

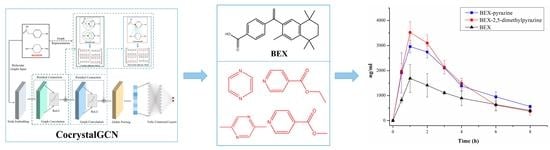
Cocrystal Prediction of Bexarotene by Graph Convolution Network and Bioavailability Improvement

Abstract
:
1. Introduction
2. Materials and Methods
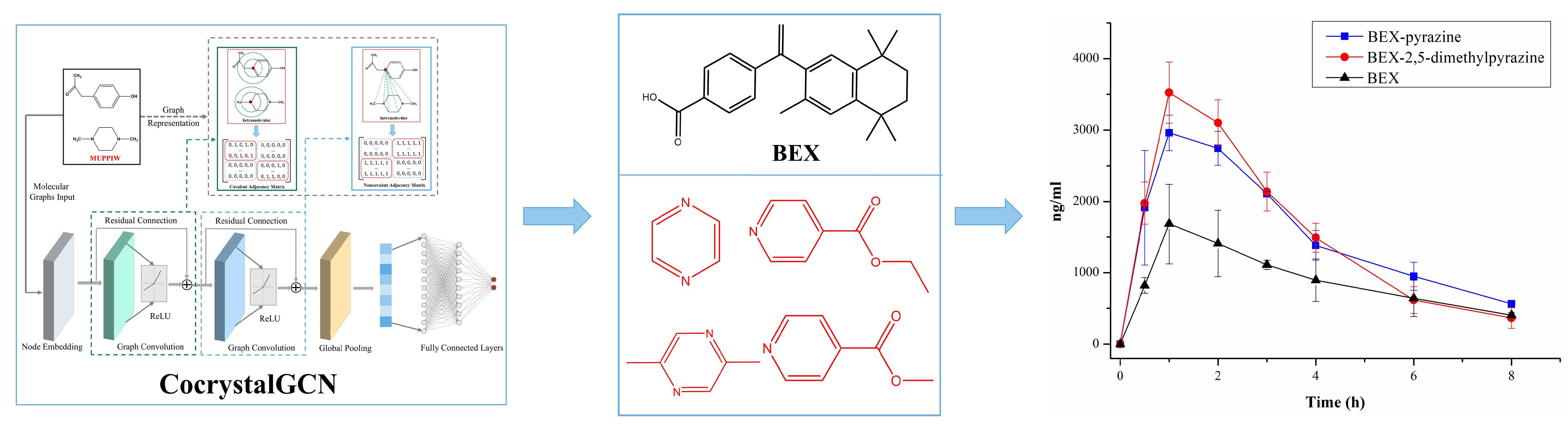
2.1. Data Collection and Processing
- Count the frequency of each compound in csd_pos and gen_neg;
- Calculate the difference between the frequencies of each compound in gen_neg and csd_pos, rank them from large to small;
- If the frequencies of top 1 compound in gen_neg and csd_pos are M and N, respectively, and M > N, use the MaxMin method [46] to select N molecule pairs from the M molecule pairs of top 1 compound in gen_neg, and delete the remaining molecules;
- Repeat step 3 to top 2, top 3, …, top n compounds until 1012 artificially generated negative samples are removed, and then add the exp_neg.
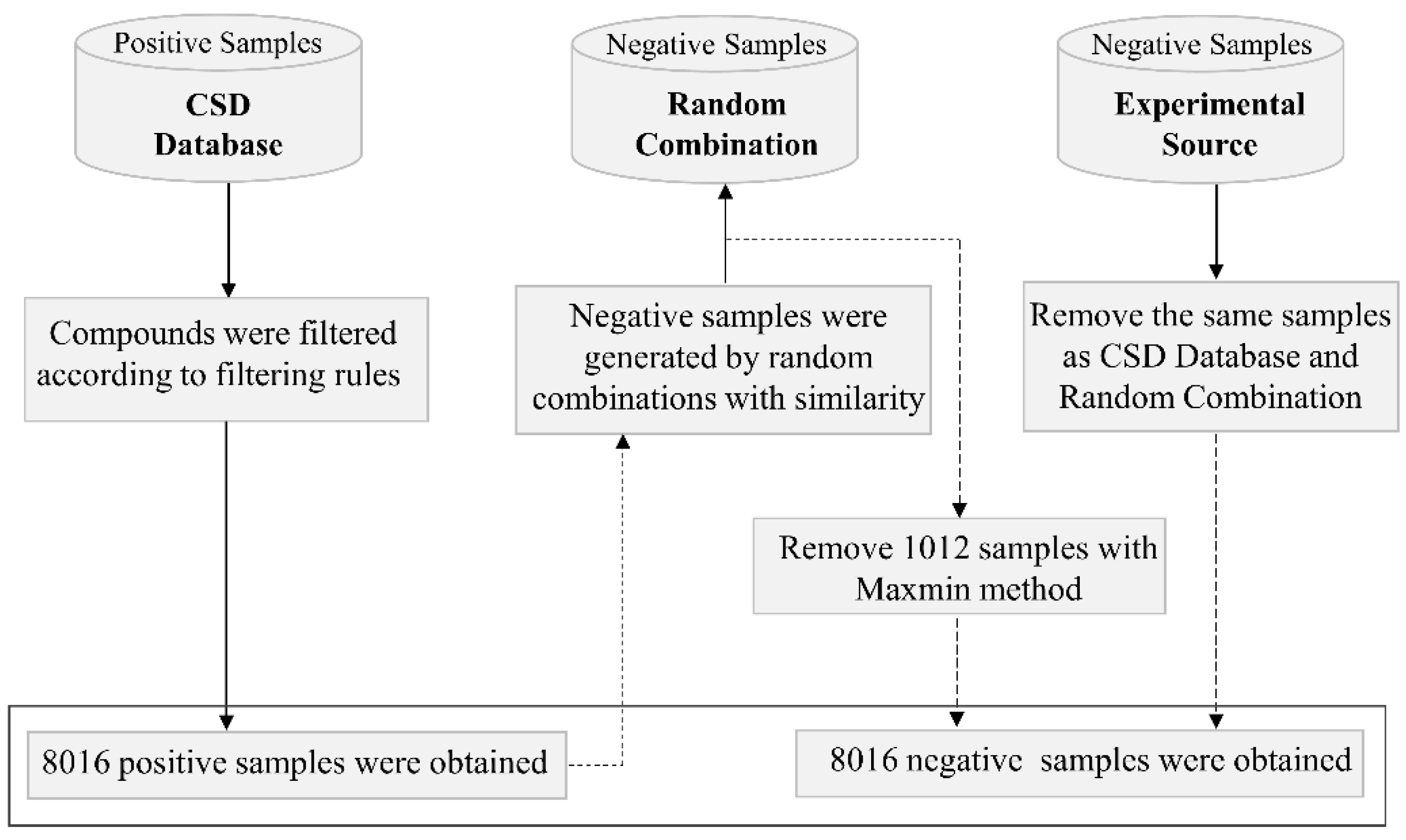
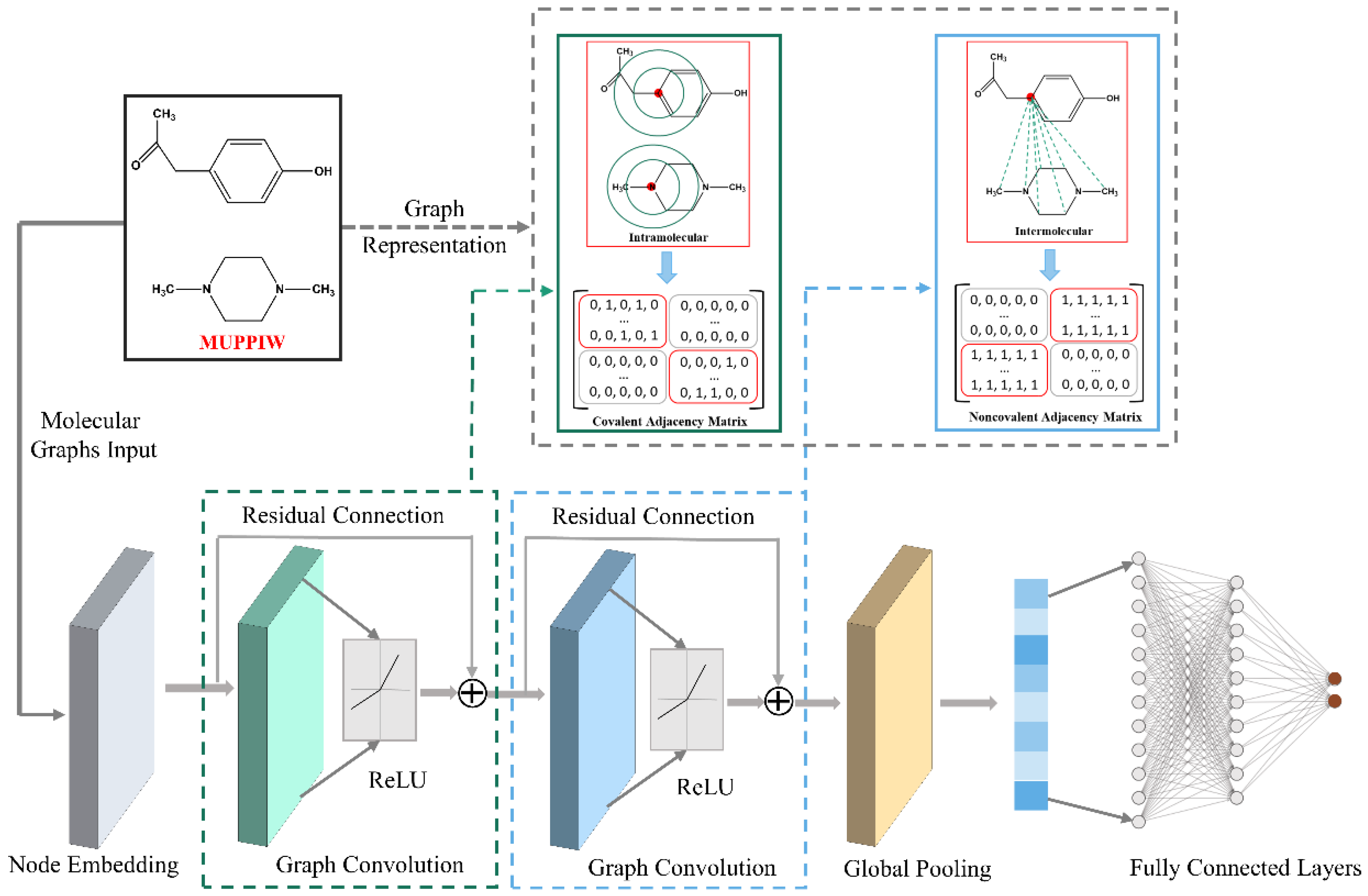
2.2. Cocrystal Representation
2.3. Architecture of the CocrystalGCN
2.4. Performance Evaluation
2.5. Model Training and Interpretation
2.6. Prediction of Cocrystal of BEX
2.7. Materials
2.8. Preparation of BEX Cocrystals
2.9. Preparation of Single Crystals
2.10. Powder X-ray Diffraction (PXRD)
2.11. Single Crystal X-ray Diffraction (SCXRD)
2.12. Thermal Analyses
2.13. Fourier Transformation Infrared (FTIR)
2.14. High-Performance Liquid Chromatography
2.15. Solubility and Powder Dissolution
2.16. Pharmacokinetics in Rats
3. Results and Discussion
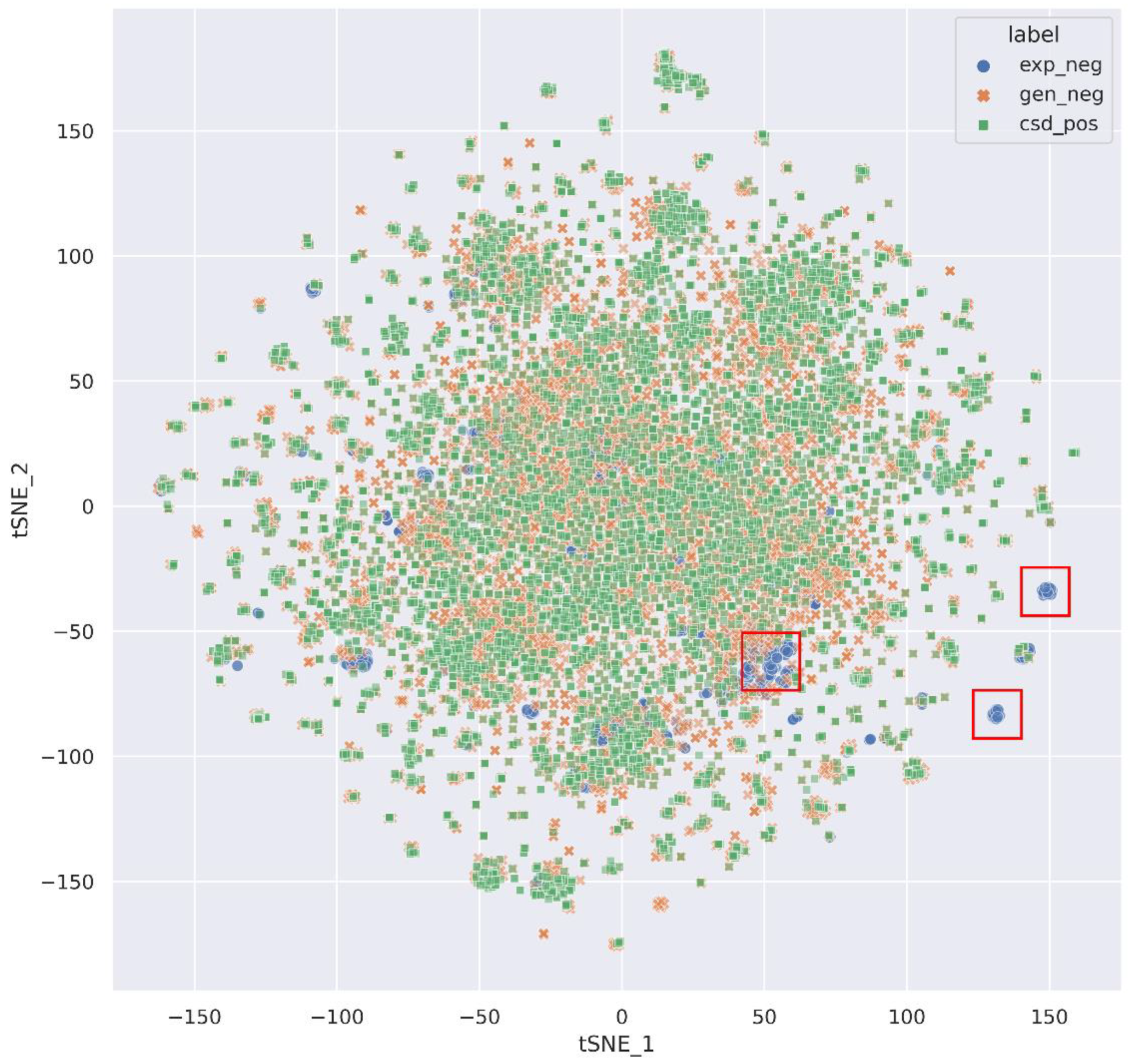
3.1. Dataset Analysis
3.2. Comparison of Prediction Performance between CocrystalGCN and Baselines
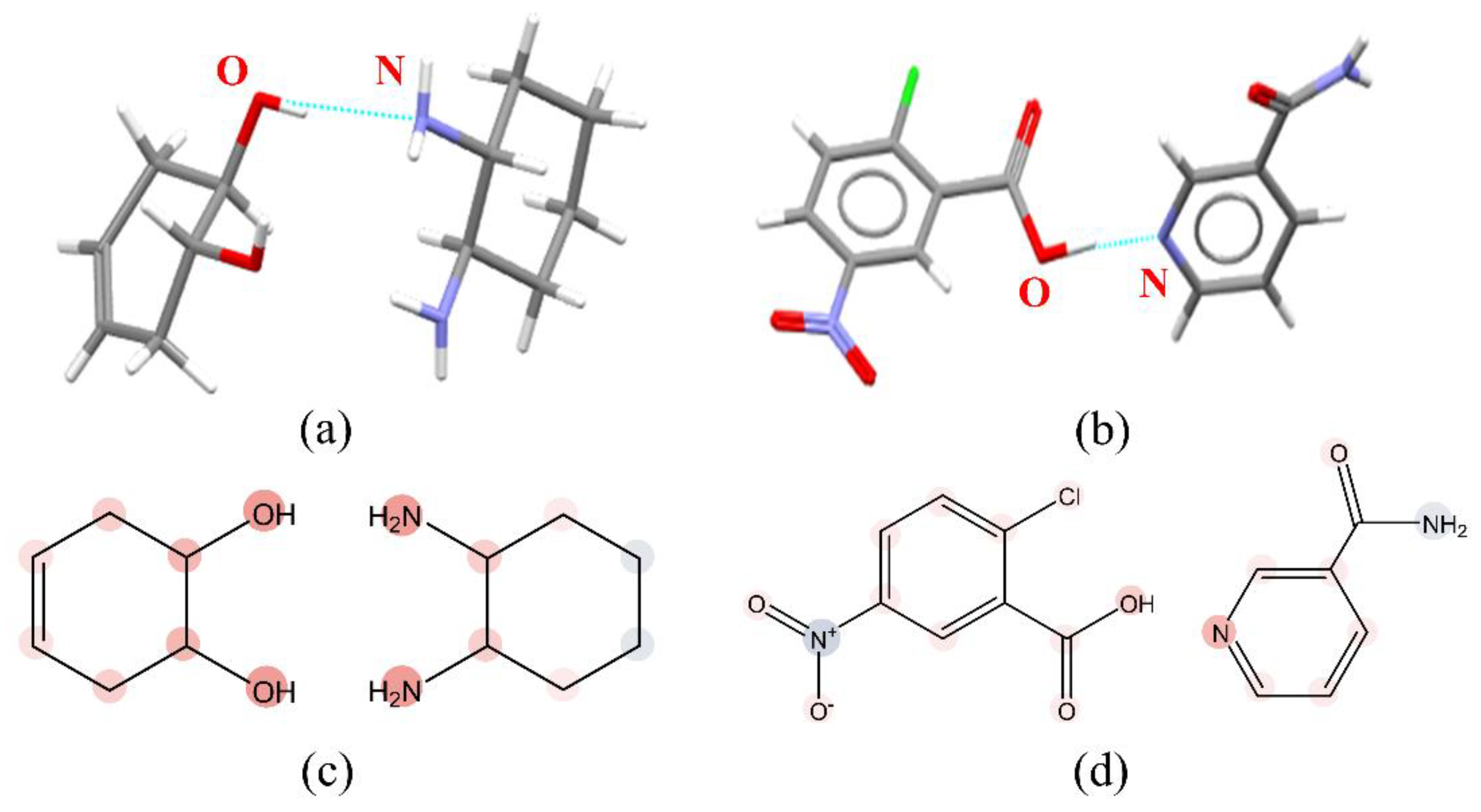
3.3. Interpretation of the CocrystalGCN
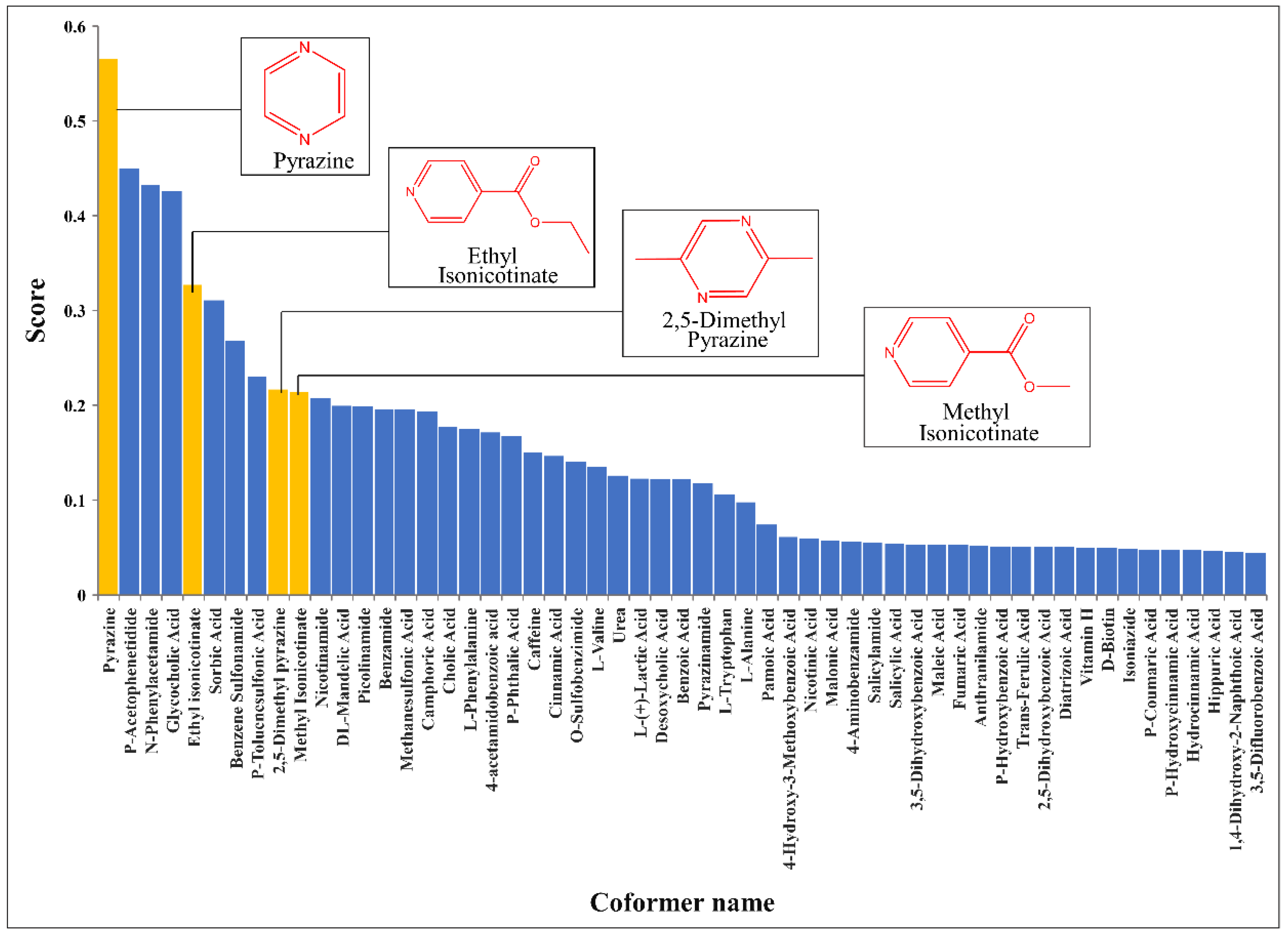
3.4. Results of Virtual Screening Based on CocrystalGCN
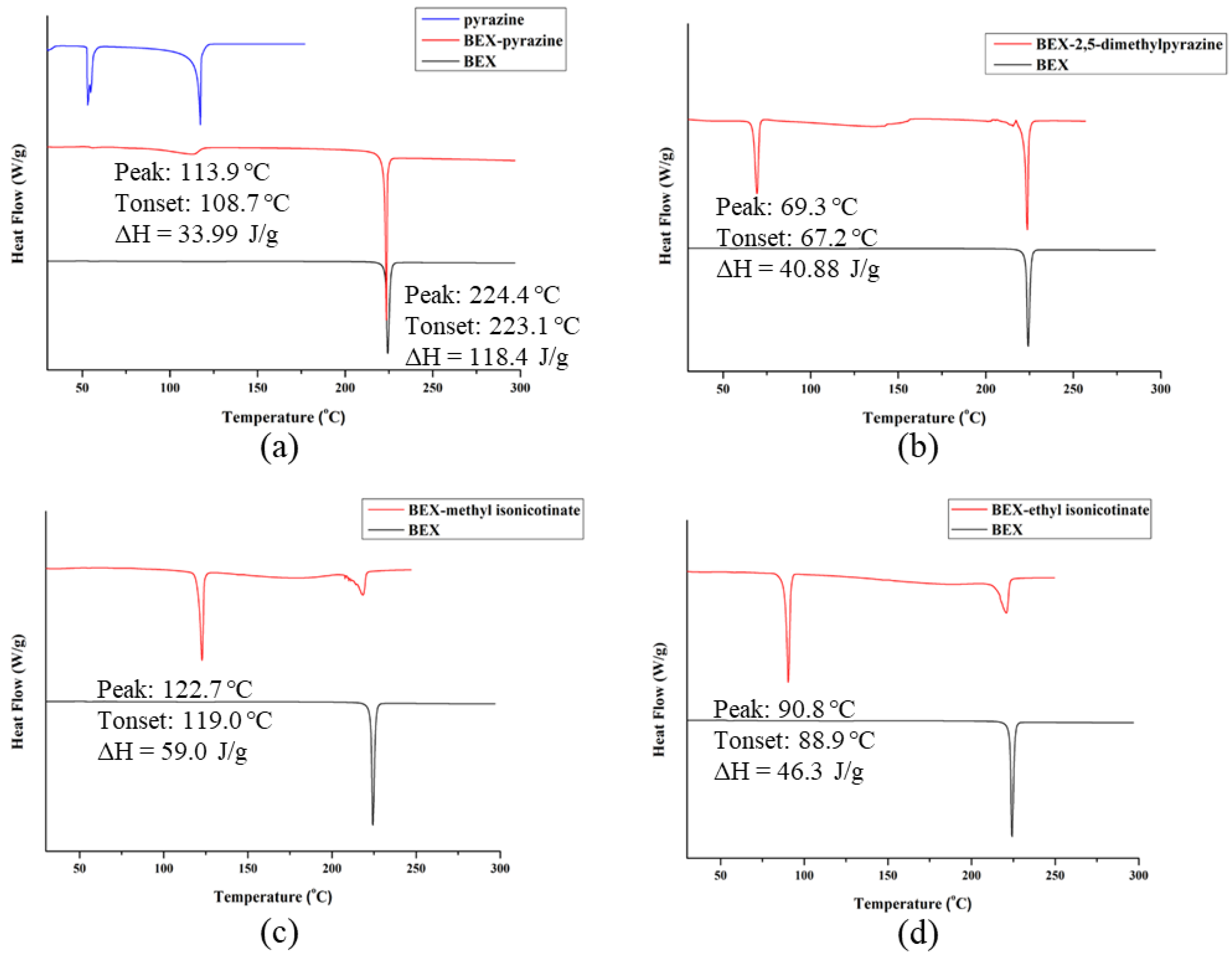
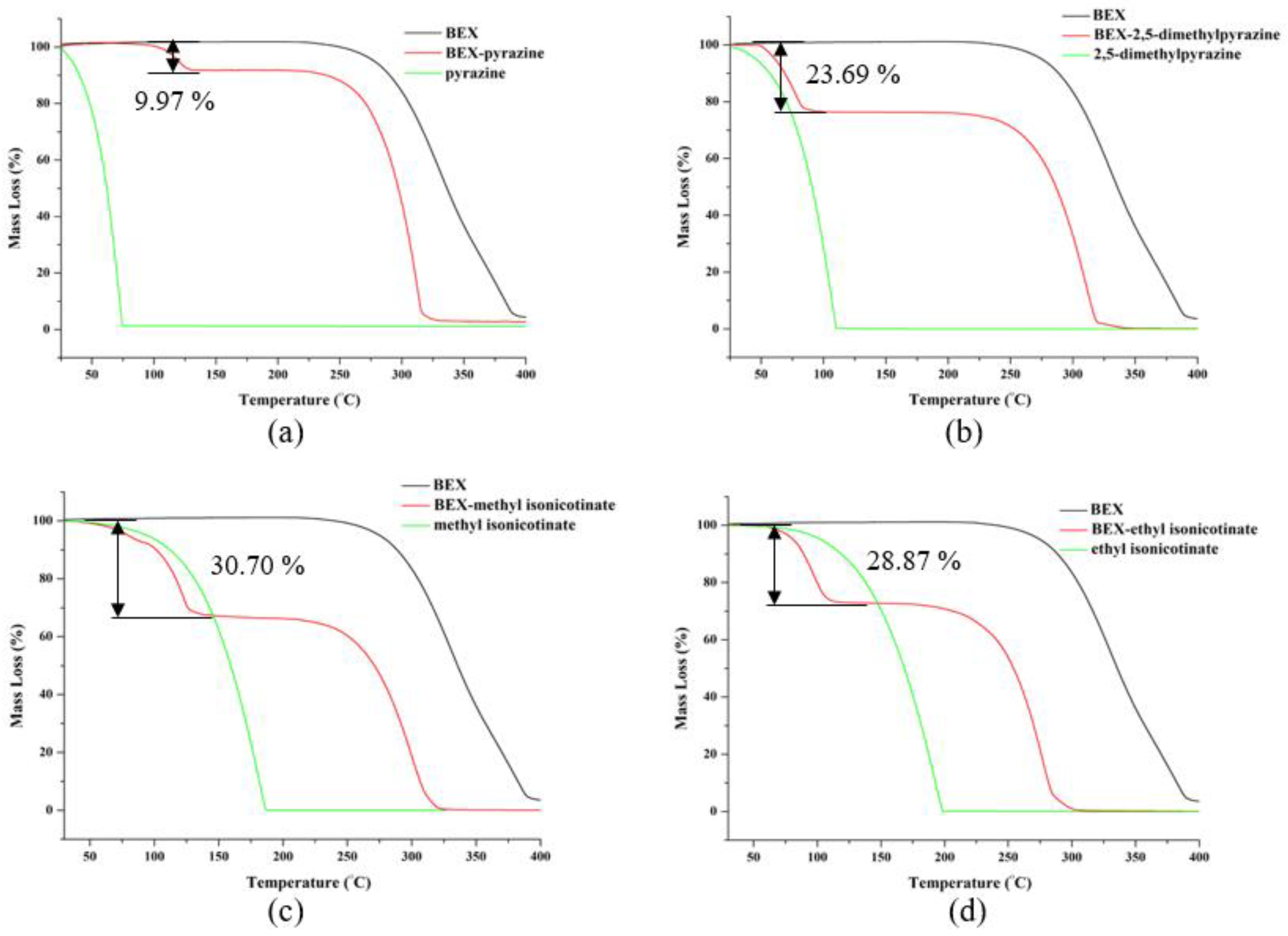
3.5. PXRD and Thermal Analysis of Cocrystals
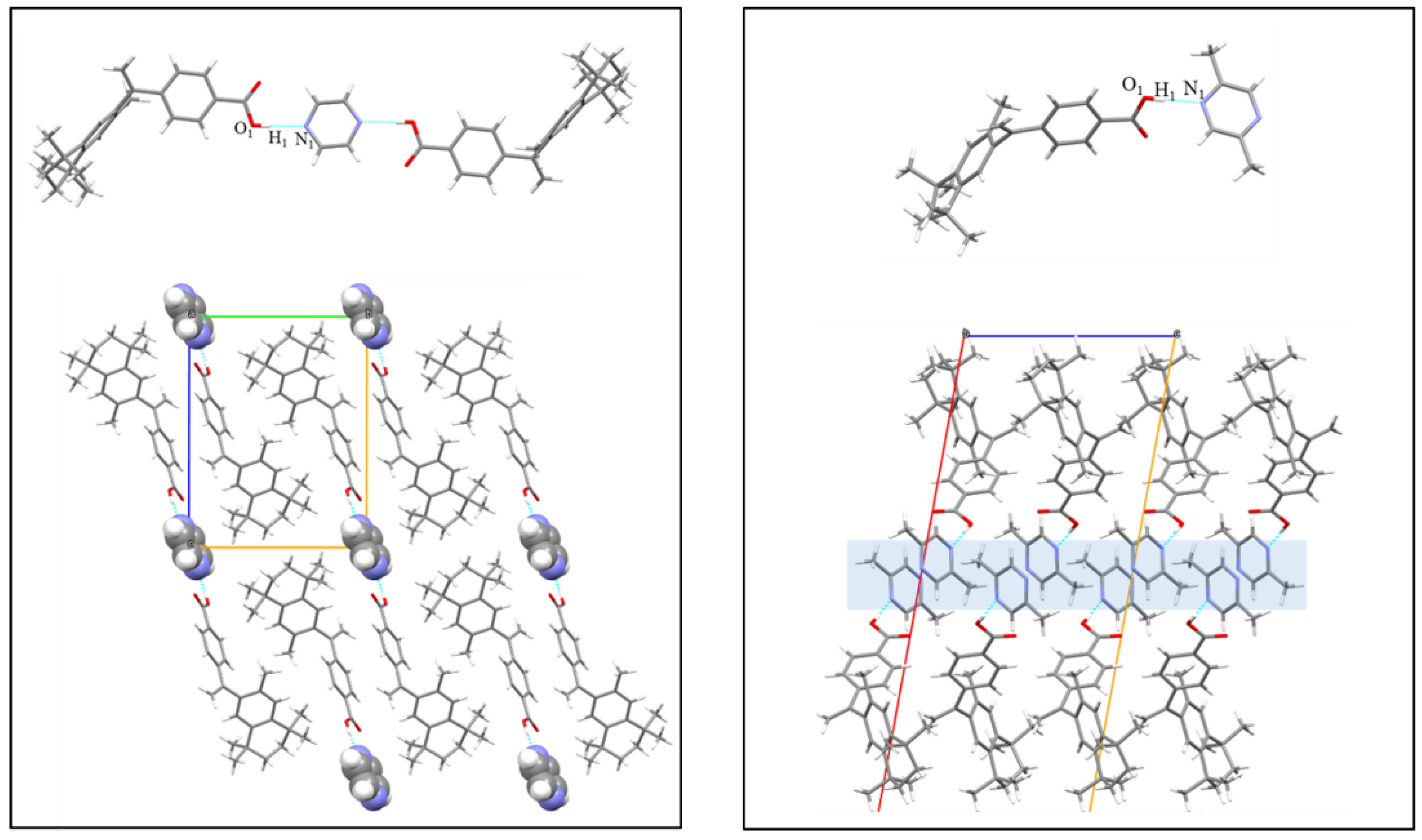
3.6. Crystal Structure Analysis
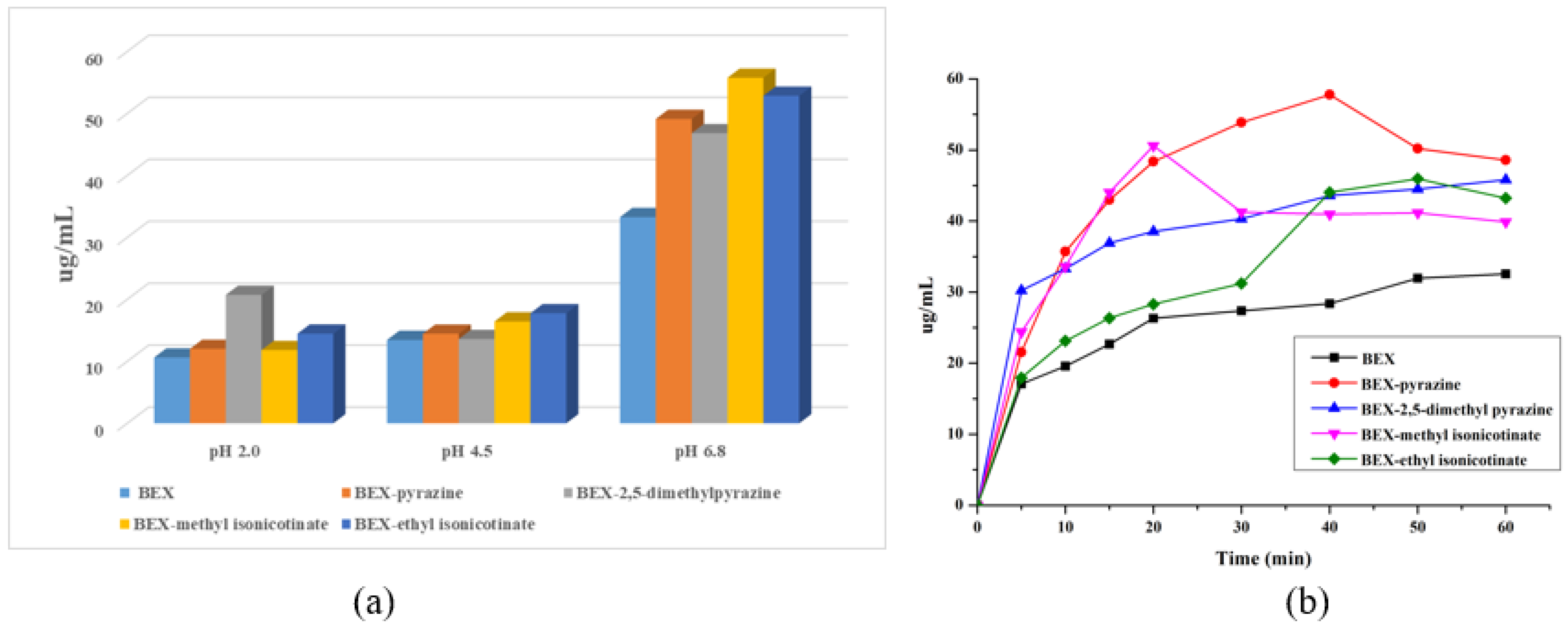
3.7. Solubility and Powder Dissolution
3.8. PK Studies in Rats
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Assaf, C.; Bagot, M.; Dummer, R.; Duvic, M.; Gniadecki, R.; Knobler, R.; Ranki, A.; Schwandt, P.; Whittaker, S. Minimizing adverse side-effects of oral bexarotene in cutaneous T-cell lymphoma: An expert opinion. Br. J. Dermatol. 2006, 155, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Panchal, M.R.; Scarisbrick, J.J. The utility of bexarotene in mycosis fungoides and Sezary syndrome. Oncotargets Ther. 2015, 8, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.J.; Marsden, J.R.; Spittle, M.; Jones, R.R. Joint British Association of Dermatologists and UK Cutaneous Lymphoma Group guidelines for the management of primary cutaneous T-cell lymphomas. Br. J. Dermatol. 2003, 149, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Farol, L.T.; Hymes, K.B. Bexarotene: A clinical review. Expert Rev. Anticancer. Ther. 2004, 4, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J.; Wang, Y.J.; Zhang, J.Z.; Hao, L.L.; Guo, H.J.; Lou, H.X.; Zhang, D.R. Bexarotene nanocrystal-Oral and parenteral formulation development, characterization and pharmacokinetic evaluation. Eur. J. Pharm. Biopharm. 2014, 87, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, Salts and Cocrystals: What’s in a Name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Bolla, G.; Sarma, B.; Nangia, A.K. Crystal Engineering of Pharmaceutical Cocrystals in the Discovery and Development of Improved Drugs. Chem. Rev. 2022, 122, 11514–11603. [Google Scholar] [CrossRef]
- Zheng, L.L.; Zhu, B.; Wu, Z.R.; Fang, X.X.; Hong, M.H.; Liu, G.X.; Li, W.H.; Ren, G.B.; Tang, Y. Strategy for Efficient Discovery of Cocrystals via a Network-Based Recommendation Model. Cryst. Growth Des. 2020, 20, 6820–6830. [Google Scholar] [CrossRef]
- Arabiani, M.R.; Bhunia, S.; Teja, P.K.; Lodagekar, A.; Chavan, R.B.; Shastri, N.R.; Reddy, C.M.; Shelat, P.; Dave, D. Brexpiprazole-catechol cocrystal: Structure elucidation, excipient compatibility and stability. CrystEngComm 2019, 21, 6703–6708. [Google Scholar] [CrossRef]
- Hong, M.; Li, S.; Ji, W.; Qi, M.-H.; Ren, G.-b. Cocrystals of Lenvatinib with Sulfamerazine and Salicylic Acid: Crystal Structure, Equilibrium Solubility, Stability Study, and Anti-Hepatoma Activity. Cryst. Growth Des. 2021, 21, 3714–3727. [Google Scholar] [CrossRef]
- Surov, A.O.; Manin, A.N.; Churakov, A.V.; Perlovich, G.L. New Solid Forms of the Antiviral Drug Arbidol: Crystal Structures, Thermodynamic Stability, and Solubility. Mol. Pharm. 2015, 12, 4154–4165. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Wu, H.; Guo, C.Y.; Zhu, B.; Ren, G.B. Enhancing the solubility of natural compound xanthotoxin by modulating stability via cocrystallization engineering. Int. J. Pharm. 2019, 572, 118776. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, L.; Yao, J.; Ma, Y.Y.; Chen, J.M.; Lu, T.B. Improving the Solubility and Bioavailability of Apixaban via Apixaban-Oxalic Acid Cocrystal. Cryst. Growth Des. 2016, 16, 2923–2930. [Google Scholar] [CrossRef]
- Drozd, K.V.; Manin, A.N.; Boycov, D.E.; Perlovich, G.L. Simultaneous Improvement of Dissolution Behavior and Oral Bioavailability of Antifungal Miconazole via Cocrystal and Salt Formation. Pharmaceutics 2022, 14, 1107. [Google Scholar] [CrossRef]
- Childs, S.L.; Rodríguez-Hornedo, N.; Reddy, L.S.; Jayasankar, A.; Maheshwari, C.; McCausland, L.; Shipplett, R.; Stahly, B.C. Screening strategies based on solubility and solution composition generate pharmaceutically acceptable cocrystals of carbamazepine. CrystEngComm 2008, 10, 856–864. [Google Scholar] [CrossRef]
- Puneeth Jayram, P.S. Pharmaceutical Co-crystals: A Systematic Review. Int. J. Pharm. Investig. 2020, 10, 246–252. [Google Scholar] [CrossRef]
- Bysouth, S.R.; Bis, J.A.; Igo, D. Cocrystallization via planetary milling: Enhancing throughput of solid-state screening methods. Int. J. Pharm. 2011, 411, 169–171. [Google Scholar] [CrossRef]
- Chun, N.-H.; Lee, M.-J.; Song, G.-H.; Chang, K.-Y.; Kim, C.-S.; Choi, G.J. Combined anti-solvent and cooling method of manufacturing indomethacin-saccharin (IMC-SAC) co-crystal powders. J. Cryst. Growth 2014, 408, 112–118. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Galek, P.T.A.; Allen, F.H.; Fábián, L.; Feeder, N. Knowledge-based H-bond prediction to aid experimental polymorph screening. CrystEngComm 2009, 11, 2634–2639. [Google Scholar] [CrossRef]
- Delori, A.; Galek, P.T.A.; Pidcock, E.; Patni, M.; Jones, W. Knowledge-based hydrogen bond prediction and the synthesis of salts and cocrystals of the anti-malarial drug pyrimethamine with various drug and GRAS molecules. CrystEngComm 2013, 15, 2916–2928. [Google Scholar] [CrossRef]
- Wood, P.A.; Feeder, N.; Furlow, M.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Knowledge-based approaches to co-crystal design. CrystEngComm 2014, 16, 5839–5848. [Google Scholar] [CrossRef]
- Kumar, A.; Nanda, A. In-silico methods of cocrystal screening: A review on tools for rational design of pharmaceutical cocrystals. J. Drug Deliv. Sci. Technol. 2021, 63, 102527. [Google Scholar] [CrossRef]
- Fábián, L. Cambridge Structural Database Analysis of Molecular Complementarity in Cocrystals. Cryst. Growth Des. 2009, 9, 1436–1443. [Google Scholar] [CrossRef]
- Devogelaer, J.-J.; Brugman, S.J.T.; Meekes, H.; Tinnemans, P.; Vlieg, E.; de Gelder, R. Cocrystal design by network-based link prediction. CrystEngComm 2019, 21, 6875–6885. [Google Scholar] [CrossRef] [Green Version]
- Devogelaer, J.J.; Charpentier, M.D.; Tijink, A.; Dupray, V.; Coquerel, G.; Johnston, K.; Meekes, H.; Tinnemans, P.; Vlieg, E.; Ter Horst, J.H.; et al. Cocrystals of Praziquantel: Discovery by Network-Based Link Prediction. Cryst. Growth Des. 2021, 21, 3428–3437. [Google Scholar] [CrossRef]
- Musumeci, D.; Hunter, C.A.; Prohens, R.; Scuderi, S.; McCabe, J.F. Virtual cocrystal screening. Chem. Sci. 2011, 2, 883–890. [Google Scholar] [CrossRef]
- McKenzie, J.; Feeder, N.; Hunter, C.A. H-bond competition experiments in solution and the solid state. CrystEngComm 2016, 18, 394–397. [Google Scholar] [CrossRef] [Green Version]
- Klamt, A. Solvent-screening and co-crystal screening for drug development with COSMO-RS. J. Cheminform. 2012, 4, 1–2. [Google Scholar] [CrossRef]
- Loschen, C.; Klamt, A. Cocrystal Ternary Phase Diagrams from Density Functional Theory and Solvation Thermodynamics. Cryst. Growth Des. 2018, 18, 5600–5608. [Google Scholar] [CrossRef]
- Bhardwaj, R.M.; Johnston, A.; Johnston, B.F.; Florence, A.J. A random forest model for predicting the crystallisability of organic molecules. CrystEngComm 2015, 17, 4272–4275. [Google Scholar] [CrossRef] [Green Version]
- Pillong, M.; Marx, C.; Piechon, P.; Wicker, J.G.P.; Cooper, R.I.; Wagner, T. A publicly available crystallisation data set and its application in machine learning. CrystEngComm 2017, 19, 3737–3745. [Google Scholar] [CrossRef]
- Ma, Y.; Gao, Z.; Shi, P.; Chen, M.; Wu, S.; Yang, C.; Wang, J.; Cheng, J.; Gong, J. Machine learning-based solubility prediction and methodology evaluation of active pharmaceutical ingredients in industrial crystallization. Front. Chem. Sci. Eng. 2021, 16, 523–535. [Google Scholar] [CrossRef]
- Bhardwaj, R.M.; Reutzel-Edens, S.M.; Johnston, B.F.; Florence, A.J. A random forest model for predicting crystal packing of olanzapine solvates. CrystEngComm 2018, 20, 3947–3950. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Li, N.; Li, S. The interplay among molecular structures, crystal symmetries and lattice energy landscapes revealed using unsupervised machine learning: A closer look at pyrrole azaphenacenes. CrystEngComm 2019, 21, 6173–6185. [Google Scholar] [CrossRef]
- Galek, P.T.A.; Pidcock, E.; Wood, P.A.; Bruno, I.J.; Groom, C.R. One in half a million a solid form informatics study of a pharmaceutical. CrystEngComm 2012, 14, 2335–2596. [Google Scholar] [CrossRef]
- Wicker, J.G.P.; Crowley, L.M.; Robshaw, O.; Little, E.J.; Stokes, S.P.; Cooper, R.I.; Lawrence, S.E. Will they co-crystallize? CrystEngComm 2017, 19, 5336–5340. [Google Scholar] [CrossRef] [Green Version]
- Przybyłek, M.; Jeliński, T.; Słabuszewska, J.; Ziółkowska, D.; Mroczyńska, K.; Cysewski, P. Application of Multivariate Adaptive Regression Splines (MARSplines) Methodology for Screening of Dicarboxylic Acid Cocrystal Using 1D and 2D Molecular Descriptors. Cryst. Growth Des. 2019, 19, 3876–3887. [Google Scholar] [CrossRef]
- Chabalenge, B.; Korde, S.; Kelly, A.L.; Neagu, D.; Paradkar, A. Understanding Matrix-Assisted Continuous Co-crystallization Using a Data Mining Approach in Quality by Design (QbD). Cryst. Growth Des. 2020, 20, 4540–4549. [Google Scholar] [CrossRef]
- Mswahili, M.E.; Lee, M.-J.; Martin, G.L.; Kim, J.; Kim, P.; Choi, G.J.; Jeong, Y.-S. Cocrystal Prediction Using Machine Learning Models and Descriptors. Appl. Sci. 2021, 11, 1323. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Z.; Zhu, B.; Mei, X.; Luo, X. Machine-Learning-Guided Cocrystal Prediction Based on Large Data Base. Cryst. Growth Des. 2020, 20, 6610–6621. [Google Scholar] [CrossRef]
- Sun, M.; Zhao, S.; Gilvary, C.; Elemento, O.; Zhou, J.; Wang, F. Graph convolutional networks for computational drug development and discovery. Brief. Bioinform. 2019, 21, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tong, H.; Xu, J.; Maciejewski, R. Graph convolutional networks: A comprehensive review. Comput. Soc. Netw. 2019, 6, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Devogelaer, J.J.; Meekes, H.; Tinnemans, P.; Vlieg, E.; de Gelder, R. Co-crystal Prediction by Artificial Neural Networks. Angew. Chem. Int. Ed. 2020, 59, 21711–21718. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, Z.; Guo, J.; Li, H.; Liu, Y.; Guo, Y.; Li, M.; Pu, X. Coupling complementary strategy to flexible graph neural network for quick discovery of coformer in diverse co-crystal materials. Nat. Commun. 2021, 12, 5950. [Google Scholar] [CrossRef]
- Ashton, M.; Barnard, J.; Casset, F.; Charlton, M.; Downs, G.; Gorse, D.; Holliday, J.; Lahana, R.; Willett, P. Identification of Diverse Database Subsets using Property-Based and Fragment-Based Molecular Descriptions. Quant. Struct. Act. Relatsh. 2002, 21, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Lee, E.K.; Choi, I.S. Layer-wise relevance propagation of InteractionNet explains protein-ligand interactions at the atom level. Sci. Rep. 2020, 10, 21155. [Google Scholar] [CrossRef]
- Mercado, R.; Rastemo, T.; Lindelöf, E.; Klambauer, G.; Engkvist, O.; Chen, H.; Jannik Bjerrum, E. Graph networks for molecular design. Mach. Learn. Sci. Technol. 2021, 2, 025023. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. A Method For Stochastic Optimization. In Proceedings of the International Conference on Learning Representations (ICLR), San Diego, CA, USA, 7–9 May 2015. [Google Scholar]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems. arXiv 2016, arXiv:1603.04467. [Google Scholar] [CrossRef]
- Polishchuk, P. Interpretation of Quantitative Structure-Activity Relationship Models: Past, Present, and Future. J. Chem. Inf. Model 2017, 57, 2618–2639. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
- Bach, S.; Binder, A.; Montavon, G.; Klauschen, F.; Muller, K.R.; Samek, W. On Pixel-Wise Explanations for Non-Linear Classifier Decisions by Layer-Wise Relevance Propagation. PLoS ONE 2015, 10, e0130140. [Google Scholar] [CrossRef] [Green Version]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Laurens van der Maaten, G.H. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Glennon, R.A.; Dukat, M. Structure-Activity Relationships of Synthetic Cathinones. Neuropharmacol. New Psychoact. Subst. 2017, 32, 19–47. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. Available online: https://scikit-learn.sourceforge.net (accessed on 10 September 2021).
- Corinna Cortes; Vapnik, V. Support Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- González, S.; García, S.; Del Ser, J.; Rokach, L.; Herrera, F. A practical tutorial on bagging and boosting based ensembles for machine learning: Algorithms, software tools, performance study, practical perspectives and opportunities. Inf. Fusion 2020, 64, 205–237. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Shen, S.; Deng, L.; Liu, H. DeepDDS: Deep graph neural network with attention mechanism to predict synergistic drug combinations. Brief. Bioinform. 2021, 23, bbab390. [Google Scholar] [CrossRef] [PubMed]
- Springuel, G.; Norberg, B.; Robeyns, K.; Wouters, J.; Leyssens, T. Advances in Pharmaceutical Co-crystal Screening: Effective Co-crystal Screening through Structural Resemblance. Cryst. Growth Des. 2011, 12, 475–484. [Google Scholar] [CrossRef]
- Cheng, Y.; Rong, X.; Xia, M.; Zhang, Z.; Wang, J.-R.; Mei, X. Conformational polymorphs of isotretinoin and their impact on physicochemical and biological properties. Int. J. Pharm. 2021, 610, 121222. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Validation Set | Test Set | ||||||
|---|---|---|---|---|---|---|---|---|
| AUC | Acc | Precision | Recall | AUC | Acc | Precision | Recall | |
| RF | 0.884 | 0.801 | 0.797 | 0.808 | 0.795 | 0.795 | 0.793 | 0.801 |
| SVM | 0.852 | 0.787 | 0.770 | 0.817 | 0.783 | 0.783 | 0.767 | 0.818 |
| XGBoost | 0.855 | 0.781 | 0.770 | 0.802 | 0.780 | 0.780 | 0.775 | 0.792 |
| DNN | 0.839 | 0.766 | 0.757 | 0.782 | 0.759 | 0.760 | 0.739 | 0.806 |
| DeepDDS | 0.879 | 0.810 | 0.789 | 0.844 | 0.871 | 0.805 | 0.787 | 0.838 |
| CocrystalGCN_C 1 | 0.855 | 0.803 | 0.782 | 0.829 | 0.854 | 0.794 | 0.784 | 0.812 |
| CocrystalGCN_NC 2 | 0.853 | 0.816 | 0.795 | 0.854 | 0.855 | 0.806 | 0.785 | 0.840 |
| CocrystalGCN 3 | 0.866 | 0.818 | 0.802 | 0.845 | 0.866 | 0.811 | 0.802 | 0.830 |
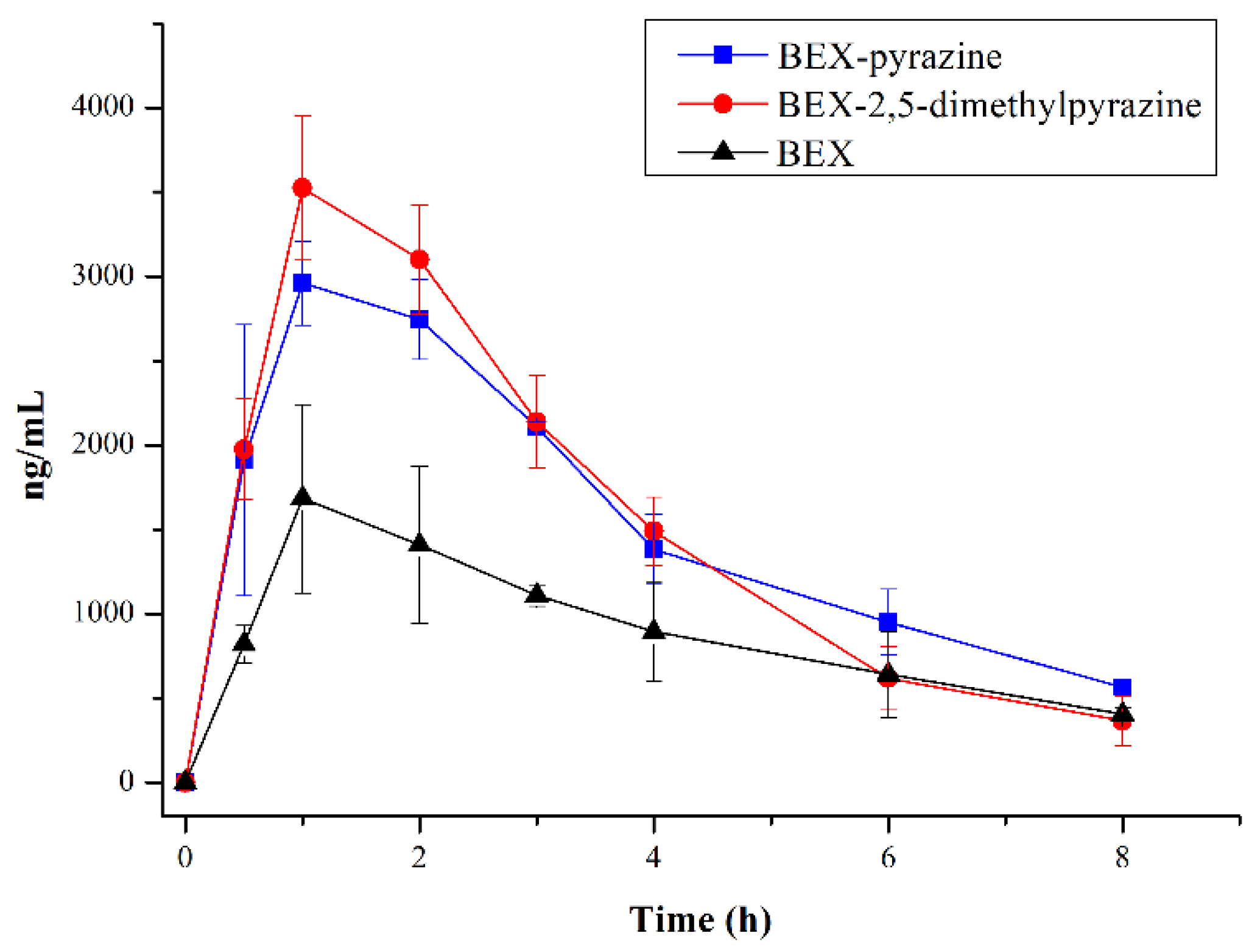
| Parameters | BEX | BEX-Pyrazine | BEX-2,5- Dimethylpyrazine |
|---|---|---|---|
| T1/2 (h) | 2.98 ± 1.18 | 2.59 ± 0.10 | 1.74 ± 0.39 |
| Cmax (μg/L) | 1682.67 ± 559.27 | 2960.33 ± 248.02 * | 3577.00 ± 387.34 * |
| AUC0−8h (h·μg/L) | 7215.17 ± 810.61 | 12561.45 ± 919.13 * | 12702.38 ± 978.30 * |
| AUCinf (h·μg/L) | 8792.75 ± 1076.36 | 14656.33 ± 1085.66 * | 13513.29 ± 1358.85 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, F.; Cheng, Y.; Wang, J.-R.; Wang, D.; Zhang, Y.; Chen, K.; Mei, X.; Luo, X. Cocrystal Prediction of Bexarotene by Graph Convolution Network and Bioavailability Improvement. Pharmaceutics 2022, 14, 2198. https://doi.org/10.3390/pharmaceutics14102198
Xiao F, Cheng Y, Wang J-R, Wang D, Zhang Y, Chen K, Mei X, Luo X. Cocrystal Prediction of Bexarotene by Graph Convolution Network and Bioavailability Improvement. Pharmaceutics. 2022; 14(10):2198. https://doi.org/10.3390/pharmaceutics14102198
Chicago/Turabian StyleXiao, Fu, Yinxiang Cheng, Jian-Rong Wang, Dingyan Wang, Yuanyuan Zhang, Kaixian Chen, Xuefeng Mei, and Xiaomin Luo. 2022. "Cocrystal Prediction of Bexarotene by Graph Convolution Network and Bioavailability Improvement" Pharmaceutics 14, no. 10: 2198. https://doi.org/10.3390/pharmaceutics14102198