
SAR Study and Molecular Mechanism Investigation of Novel Naphthoquinone-furan-2-cyanoacryloyl Hybrids with Antitumor Activity

Abstract
:1. Introduction
2. Results and Discussion
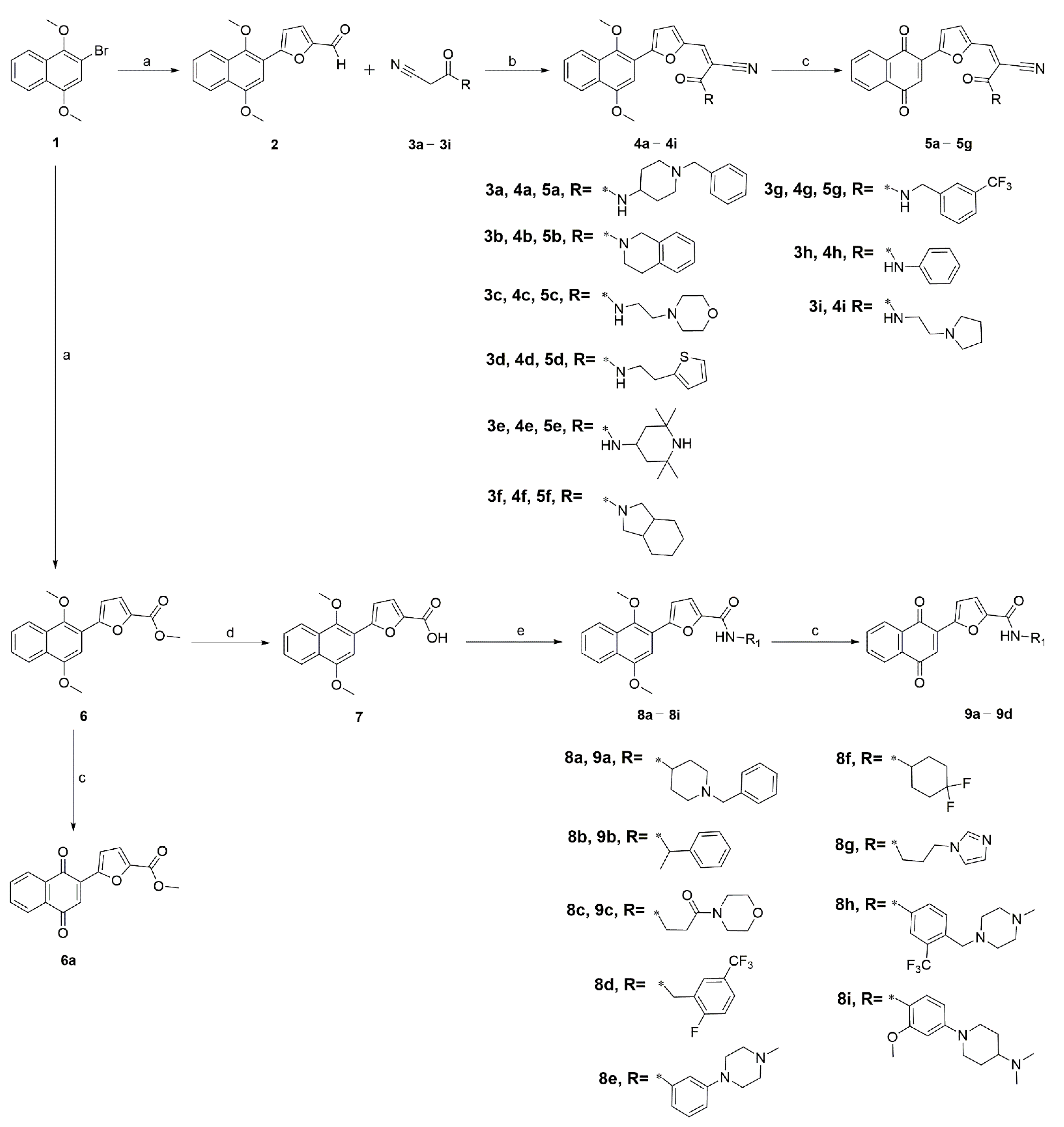
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Evaluation of In Vitro Antitumor Activity and Safety
2.2.2. Compound 5c Inhibited HeLa Cell Migration
2.2.3. Compound 5c Inhibited the Colony Formation of HeLa Cells
2.2.4. Compound 5c Blocks HeLa Cells in G2/M Phase
2.2.5. Compound 5c Induced the Generation of Intracellular Reactive Oxygen Species
2.2.6. Effect of Compound 5c on HeLa Cell Apoptosis
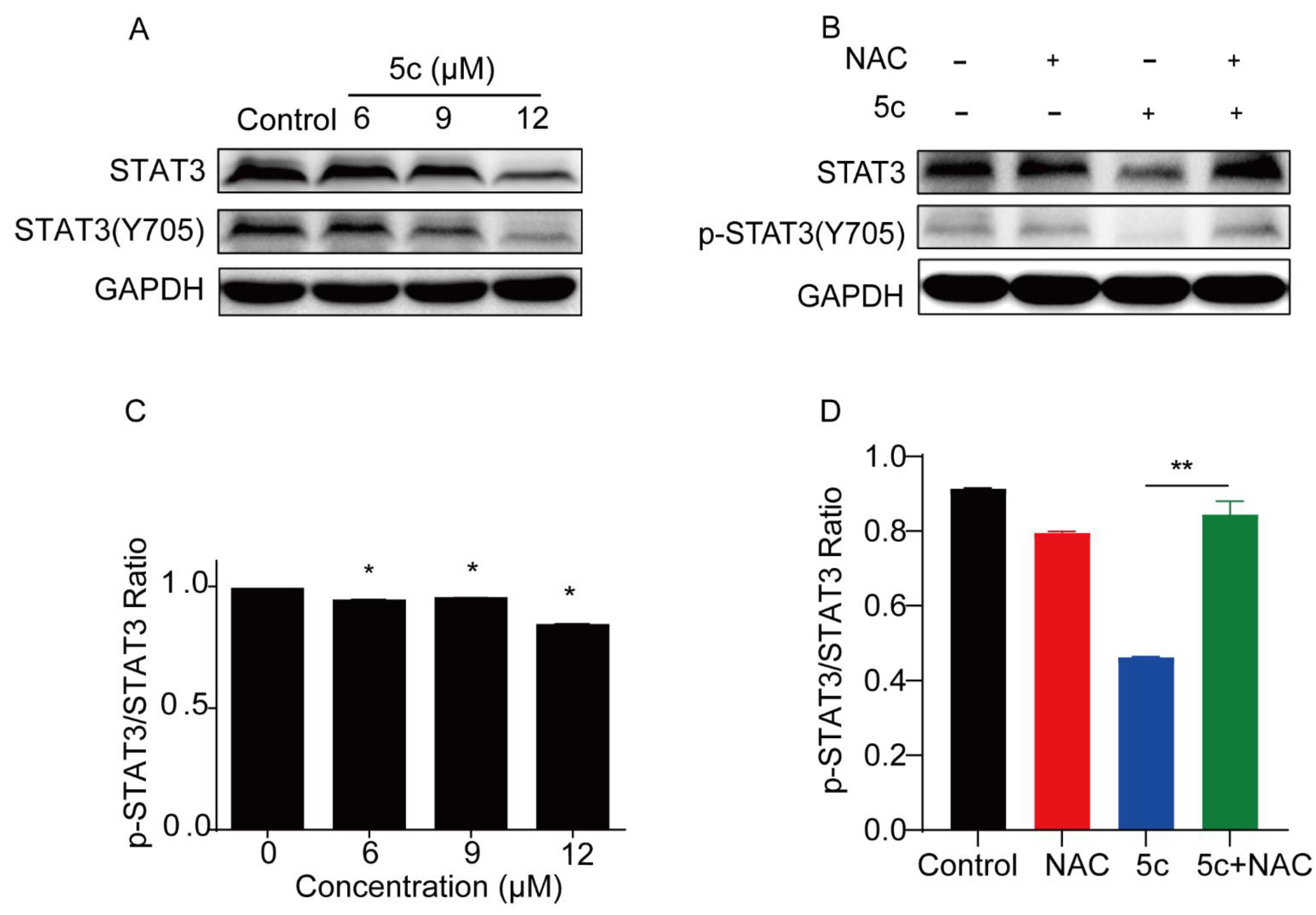
2.2.7. Compound 5c Inhibited STAT3 Y705 Phosphorylation in Cell-Based Assays
2.2.8. Kinetic Affinity of Compound 5c against STAT3
2.3. Docking Study of Compound 5c with the STAT3 SH2 Domain
2.4. Molecular Dynamics Simulation Analyses
3. Conclusions
4. Experimental
4.1. Chemistry
4.2. Biological Evaluation
4.2.1. Cell Lines and Cell Culture
4.2.2. Cell Counting Kit-8 (CCK-8) Assay
4.2.3. Hoechst-33342 Staining
4.2.4. Western Blotting Assays
4.2.5. Cell Cycle and Annexin V apoptosis Assays
4.2.6. Wound Healing Assay
4.2.7. Colony Formation Assay
4.2.8. Cellular ROS Detection In Vitro
4.2.9. Biolayer Interferometry (BLI) Assay
4.2.10. Molecular Docking
4.2.11. Molecular Dynamics Simulation
4.3. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. World Cancer Report: Cancer Research for Cancer Prevention; The World Health Organization(WHO): Geneva, Switzerland, 2020. [Google Scholar]
- Majolo, F.; Knabben, D.; Marmitt, D.J.; Bustamante-Filho, I.C.; Goettert, M.I. Medicinal plants and bioactive natural compounds for cancer treatment: Important advances for drug discovery. Phytochem. Lett. 2019, 31, 196–207. [Google Scholar] [CrossRef]
- Pan, L.; Chai, H.B.; Kinghorn, A.D. Discovery of new anticancer agents from higher plants. Front. Biosci.-Landmark 2011, 4, 142–156. [Google Scholar]
- Xu, Y.; Xu, X.; Gao, X.; Chen, H.; Geng, L. Shikonin suppresses IL-17-induced VEGF expression via blockage of JAK2/STAT3 pathway. Int. Immunopharmacol. 2014, 19, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, Y.; Cao, K.; Xu, Y.Y.; Chen, H.D. Shikonin inhibits IFN-γ- induced K17 over-expression of HaCaT cells by interfering with STAT3 signaling. Int. J. Clin. Exp. Pathol. 2015, 8, 9202–9207. [Google Scholar]
- Hubbard, J.M.; Grothey, A. Napabucasin: An Update on the First-in-Class Cancer Stemness Inhibitor. Drugs 2017, 77, 1091–1103. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; El-Rayes, B. Identifying and targeting cancer stem cells in the treatment of gastric cancer. Cancer 2017, 123, 1303–1312. [Google Scholar] [CrossRef] [Green Version]
- Xiang, M.; Kim, H.; Ho, V.T.; Walker, S.R.; Bar-Natan, M.; Anahtar, M.; Liu, S.; Toniolo, P.A.; Kroll, Y.; Jones, N.; et al. Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood 2016, 128, 1845–1853. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Xiao, H.; Lin, J.; Li, C. Discovery of novel STAT3 small molecule inhibitors via in silico site-directed fragment-based drug design. J. Med. Chem. 2013, 56, 4402–4412. [Google Scholar] [CrossRef]
- Qiu, H.Y.; Fu, J.Y.; Yang, M.K.; Han, H.W.; Wang, P.F. Identification of new shikonin derivatives as STAT3 inhibitors. Biochem. Pharmacol. 2017, 146, 74–86. [Google Scholar] [CrossRef]
- Qiu, H.Y.; Zhu, X.; Luo, Y.L.; Lin, H.Y.; Tang, C.Y.; Qi, J.L.; Pang, Y.J.; Yang, R.W.; Lu, G.H.; Wang, X.M. Identification of New Shikonin Derivatives as Antitumor Agents Targeting STAT3 SH2 Domain. Sci. Rep. 2017, 7, 2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereyra, C.E.; Dantas, R.F.; Ferreira, S.B.; Gomes, L.P.; Silva, F.P., Jr. The diverse mechanisms and anticancer potential of naphthoquinones. Cancer Cell Int. 2019, 19, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, P.; Baltas, M.; Bedos-Belval, F. Cinnamic Acid Derivatives as Anticancer Agents-A Review. Curr. Med. Chem. 2011, 18, 1672–1703. [Google Scholar] [CrossRef] [PubMed]
- Swiatek-Machado, K.; Mieczkowski, J.; Ellert-Miklaszewska, A.; Swierk, P.; Fokt, I.; Szymanski, S.; Skora, S.; Szeja, W.; Grynkiewicz, G.; Lesyng, B.; et al. Novel small molecular inhibitors disrupt the JAK/STAT3 and FAK signaling pathways and exhibit a potent antitumor activity in glioma cells. Cancer Biol. Ther. 2012, 13, 657–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Pal, A.; Han, D.; Wang, S.; Maxwell, D.; Levitzki, A.; Talpaz, M.; Donato, N.J.; Bornmann, W. Tyrphostin-like compounds with ubiquitin modulatory activity as possible therapeutic agents for multiple myeloma. Bioorg. Med. Chem. 2011, 19, 7194–7204. [Google Scholar] [CrossRef]
- Verstovsek, S.; Manshouri, T.; Quintas-Cardama, A.; Harris, D.; Cortes, J.; Giles, F.J.; Kantarjian, H.; Priebe, W.; Estrov, Z. WP1066, a Novel JAK2 Inhibitor, Suppresses Proliferation and Induces Apoptosis in Erythroid Human Cells Carrying the JAK2 V617F Mutation. Clin. Cancer Res. 2015, 14, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Mielecki, M.; Krawiec, K.; Kiburu, I.; Grzelak, K.; Zagorski, W.; Kierdaszuk, B.; Kowa, K.; Fokt, I.; Szymanski, S.; Swierk, P.; et al. Development of novel molecular probes of the Rio1 atypical protein kinase. Biochim. Biophys. Acta 2013, 1834, 1292–1301. [Google Scholar] [CrossRef]
- Jin, W.; Zhang, T.; Zhou, W.; He, P.; Sun, Y.; Hu, S.; Chen, H.; Ma, X.; Peng, Y.; Yi, Z.; et al. Discovery of 2-Amino-3-cyanothiophene Derivatives as Potent STAT3 Inhibitors for the Treatment of Osteosarcoma Growth and Metastasis. J. Med. Chem. 2022, 65, 6710–6728. [Google Scholar] [CrossRef]
- Krause, T.; Gerbershagen, M.U.; Fiege, M.; Weisshorn, R.; Wappler, F. Dantrolene—A review of its pharmacology, therapeutic use and new developments. Anaesthesia 2015, 59, 364–373. [Google Scholar] [CrossRef]
- Fuentes, B.R.; Maturana, A.M.; de la Cruz, M.R. Efficacy of nifurtimox for the treatment of chronic Chagas disease. Rev. Chil. Infectol. 2012, 29, 82–86. [Google Scholar]
- Kong, E.; Zhu, J.; Wu, W.; Ren, H.; Jiao, X.; Wang, H.; Zhang, Z. Nifurtimox Inhibits the Progression of Neuroblastoma in vivo. J. Cancer 2019, 10, 2194–2204. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Yang, Z.; Ding, C.; Chu, L.; Zhang, Y.; Terry, K.; Liu, H.; Shen, Q.; Zhou, J. Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. Eur. J. Med. Chem. 2013, 62, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zhou, X.; Wang, A.; Zheng, Y.; Gao, Y.; Zhou, J. Evolutions in fragment-based drug design: The deconstruction-reconstruction approach. Drug Discov Today 2015, 20, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.L.; Zhang, J.; Liu, C.L.; Liu, C.; Zhu, K.K.; Yang, F.F.; Liu, X.G.; Figueiro Longo, J.P.; Alexandre Muehlmann, L.; Azevedo, R.B.; et al. Design and synthesis of pregnenolone/2-cyanoacryloyl conjugates with dual NF-kappaB inhibitory and anti-proliferative activities. Bioorg. Med. Chem. Lett. 2017, 27, 4682–4686. [Google Scholar] [CrossRef] [PubMed]
- Marcin Mielecki, M.M.-K.; Grzelak, K.; Mielecki, D.; Krzyśko, A.K.; Lesyng, B.; Priebe, W. Analogs of Cinnamic Acid Benzyl Amide As Nonclassical Inhibitors of Activated JAK2 Kinase. Curr. Cancer Drug Targets 2014, 14, 638–651. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shi, C.; He, Z.; Zhu, F.; Qin, R. Inhibition of PI3K/AKT signaling via ROS regulation is involved in Rhein-induced apoptosis and enhancement of oxaliplatin sensitivity in pancreatic cancer cells. Int. J. Biol. Sci. 2021, 17, 589–602. [Google Scholar] [CrossRef]
- Su, X.; Shen, Z.; Yang, Q.; Sui, F.; Pu, J.; Ma, J.J.; Ma, S.R.; Yao, D.M.; Ji, M.J.; Hou, P. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics 2019, 9, 4461–4473. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, T.; Nie, G.; Hu, R.; Hu, G. Cadmium and Molybdenum co-induce pyroptosis via ROS/PTEN/PI3K/AKT axis in duck renal tubular epithelial cells. Environ. Pollut. 2020, 272, 116403. [Google Scholar] [CrossRef]
- Biasutto, L.; Chiechi, A.; Couch, R.; Liotta, L.A.; Espina, V. Retinal pigment epithelium (RPE) exosomes contain signaling phosphoproteins affected by oxidative stress. Exp. Cell Res. 2013, 319, 2113–2123. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tan, X.; Yang, Q.; Zeng, X.; Zhou, Y. Inhibition of autophagy promotes apoptosis and enhances anticancer efficacy of adriamycin via augmented ROS generation in prostate cancer cells. Int. J. Biochem. Cell Biol. 2016, 77, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Sánchez, N. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ROS-induced apoptotic trigger. Mol. Asp. Med. 2010, 31, 29–59. [Google Scholar]
- Cui, Q.; Wang, J.Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.H.; Chen, Z.S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Xia, Y.; He, W.; Zhang, T.; Hong, L.; Zheng, P.; Shen, X.; Liang, G.; Cui, R.; Zou, P. Enhancement of oxaliplatin-induced colon cancer cell apoptosis by alantolactone, a natural product inducer of ROS. Int. J. Biol. Sci. 2019, 15, 1676–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Cheng, X.D.; Zhang, W.D.; Qin, J.J. Recent Update on Development of Small-Molecule STAT3 Inhibitors for Cancer Therapy: From Phosphorylation Inhibition to Protein Degradation. J. Med. Chem. 2021, 64, 8884–8915. [Google Scholar] [CrossRef] [PubMed]
- Dixit, D.; Sharma, V.; Ghosh, S.; Koul, N.; Mishra, P.K.; Sen, E. Manumycin inhibits STAT3, telomerase activity, and growth of glioma cells by elevating intracellular reactive oxygen species generation. Free Radic. Biol. Med. 2009, 47, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, M.; Li, G.; Cai, R. Raddeanin A induced apoptosis of non-small cell lung cancer cells by promoting ROS-mediated STAT3 inactivation. Tissue Cell 2021, 71, 101577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Guo, J.; Li, S.; Ma, T.; Xu, D.; Han, C.; Liu, F.; Yu, W.; Kong, L. Discovery of monocarbonyl curcumin-BTP hybrids as STAT3 inhibitors for drug-sensitive and drug-resistant breast cancer therapy. Sci. Rep. 2017, 7, 46352. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Park, A.; Hwang, J.; Zhao, X.; Kwak, J.; Kim, H.W.; Ku, M.; Yang, J.; Kim, T.I.; Jeong, K.S.; et al. KS10076, a chelator for redox-active metal ions, induces ROS-mediated STAT3 degradation in autophagic cell death and eliminates ALDH1(+) stem cells. Cell Rep. 2022, 40, 111077. [Google Scholar] [CrossRef]
- Debnath, B.; Xu, S.; Neamati, N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J. Med. Chem. 2012, 55, 6645–6668. [Google Scholar] [CrossRef]
- Gilardini Montani, M.S.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; D’Orazi, G.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell. Oncol. 2017, 40, 1–14. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibition Rate a (%) at 20 μM | |||
|---|---|---|---|---|
| HeLa b | PC3 c | A549 d | HCT116 e | |
| 4a | 13.64 ± 0.15 | 15.47 ± 0.17 | 16.30 ± 0.22 | 5.69 ± 0.23 |
| 4b | 11.71 ± 0.04 | 3.88 ± 0.13 | 24.2 ± 0.31 | 19.55 ± 0.65 |
| 4c | 24.23 ± 0.01 | 8.99 ± 0.09 | 11.25 ± 0.05 | 18.78 ± 0.04 |
| 4d | 11.60 ± 0.79 | 7.17 ± 0.31 | 21.58 ± 0.02 | 10.77 ± 0.49 |
| 4e | 59.10 ± 0.21 | 15.84 ± 0.33 | 59.49 ± 0.34 | 40.53 ± 0.03 |
| 4f | 11.49 ± 0.83 | 11.75 ± 0.79 | 25.11 ± 0.79 | 43.69 ± 0.84 |
| 4g | 16.12 ± 0.17 | 15.38 ± 0.01 | 4.87 ± 0.30 | 7.69 ± 0.02 |
| 4h | 9.49 ± 0.16 | 19.53 ± 0.17 | 0.25 ± 0.13 | 11.54 ± 0.14 |
| 4i | 34.99 ± 0.41 | 12.29 ± 0.12 | 13.95 ± 0.87 | 29.05 ± 0.08 |
| 5a | 10.43 ± 0.41 | 3.70 ± 0.19 | 13.28 ± 0.17 | 8.36 ± 0.26 |
| 5b | 20.50 ± 0.28 | 17.83 ± 0.33 | 18.40 ± 0.82 | 8.83 ± 0.10 |
| 5c | 75.78 ± 0.05 | 25.54 ± 0.06 | 24.69 ± 0.04 | 67.67 ± 0.01 |
| 5d | 5.61 ± 0.24 | 15.51 ± 0.20 | 17.24 ± 0.35 | 26.77 ± 0.94 |
| 5e | 29.73 ± 0.07 | 19.33 ± 0.28 | 19.84 ± 0.32 | 5.97 ± 95 |
| 5f | 1.90 ± 0.72 | 11.13 ± 0.03 | 13.88 ± 0.14 | 3.41 ± 0.26 |
| 5g | 27.98 ± 0.36 | 11.20 ± 0.02 | 32.35 ± 0.95 | 28.25 ± 0.04 |
| 5h | 17.23 ± 0.11 | 10.12 ± 0.85 | 9.91 ± 0.06 | 3.94 ± 0.03 |
| 5i | 30.49 ± 0.03 | 16.25 ± 0.19 | 25.03 ± 0.22 | 10.25 ± 0.04 |
| 6 | 27.00 ± 0.32 | 21.58 ± 0.83 | 21.31 ± 0.45 | 16.78 ± 0.07 |
| 6a | 26.09 ± 0.66 | 6.93 ± 0.79 | 2.15 ± 0.37 | 17.67 ± 0.06 |
| 8a | 20.86 ± 0.18 | 15.47 ± 0.29 | 24.06 ± 0.35 | 12.40 ± 0.15 |
| 8b | 19.33 ± 0.18 | 24.99 ± 0.19 | 7.87 ± 0.11 | 2.17 ± 0.02 |
| 8c | 9.10 ± 0.21 | 4.65 ± 0.56 | 14.11 ± 0.32 | 4.73 ± 0.47 |
| 8d | 22.70 ± 0.27 | 22.45 ± 0.05 | 19.35 ± 0.07 | 14.48 ± 0.24 |
| 8e | 40.58 ± 0.36 | 11.50 ± 0.17 | 14.52 ± 0.25 | 7.70 ± 0.27 |
| 8f | 30.10 ± 0.39 | 17.72 ± 0.12 | 28.29 ± 0.33 | 27.19 ± 0.15 |
| 8g | 18.28 ± 0.03 | 7.59 ± 0.72 | 26.89 ± 0.16 | 27.46 ± 0.16 |
| 8h | 28.47 ± 0.10 | 16.50 ± 0.97 | 21.64 ± 0.39 | 19.61 ± 0.34 |
| 8i | 55.62 ± 0.12 | 25.11 ± 0.05 | 31.67 ± 0.07 | 47.78 ± 0.19 |
| 9a | 42.87 ± 0.22 | 2.39 ± 0.09 | 20.73 ± 0.49 | 33.71 ± 0.13 |
| 9b | 18.65 ± 0.16 | 12.82 ± 0.01 | 8.80 ± 0.04 | 7.22 ± 0.34 |
| 9c | 5.905 ± 0.29 | 1.45 ± 0.86 | 15.11 ± 0.31 | 37.21 ± 0.08 |
| Cell Lines | IC50 (μM) j | |
|---|---|---|
| 5c | WP1066 k | |
| HeLa a | 3.10 ± 0.02 | 7.06 ± 1.17 |
| PC3 b | 20.50 ± 0.05 | 1.202 ± 0.03 |
| HCT116 c | 13.38 ± 0.04 | 7.22 ± 3.14 |
| A549 d | 35.27 ± 0.07 | 7.55 ± 0.06 |
| MDA-MB-231 e | 40.74 ± 0.12 | 8.17 ± 0.16 |
| B16 f | >80 | 24.09 ± 0.70 |
| SKOV3 g | 44.6 ± 0.01 | 4.66 ± 0.13 |
| Hep3B h | 34.77 ± 0.03 | 4.36 ± 0.05 |
| L02 i | 51.7 ± 0.10 | 3.45 ± 0.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, P.; Fan, D.; Qiao, W.; He, X.; Zhang, L.; Jiang, Y.; Yang, T. SAR Study and Molecular Mechanism Investigation of Novel Naphthoquinone-furan-2-cyanoacryloyl Hybrids with Antitumor Activity. Pharmaceutics 2022, 14, 2104. https://doi.org/10.3390/pharmaceutics14102104
Liu P, Fan D, Qiao W, He X, Zhang L, Jiang Y, Yang T. SAR Study and Molecular Mechanism Investigation of Novel Naphthoquinone-furan-2-cyanoacryloyl Hybrids with Antitumor Activity. Pharmaceutics. 2022; 14(10):2104. https://doi.org/10.3390/pharmaceutics14102104
Chicago/Turabian StyleLiu, Pingxian, Dongmei Fan, Wenliang Qiao, Xinlian He, Lidan Zhang, Yunhan Jiang, and Tao Yang. 2022. "SAR Study and Molecular Mechanism Investigation of Novel Naphthoquinone-furan-2-cyanoacryloyl Hybrids with Antitumor Activity" Pharmaceutics 14, no. 10: 2104. https://doi.org/10.3390/pharmaceutics14102104