021, 278, 121137. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Xiao, F.; Chronopoulos, A.; LeBleu, V.S.; Kugeratski, F.G.; Kalluri, R. Exosome-mediated delivery of CRISPR/Cas9 for targeting of oncogenic Kras(G12D) in pancreatic cancer. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef]
- Jeong, Y.; Yu, Y.O.J.; You, A.E.Y.; Rhee, W.O.; Kim, E.O.A. Exosome-mediated microRNA-497 delivery for anti-cancer therapy in a microfluidic 3D lung cancer model. Lab Chip 2020, 20, 548–557. [Google Scholar] [CrossRef]
- Nie, H.F.; Xie, X.D.; Zhang, D.D.; Zhou, Y.; Li, B.F.; Li, F.Q.; Li, F.Y.; Cheng, Y.L.; Mei, H.; Meng, H.; et al. Use of lung-specific exosomes for miRNA-126 delivery in non-small cell lung cancer. Nanoscale 2020, 12, 877–887. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Codiak BioSciences, Inc. Available online: https://www.codiakbio.com/ (accessed on 25 August 2022).
- Evox Therapeutics Co., Ltd. Available online: https://www.evoxtherapeutics.com/ (accessed on 25 August 2022).
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Echo Biotech Co., Ltd. Available online: http://www.echobiotech.com/ (accessed on 25 August 2022).
- VesiCURE Co., Ltd. Available online: https://vesicure.com/ (accessed on 25 August 2022).
- Liu, C.; He, D.; Cen, H.; Chen, H.; Li, L.; Nie, G.; Zhong, Z.; He, Q.; Yang, X.; Guo, S.; et al. Nucleic acid functionalized extracellular vesicles as promising therapeutic systems for nanomedicine. Extracell. Vesicles Circ. Nucleic Acids 2022, 3, 14–30. [Google Scholar] [CrossRef]
- Kimiz-Gebologlu, I.; Oncel, S.S. Exosomes: Large-scale production, isolation, drug loading efficiency, and biodistribution and uptake. J. Control Release 2022, 347, 533–543. [Google Scholar] [CrossRef]
- Butreddy, A.; Kommineni, N.; Dudhipala, N. Exosomes as Naturally Occurring Vehicles for Delivery of Biopharmaceuticals: Insights from Drug Delivery to Clinical Perspectives. Nanomaterials 2021, 11, 1481. [Google Scholar] [CrossRef]
- Zhang, Y.; Bi, J.; Huang, J.; Tang, Y.; Du, S.; Li, P. Exosome: A Review of Its Classification, Isolation Techniques, Storage, Diagnostic and Targeted Therapy Applications. Int. J. Nanomed. 2020, 15, 6917–6934. [Google Scholar] [CrossRef]
- Lazaro-Ibanez, E.; Faruqu, F.N.; Saleh, A.F.; Silva, A.M.; Tzu-Wen Wang, J.; Rak, J.; Al-Jamal, K.T.; Dekker, N. Selection of Fluorescent, Bioluminescent, and Radioactive Tracers to Accurately Reflect Extracellular Vesicle Biodistribution in Vivo. ACS Nano 2021, 15, 3212–3227. [Google Scholar] [CrossRef]
- Wang, J.; Chen, D.; Ho, E.A. Challenges in the development and establishment of exosome-based drug delivery systems. J. Control Release 2021, 329, 894–906. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Purposes | Strategies | References |
|---|---|---|
| To increase EV production | Changes in cell culture methods (specially made culture flasks or 3D culture) | [42,43,44,45,46,47] |
| Ca2+-dependent regulation induction | [48,49,50,51] | |
| Stressed culture conditions stimulation (hypoxia, low pH, electrical stimulation, liposome stimulation, and drug stimulation) | [52,53,54,55,56,57] | |
| Changes in EV separation methods (tangential flow filtration) | [58,59,60] | |
| To improve nucleic acid drug loading efficiency | Physical strategies (1. cellular nanoporation biochips; 2. pH gradient; 3. extrusion) | [38,52,61,62] |
| Chemical strategies (hydrophobic modification of nucleic acids) | [63,64] | |
| Biological strategies (fusion with liposomes and RNA fusion proteins) | [65,66] | |
| To extend circulation time | Immune checkpoint strategies (CD47, CD24, MHC, PD-1/PD-L1) | [31,67,68,69,70,71,72,73] |
| Biochemical strategies (PEGylation and complement factor H) | [74,75] | |
| To improve targeting capability | Organ targeting | [76,77] |
| Molecular targeting (EGFR, A33, CD44, HER2, et al.) | [78,79,80,81,82,83,84] |
| Loading Strategies | Loaded Drugs | Notable Details | References |
|---|---|---|---|
| Electroporation | ASO | Target genes can be inhibited by up to 95% at the mRNA level. | [29] |
| siRNA | The drug loading rate is close to 60%. | [41] | |
| ASO | Upregulation of tumor suppressor genes and tumor suppressors. | [42] | |
| CRISPR/Cas9 | Electroporation of Cas9-/sgRNA-expressing plasmids into EVs. | [61,62,63,64] | |
| siRNA | EV transfects siRNA better than commercial transfection reagents LipofectamineTM and polyethyleneimine (PEI). | [94] | |
| siRNA | EVs are hybridized with liposomes through membrane extrusion technology. | [103] | |
| Incubation | siRNA | EVs were briefly incubated with siRNA in the presence of PEI. | [94] |
| ASO | ASO is attached to the membrane of EVs. | [114] | |
| miRNA and DOX | EVs were first mixed with DOX and then incubated with miRNA with shaking. | [122] | |
| Pre-transfected source cells | mRNA | Introducing plasmids into cells to obtain mRNA-carrying EVs. | [131,132] |
| Repeated freeze–thaw cycles | CRISPR/Cas9 | Cas9 protein was incubated with sgRNA to form RNP | [145] |
| Using transfection reagents | siRNA and miRNA | Using Exo-FectTM Exosome Transfection Kit | [147] |
| CRISPR/Cas9 | Transfection of encoding plasmids using the Exo-FectTM Exosome Transfection Kit. | [148] | |
| miRNA | Using LipofectamineTM RNAiMAX Reagent Kit | [149] | |
| miRNA | Using ExoFectin® Kit | [150] |
| Country | Institution | Research Content | Payload | Clinical Trial Number |
|---|---|---|---|---|
| United States | Codiak BioSciences | exoASOTM-STAT6 | ASO | NCT05375604 (Phase I) |
| United States | M.D. Anderson Cancer Center | iExosomes | KRASG12D siRNA | NCT03608631 (Phase I) |
| United States | Vesigen Therapeutics | ARMMs technology | RNA/Gene editing | / |
| United States | Carmine Therapeutics | REGENT® platform | RNA/Gene editing | / |
| England | EVOX Therapeutics | DeliverEXTM platform | mRNA/Gene editing | / |
| China | Echo Biotech | Echosome® platform | / | / |
| China | VesiCURE | modEXOTM | / | / |
| Korea | EXOSOME plus | ExoTheraTM | / | / |
| Switzerland | Anjarium Biosciences | Hybridosome® delivery technology | DNA | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, R.; Wang, C.; Zhu, L.; Yang, Y. Extracellular Vesicles as Delivery Vehicles for Therapeutic Nucleic Acids in Cancer Gene Therapy: Progress and Challenges. Pharmaceutics 2022, 14, 2236. https://doi.org/10.3390/pharmaceutics14102236
Du R, Wang C, Zhu L, Yang Y. Extracellular Vesicles as Delivery Vehicles for Therapeutic Nucleic Acids in Cancer Gene Therapy: Progress and Challenges. Pharmaceutics. 2022; 14(10):2236. https://doi.org/10.3390/pharmaceutics14102236
Chicago/Turabian StyleDu, Rong, Chen Wang, Ling Zhu, and Yanlian Yang. 2022. "Extracellular Vesicles as Delivery Vehicles for Therapeutic Nucleic Acids in Cancer Gene Therapy: Progress and Challenges" Pharmaceutics 14, no. 10: 2236. https://doi.org/10.3390/pharmaceutics14102236