
Disrupting GPCR Complexes with Smart Drug-like Peptides


Abstract
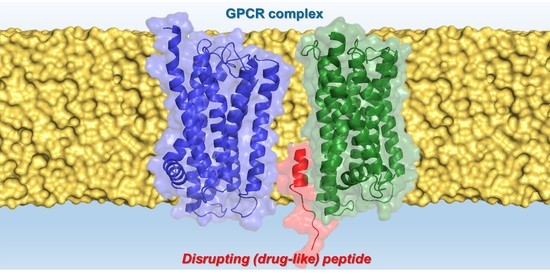
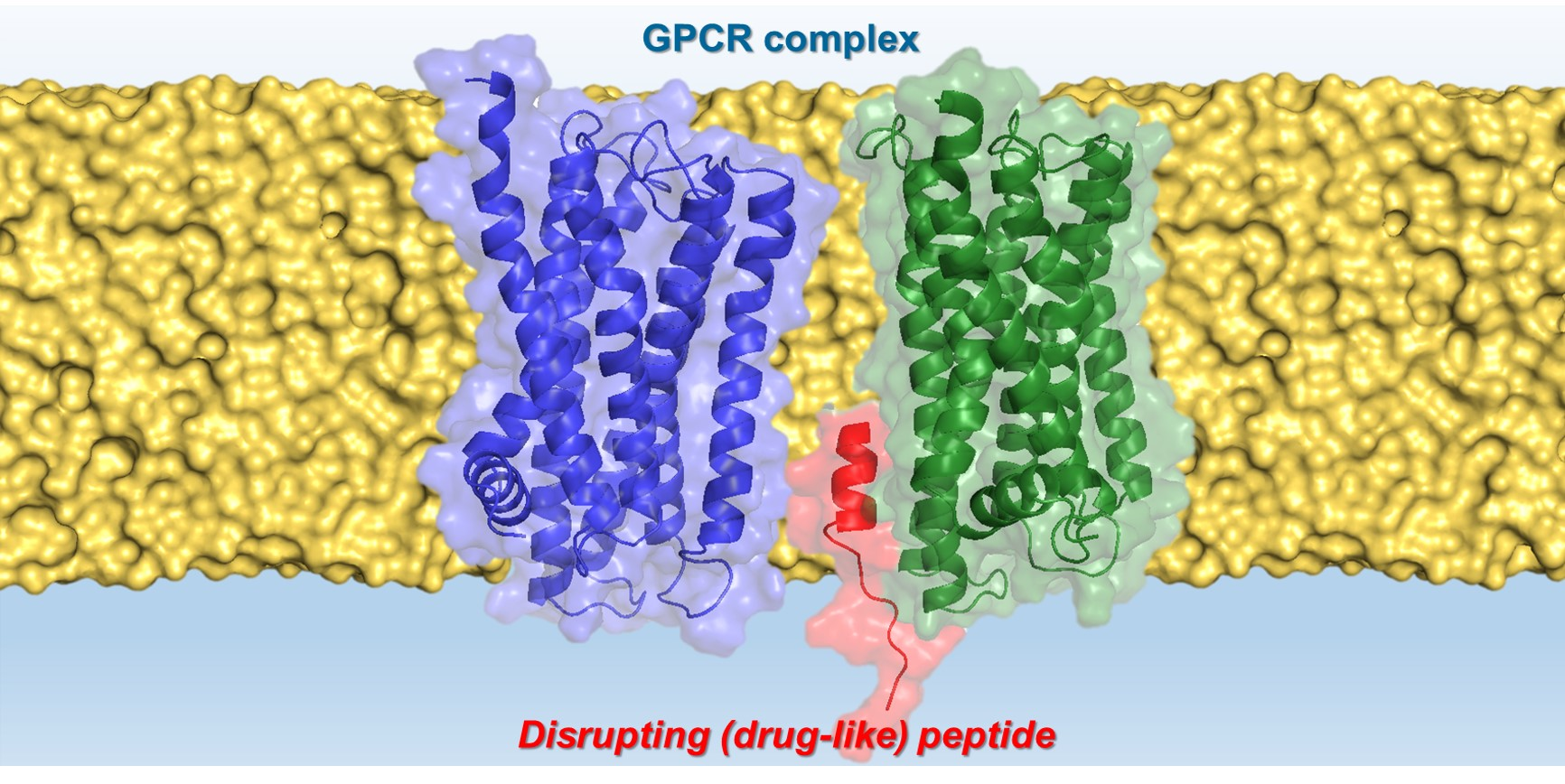
:
1. Introduction
2. GPCR Oligomers
3. Synthetic TM Peptides as Tools for GPCR Complex Exploration
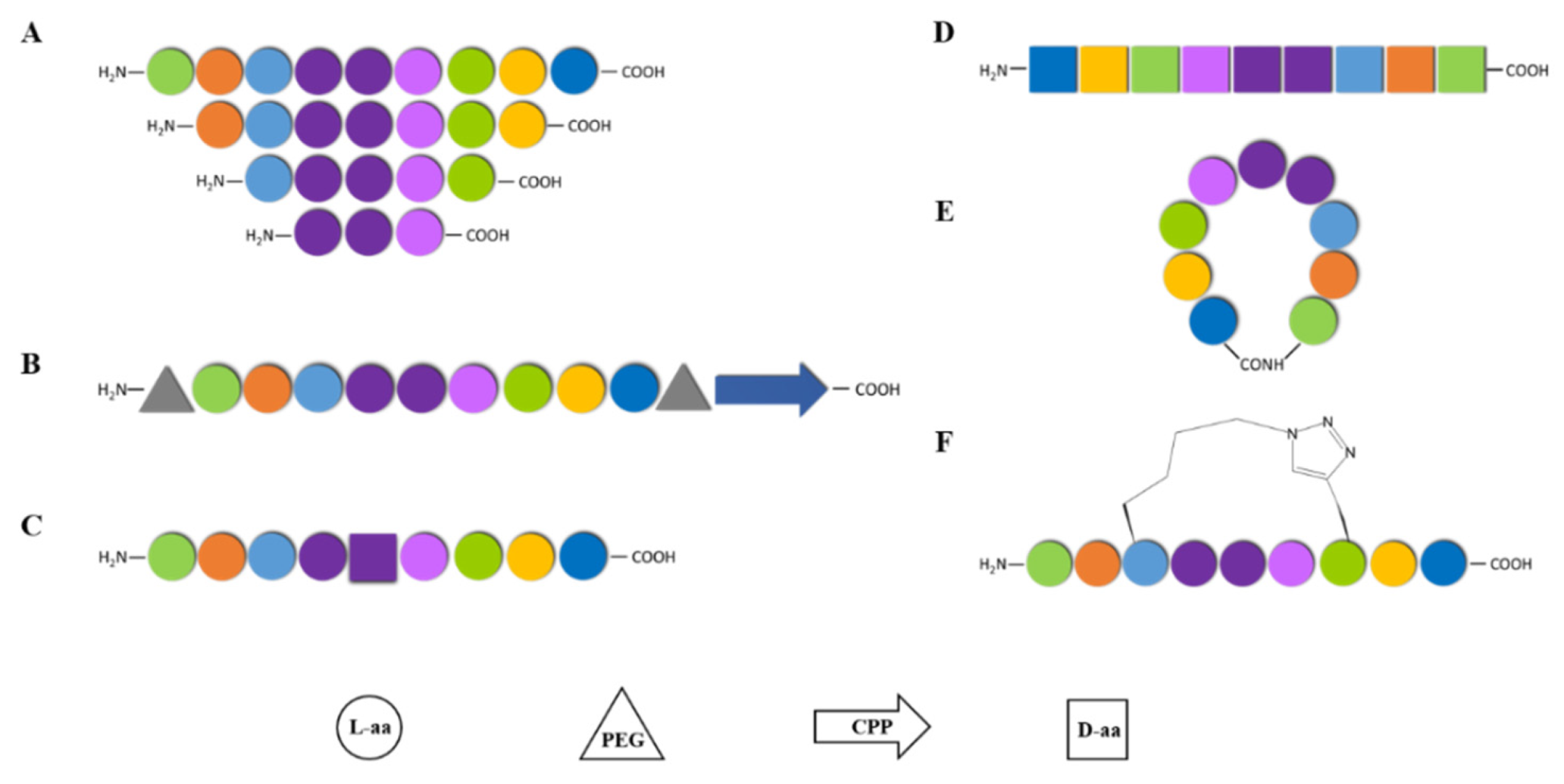
3.1. TM Peptides: Challenges and Opportunities to Drug the Undruggable
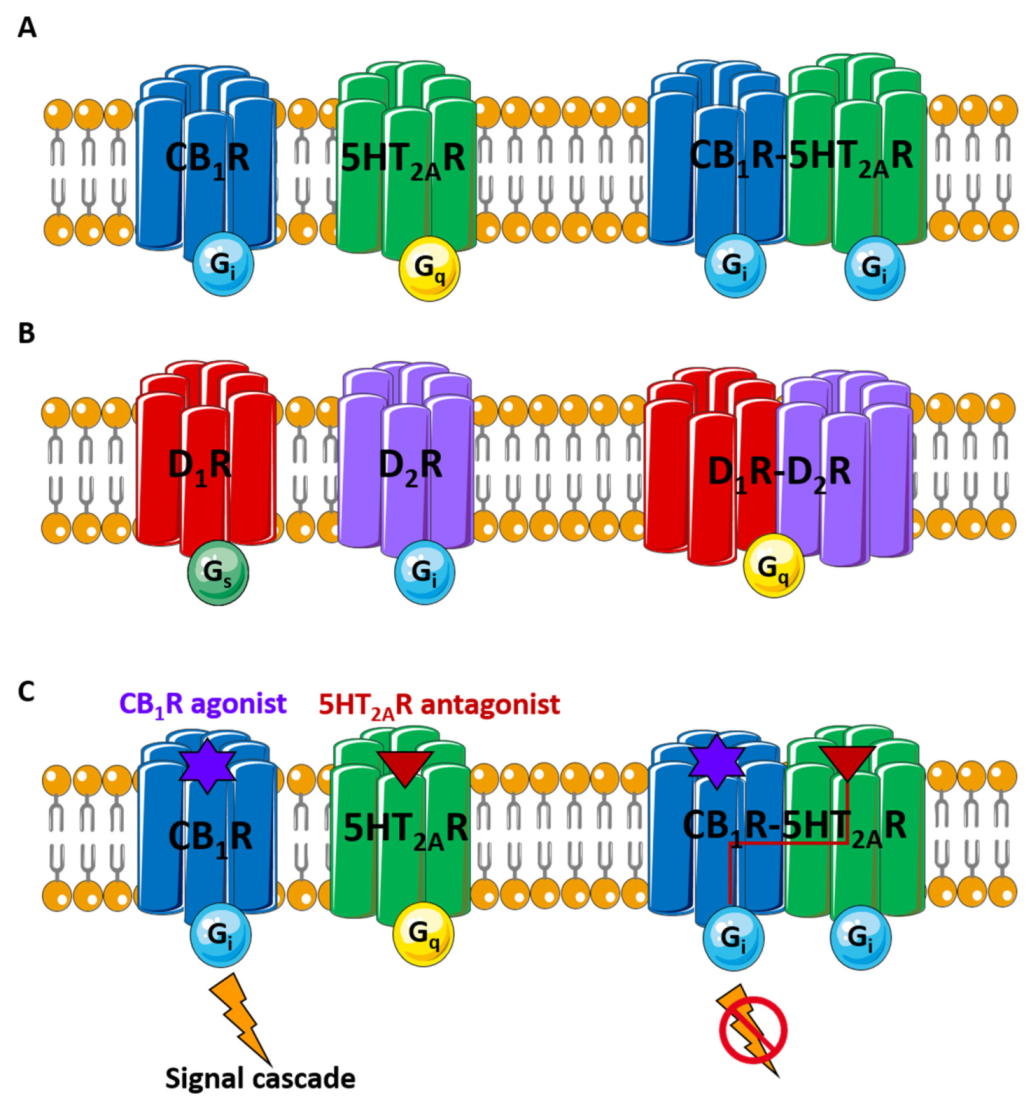
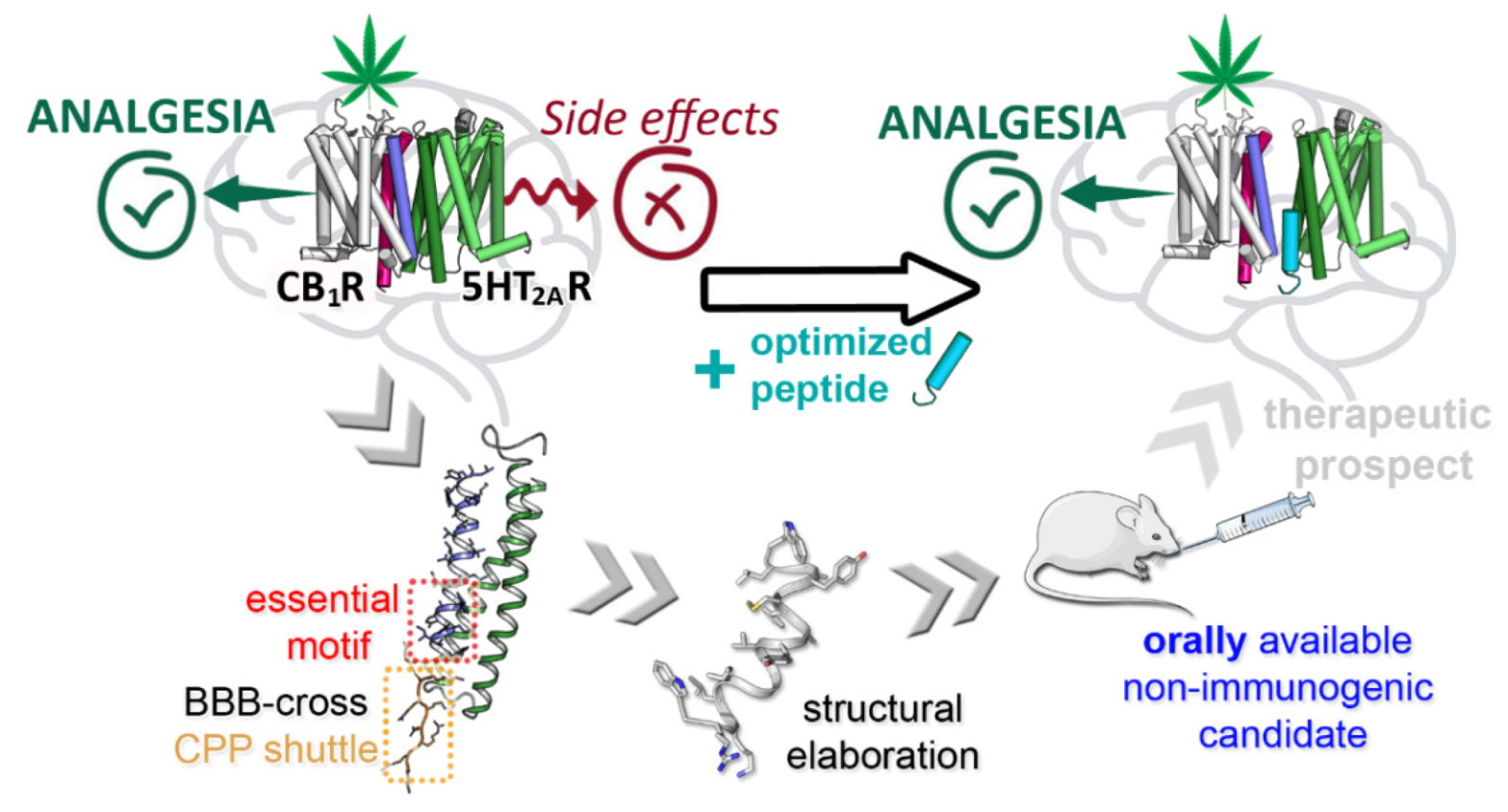
3.2. TM Peptides Restricting CB1R-5HT2AR Dimer for Cannabinoid Management in Pain Therapy
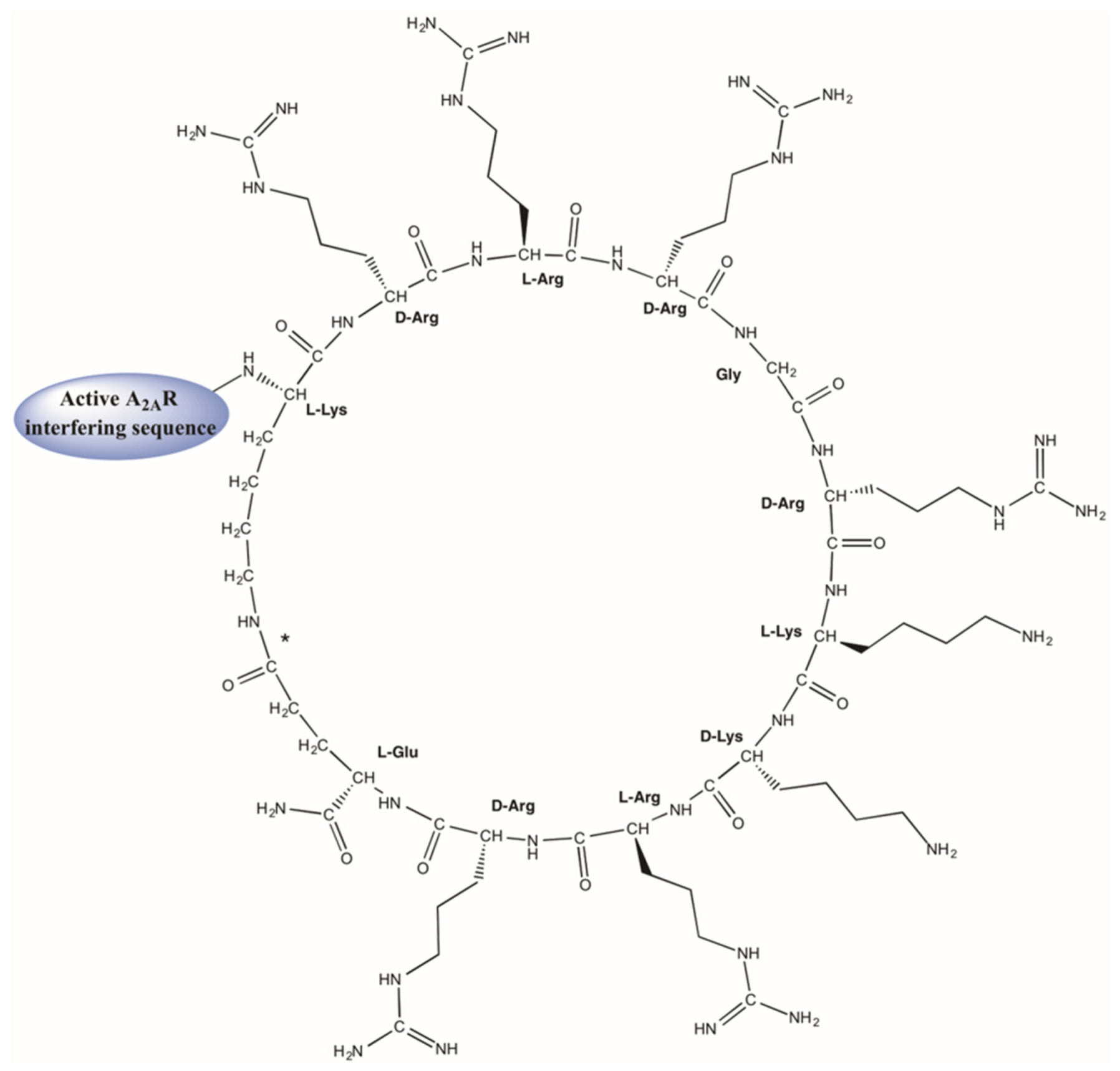
3.3. TM Peptide Restricting A2AR-A2AR Dimer for CNS Disorders
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. Historical review: A brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 2004, 25, 413–422. [Google Scholar] [CrossRef]
- Limbird, L.E.; Meyts, P.D.; Lefkowitz, R.J. Beta-adrenergic receptors: Evidence for negative cooperativity. Biochem. Biophys. Res. Commun. 1975, 64, 1160–1168. [Google Scholar] [CrossRef]
- Wettschureck, N.; Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwood, T.K.; Findlay, J.B. Fingerprinting G-protein-coupled receptors. Protein Eng. 1994, 7, 195–203. [Google Scholar] [CrossRef]
- Tuteja, N. Signaling through G protein coupled receptors. Plant Signal. Behav. 2009, 4, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Ferré, S.; Bonaventura, J.; Tomasi, D.; Navarro, G.; Moreno, E.; Cortés, A.; Lluís, C.; Casadó, V.; Volkow, N.D. Allosteric mechanisms within the adenosine A2A-dopamine D2 receptor heterotetramer. Neuropharmacology 2016, 104, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.L. G protein-coupled receptors as disease targets: Emerging paradigms. Ochsner J. 2010, 10, 2–7. [Google Scholar]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A. New paradigms in GPCR drug discovery. Biochem. Pharmacol. 2015, 98, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto Gutierrez, A.; McDonald, P.H. GPCRs: Emerging anti-cancer drug targets. Cell. Signal. 2018, 41, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.e19. [Google Scholar] [CrossRef] [Green Version]
- Glukhova, A.; Draper-Joyce, C.J.; Sunahara, R.K.; Christopoulos, A.; Wootten, D.; Sexton, P.M. Rules of Engagement: GPCRs and G Proteins. ACS Pharmacol. Transl. Sci. 2018, 1, 73–83. [Google Scholar] [CrossRef]
- Milligan, G.; Ward, R.J.; Marsango, S. GPCR homo-oligomerization. Curr. Opin. Cell Biol. 2019, 57, 40–47. [Google Scholar] [CrossRef]
- Fuxe, K.; Borroto-Escuela, D.O.; Marcellino, D.; Romero-Fernandez, W.; Frankowska, M.; Guidolin, D.; Filip, M.; Ferraro, L.; Woods, A.S.; Tarakanov, A.; et al. GPCR heteromers and their allosteric receptor-receptor interactions. Curr. Med. Chem. 2012, 19, 356–363. [Google Scholar] [CrossRef]
- Hebert, T.E.; Moffett, S.; Morello, J.P.; Loisel, T.P.; Bichet, D.G.; Barret, C.; Bouvier, M. A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J. Biol. Chem. 1996, 271, 16384–16392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhou, M.; Huang, W.; Yang, H. N-glycosylation of the β(2) adrenergic receptor regulates receptor function by modulating dimerization. FEBS J. 2017, 284, 2004–2018. [Google Scholar] [CrossRef] [Green Version]
- Bagher, A.M.; Young, A.P.; Laprairie, R.B.; Toguri, J.T.; Kelly, M.E.M.; Denovan-Wright, E.M. Heteromer formation between cannabinoid type 1 and dopamine type 2 receptors is altered by combination cannabinoid and antipsychotic treatments. J. Neurosci. Res. 2020, 98, 2496–2509. [Google Scholar] [CrossRef]
- Ferré, S.; Ciruela, F. Functional and Neuroprotective Role of Striatal Adenosine A(2A) Receptor Heterotetramers. J. Caffeine Adenosine Res. 2019, 9, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bono, F.; Mutti, V.; Fiorentini, C.; Missale, C. Dopamine D3 Receptor Heteromerization: Implications for Neuroplasticity and Neuroprotection. Biomolecules 2020, 10, 1016. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Tarakanov, A.O.; Brito, I.; Fuxe, K. Glutamate heteroreceptor complexes in the brain. Pharmacol. Rep. 2018, 70, 936–950. [Google Scholar] [CrossRef]
- Bontempi, L.; Savoia, P.; Bono, F.; Fiorentini, C.; Missale, C. Dopamine D3 and acetylcholine nicotinic receptor heteromerization in midbrain dopamine neurons: Relevance for neuroplasticity. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2017, 27, 313–324. [Google Scholar] [CrossRef]
- Guitart, X.; Navarro, G.; Moreno, E.; Yano, H.; Cai, N.-S.; Sánchez-Soto, M.; Kumar-Barodia, S.; Naidu, Y.T.; Mallol, J.; Cortés, A.; et al. Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: The dopamine D1-D3 receptor heterotetramer. Mol. Pharmacol. 2014, 86, 417–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, N.-S.; Quiroz, C.; Bonaventura, J.; Bonifazi, A.; Cole, T.O.; Purks, J.; Billing, A.S.; Massey, E.; Wagner, M.; Wish, E.D.; et al. Opioid-galanin receptor heteromers mediate the dopaminergic effects of opioids. J. Clin. Investig. 2019, 129, 2730–2744. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hua, T.; Liu, Z.-J. Structural features of activated GPCR signaling complexes. Curr. Opin. Struct. Biol. 2020, 63, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Ferraro, L.; Narvaez, M.; Tanganelli, S.; Beggiato, S.; Liu, F.; Rivera, A.; Fuxe, K. Multiple Adenosine-Dopamine (A2A-D2 Like) Heteroreceptor Complexes in the Brain and Their Role in Schizophrenia. Cells 2020, 9, 1077. [Google Scholar] [CrossRef]
- Perreault, M.L.; Hasbi, A.; O’Dowd, B.F.; George, S.R. Heteromeric dopamine receptor signaling complexes: Emerging neurobiology and disease relevance. Neuropsychopharmacology 2014, 39, 156–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farran, B. An update on the physiological and therapeutic relevance of GPCR oligomers. Pharmacol. Res. 2017, 117, 303–327. [Google Scholar] [CrossRef]
- Jordan, B.A.; Devi, L.A. G-protein-coupled receptor heterodimerization modulates receptor function. Nature 1999, 399, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Sun, Q.; Zhao, H.; Rovira, X.; Gai, S.; He, Q.; Pin, J.-P.; Liu, J.; Rondard, P. Rearrangement of the transmembrane domain interfaces associated with the activation of a GPCR hetero-oligomer. Nat. Commun. 2019, 10, 2765. [Google Scholar] [CrossRef] [Green Version]
- Kasai, R.S.; Ito, S.V.; Awane, R.M.; Fujiwara, T.K.; Kusumi, A. The Class-A GPCR Dopamine D2 Receptor Forms Transient Dimers Stabilized by Agonists: Detection by Single-Molecule Tracking. Cell Biochem. Biophys. 2018, 76, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabor, A.; Weisenburger, S.; Banerjee, A.; Purkayastha, N.; Kaindl, J.M.; Hübner, H.; Wei, L.; Grömer, T.W.; Kornhuber, J.; Tschammer, N.; et al. Visualization and ligand-induced modulation of dopamine receptor dimerization at the single molecule level. Sci. Rep. 2016, 6, 33233. [Google Scholar] [CrossRef]
- Aslanoglou, D.; Alvarez-Curto, E.; Marsango, S.; Milligan, G. Distinct Agonist Regulation of Muscarinic Acetylcholine M2-M3 Heteromers and Their Corresponding Homomers. J. Biol. Chem. 2015, 290, 14785–14796. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Westfield, G.; Erickson, J.W.; Cerione, R.A.; Skiniotis, G.; Ramachandran, S. Isolation and structure-function characterization of a signaling-active rhodopsin-G protein complex. J. Biol. Chem. 2017, 292, 14280–14289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, G.; Cordomi, A.; Zelman-Femiak, M.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Perez-Benito, L.; Cortes, A.; Casado, V.; Mallol, J.; et al. Quaternary structure of a G-protein-coupled receptor heterotetramer in complex with Gi and Gs. BMC Biol. 2016, 14, 26. [Google Scholar] [CrossRef] [Green Version]
- Cordomí, A.; Navarro, G.; Aymerich, M.S.; Franco, R. Structures for G-Protein-Coupled Receptor Tetramers in Complex with G Proteins. Trends Biochem. Sci. 2015, 40, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Deganutti, G.; Salmaso, V.; Moro, S. Could Adenosine Recognize its Receptors with a Stoichiometry Other than 1:1? Mol. Inform. 2018, 37, e1800009. [Google Scholar] [CrossRef]
- Vinals, X.; Moreno, E.; Lanfumey, L.; Cordomi, A.; Pastor, A.; de La Torre, R.; Gasperini, P.; Navarro, G.; Howell, L.A.; Pardo, L.; et al. Cognitive Impairment Induced by Delta9-tetrahydrocannabinol Occurs through Heteromers between Cannabinoid CB1 and Serotonin 5-HT2A Receptors. PLoS Biol. 2015, 13, e1002194. [Google Scholar] [CrossRef] [Green Version]
- Rashid, A.J.; So, C.H.; Kong, M.M.C.; Furtak, T.; El-Ghundi, M.; Cheng, R.; O’Dowd, B.F.; George, S.R. D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc. Natl. Acad. Sci. USA 2007, 104, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Bellot, M.; Galandrin, S.; Boularan, C.; Matthies, H.J.; Despas, F.; Denis, C.; Javitch, J.; Mazères, S.; Sanni, S.J.; Pons, V.; et al. Dual agonist occupancy of AT1-R-α2C-AR heterodimers results in atypical Gs-PKA signaling. Nat. Chem. Biol. 2015, 11, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, K.; Benleulmi-Chaachoua, A.; Journé, A.-S.; Kamal, M.; Guillaume, J.-L.; Dussaud, S.; Gbahou, F.; Yettou, K.; Liu, C.; Contreras-Alcantara, S.; et al. Heteromeric MT1/MT2 melatonin receptors modulate photoreceptor function. Sci. Signal. 2013, 6, ra89. [Google Scholar] [CrossRef] [Green Version]
- Smith, N.J.; Milligan, G. Allostery at G protein-coupled receptor homo- and heteromers: Uncharted pharmacological landscapes. Pharmacol. Rev. 2010, 62, 701–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León-Navarro, D.A.; Albasanz, J.L.; Martín, M. Functional Cross-Talk between Adenosine and Metabotropic Glutamate Receptors. Curr. Neuropharmacol. 2019, 17, 422–437. [Google Scholar] [CrossRef]
- Galvez, T.; Duthey, B.; Kniazeff, J.; Blahos, J.; Rovelli, G.; Bettler, B.; Prézeau, L.; Pin, J.P. Allosteric interactions between GB1 and GB2 subunits are required for optimal GABA(B) receptor function. EMBO J. 2001, 20, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Jordan, B.A.; Gupta, A.; Trapaidze, N.; Nagy, V.; Devi, L.A. Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J. Neurosci. 2000, 20, RC110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocheville, M.; Lange, D.C.; Kumar, U.; Patel, S.C.; Patel, R.C.; Patel, Y.C. Receptors for dopamine and somatostatin: Formation of hetero-oligomers with enhanced functional activity. Science 2000, 288, 154–157. [Google Scholar] [CrossRef]
- Franco, R.; Ferre, S.; Agnati, L.; Torvinen, M.; Gines, S.; Hillion, J.; Casado, V.; Lledo, P.-M.; Zoli, M.; Lluis, C.; et al. Evidence for Adenosine/Dopamine Receptor Interactions: Indications for Heteromerization. Neuropsychopharmacology 2000, 23, S50–S59. [Google Scholar] [CrossRef]
- Navarro, G.; Quiroz, C.; Moreno-Delgado, D.; Sierakowiak, A.; McDowell, K.; Moreno, E.; Rea, W.; Cai, N.-S.; Aguinaga, D.; Howell, L.A.; et al. Orexin-corticotropin-releasing factor receptor heteromers in the ventral tegmental area as targets for cocaine. J. Neurosci. 2015, 35, 6639–6653. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Pinilla, E.; Rodríguez-Pérez, A.I.; Navarro, G.; Aguinaga, D.; Moreno, E.; Lanciego, J.L.; Labandeira-García, J.L.; Franco, R. Dopamine D2 and angiotensin II type 1 receptors form functional heteromers in rat striatum. Biochem. Pharmacol. 2015, 96, 131–142. [Google Scholar] [CrossRef]
- Young, B.M.; Nguyen, E.; Chedrawe, M.A.J.; Rainey, J.K.; Dupré, D.J. Differential Contribution of Transmembrane Domains IV, V, VI, and VII to Human Angiotensin II Type 1 Receptor Homomer Formation. J. Biol. Chem. 2017, 292, 3341–3350. [Google Scholar] [CrossRef] [Green Version]
- Borroto-Escuela, D.O.; Wydra, K.; Li, X.; Rodriguez, D.; Carlsson, J.; Jastrzębska, J.; Filip, M.; Fuxe, K. Disruption of A2AR-D2R Heteroreceptor Complexes After A2AR Transmembrane 5 Peptide Administration Enhances Cocaine Self-Administration in Rats. Mol. Neurobiol. 2018, 55, 7038–7048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.T.O.; Ng, S.Y.L.; Chu, J.Y.S.; Sekar, R.; Harikumar, K.G.; Miller, L.J.; Chow, B.K.C. Transmembrane peptides as unique tools to demonstrate the in vivo action of a cross-class GPCR heterocomplex. FASEB J. 2014, 28, 2632–2644. [Google Scholar] [CrossRef] [Green Version]
- Harikumar, K.G.; Pinon, D.I.; Miller, L.J. Transmembrane segment IV contributes a functionally important interface for oligomerization of the Class II G protein-coupled secretin receptor. J. Biol. Chem. 2007, 282, 30363–30372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidolin, D.; Marcoli, M.; Tortorella, C.; Maura, G.; Agnati, L.F. Receptor-Receptor Interactions as a Widespread Phenomenon: Novel Targets for Drug Development? Front. Endocrinol. 2019, 10, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, T.C.; Hottin, J.; Clerté, C.; Zwier, J.M.; Durroux, T.; Rondard, P.; Prézeau, L.; Royer, C.A.; Pin, J.-P.; Margeat, E.; et al. Oligomerization of a G protein-coupled receptor in neurons controlled by its structural dynamics. Sci. Rep. 2018, 8, 10414. [Google Scholar] [CrossRef] [Green Version]
- Guidolin, D.; Marcoli, M.; Tortorella, C.; Maura, G.; Agnati, L.F. G protein-coupled receptor-receptor interactions give integrative dynamics to intercellular communication. Rev. Neurosci. 2018, 29, 703–726. [Google Scholar] [CrossRef]
- Köfalvi, A.; Moreno, E.; Cordomí, A.; Cai, N.-S.; Fernández-Dueñas, V.; Ferreira, S.G.; Guixà-González, R.; Sánchez-Soto, M.; Yano, H.; Casadó-Anguera, V.; et al. Control of glutamate release by complexes of adenosine and cannabinoid receptors. BMC Biol. 2020, 18, 9. [Google Scholar] [CrossRef]
- Wan, L.; Xu, F.; Liu, C.; Ji, B.; Zhang, R.; Wang, P.; Wu, F.; Pan, Y.; Yang, C.; Wang, C.; et al. Transmembrane peptide 4 and 5 of APJ are essential for its heterodimerization with OX1R. Biochem. Biophys. Res. Commun. 2020, 521, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Bai, B.; Zhang, R.; Wang, C.; Chen, J. Apelin receptor homodimer-oligomers revealed by single-molecule imaging and novel G protein-dependent signaling. Sci. Rep. 2017, 7, 40335. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Cordomí, A.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Pérez-Benito, L.; Ferre, S.; Cortés, A.; Casadó, V.; Mallol, J.; et al. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- McMillin, S.M.; Heusel, M.; Liu, T.; Costanzi, S.; Wess, J. Structural basis of M3 muscarinic receptor dimer/oligomer formation. J. Biol. Chem. 2011, 286, 28584–28598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harikumar, K.G.; Dong, M.; Cheng, Z.; Pinon, D.I.; Lybrand, T.P.; Miller, L.J. Transmembrane Segment Peptides Can Disrupt Cholecystokinin Receptor Oligomerization without affecting Receptor Function. Biochemistry 2006, 45, 14706–14716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernanz-Falcón, P.; Rodríguez-Frade, J.M.; Serrano, A.; Juan, D.; del Sol, A.; Soriano, S.F.; Roncal, F.; Gómez, L.; Valencia, A.; Martínez-A, C.; et al. Identification of amino acid residues crucial for chemokine receptor dimerization. Nat. Immunol. 2004, 5, 216–223. [Google Scholar] [CrossRef]
- Jastrzebska, B.; Chen, Y.; Orban, T.; Jin, H.; Hofmann, L.; Palczewski, K. Disruption of Rhodopsin Dimerization with Synthetic Peptides Targeting an Interaction Interface. J. Biol. Chem. 2015, 290, 25728–25744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, J.; Wright, S.C.; Rodríguez, D.; Matricon, P.; Lahav, N.; Vromen, A.; Friedler, A.; Strömqvist, J.; Wennmalm, S.; Carlsson, J.; et al. Agonist-induced dimer dissociation as a macromolecular step in G protein-coupled receptor signaling. Nat. Commun. 2017, 8, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.-Q.; Zhang, Z.-N.; Guan, J.-S.; Liu, H.-R.; Zhao, B.; Wang, H.-B.; Li, Q.; Yang, H.; Luo, J.; Li, Z.-Y.; et al. Facilitation of mu-opioid receptor activity by preventing delta-opioid receptor-mediated codegradation. Neuron 2011, 69, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide chemistry toolbox—Transforming natural peptides into peptide therapeutics. Bioorg. Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef]
- Ross, J.N.; Fields, F.R.; Kalwajtys, V.R.; Gonzalez, A.J.; O’Connor, S.; Zhang, A.; Moran, T.E.; Hammers, D.E.; Carothers, K.E.; Lee, S.W. Synthetic Peptide Libraries Designed From a Minimal Alpha-Helical Domain of AS-48-Bacteriocin Homologs Exhibit Potent Antibacterial Activity. Front. Microbiol. 2020, 11, 2714. [Google Scholar] [CrossRef]
- Zhang, X.; Bathgate, R.A.D.; Hossain, M.A. Human Insulin-like Peptide 5 (INSL5). Identification of a Simplified Version of Two-Chain Analog A13. ACS Med. Chem. Lett. 2020, 11, 2455–2460. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, J.; Rossi, G.C.; Majumdar, S.; Pasternak, G.W.; Pan, Y.-X. Mediation of opioid analgesia by a truncated 6-transmembrane GPCR. J. Clin. Investig. 2015, 125, 2626–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, D.; Dewangan, H.K. PEGYLATION: An important approach for novel drug delivery system. J. Biomater. Sci. Polym. Ed. 2021, 32, 266–280. [Google Scholar] [CrossRef]
- Manteghi, R.; Pallagi, E.; Olajos, G.; Csóka, I. Pegylation and formulation strategy of Anti-Microbial Peptide (AMP) according to the quality by design approach. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2020, 144, 105197. [Google Scholar] [CrossRef] [PubMed]
- Bhosle, G.S.; Nawale, L.; Yeware, A.M.; Sarkar, D.; Fernandes, M. Antibacterial and anti-TB tat-peptidomimetics with improved efficacy and half-life. Eur. J. Med. Chem. 2018, 152, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xu, H.; Xia, J.; Ma, J.; Xu, J.; Li, Y.; Feng, J. D- and Unnatural Amino Acid Substituted Antimicrobial Peptides With Improved Proteolytic Resistance and Their Proteolytic Degradation Characteristics. Front. Microbiol. 2020, 11, 2869. [Google Scholar] [CrossRef] [PubMed]
- Doti, N.; Mardirossian, M.; Sandomenico, A.; Ruvo, M.; Caporale, A. Recent Applications of Retro-Inverso Peptides. Int. J. Mol. Sci. 2021, 22, 8677. [Google Scholar] [CrossRef]
- Arranz-Gibert, P.; Ciudad, S.; Seco, J.; García, J.; Giralt, E.; Teixidó, M. Immunosilencing peptides by stereochemical inversion and sequence reversal: Retro-D-peptides. Sci. Rep. 2018, 8, 6446. [Google Scholar] [CrossRef]
- Li, X.; Wang, S.; Zhu, X.; Zhangsun, D.; Wu, Y.; Luo, S. Effects of Cyclization on Activity and Stability of α-Conotoxin TxIB. Mar. Drugs 2020, 18, 180. [Google Scholar] [CrossRef] [Green Version]
- Gallo, M.; Defaus, S.; Andreu, D. 1988–2018: Thirty years of drug smuggling at the nano scale. Challenges and opportunities of cell-penetrating peptides in biomedical research. Arch. Biochem. Biophys. 2018, 661, 74–86. [Google Scholar] [CrossRef]
- Aziz, Z.A.B.A.; Ahmad, A.; Mohd-Setapar, S.H.; Hassan, H.; Lokhat, D.; Kamal, M.A.; Ashraf, G.M. Recent Advances in Drug Delivery of Polymeric Nano-Micelles. Curr. Drug Metab. 2017, 18, 16–29. [Google Scholar] [CrossRef]
- Cao, S.-J.; Xu, S.; Wang, H.-M.; Ling, Y.; Dong, J.; Xia, R.-D.; Sun, X.-H. Nanoparticles: Oral Delivery for Protein and Peptide Drugs. AAPS PharmSciTech 2019, 20, 190. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, A.; Raines, R.T. An evaluation of peptide-bond isosteres. Chembiochem 2011, 12, 1801–1807. [Google Scholar] [CrossRef] [Green Version]
- Altman, R.A.; Sharma, K.K.; Rajewski, L.G.; Toren, P.C.; Baltezor, M.J.; Pal, M.; Karad, S.N. Tyr(1)-ψ[(Z)CF═CH]-Gly(2) Fluorinated Peptidomimetic Improves Distribution and Metabolism Properties of Leu-Enkephalin. ACS Chem. Neurosci. 2018, 9, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Maisel, S.A.; Broka, D.; Atwell, B.; Bunch, T.; Kupp, R.; Singh, S.K.; Mehta, S.; Schroeder, J. Stapled EGFR peptide reduces inflammatory breast cancer and inhibits additional HER-driven models of cancer. J. Transl. Med. 2019, 17, 201. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, F.; Campiglio, M.; Jo, H.; Abderemane-Ali, F.; Rumpf, C.H.; Pope, L.; Rossen, N.D.; Flucher, B.E.; DeGrado, W.F.; Minor, D.L.J. Stapled Voltage-Gated Calcium Channel (Ca(V)) α-Interaction Domain (AID) Peptides Act As Selective Protein-Protein Interaction Inhibitors of Ca(V) Function. ACS Chem. Neurosci. 2017, 8, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Moiola, M.; Memeo, M.G.; Quadrelli, P. Stapled Peptides-A Useful Improvement for Peptide-Based Drugs. Molecules 2019, 24, 3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botta, J.; Bibic, L.; Killoran, P.; McCormick, P.J.; Howell, L.A. Design and development of stapled transmembrane peptides that disrupt the activity of G-protein-coupled receptor oligomers. J. Biol. Chem. 2019, 294, 16587–16603. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.E.; Campbell, F. Cannabinoids for treatment of chronic non-cancer pain; a systematic review of randomized trials. Br. J. Clin. Pharmacol. 2011, 72, 735–744. [Google Scholar] [CrossRef]
- Skrabek, R.Q.; Galimova, L.; Ethans, K.; Perry, D. Nabilone for the treatment of pain in fibromyalgia. J. Pain 2008, 9, 164–173. [Google Scholar] [CrossRef]
- Ware, M.A.; Fitzcharles, M.-A.; Joseph, L.; Shir, Y. The effects of nabilone on sleep in fibromyalgia: Results of a randomized controlled trial. Anesth. Analg. 2010, 110, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Hampson, R.E.; Deadwyler, S.A. Cannabinoids, hippocampal function and memory. Life Sci. 1999, 65, 715–723. [Google Scholar] [CrossRef]
- Busquets-Garcia, A.; Puighermanal, E.; Pastor, A.; de la Torre, R.; Maldonado, R.; Ozaita, A. Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol. Psychiatry 2011, 70, 479–486. [Google Scholar] [CrossRef]
- Puighermanal, E.; Busquets-Garcia, A.; Maldonado, R.; Ozaita, A. Cellular and intracellular mechanisms involved in the cognitive impairment of cannabinoids. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3254–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, F.A.; Lutz, B. The endocannabinoid system: Emotion, learning and addiction. Addict. Biol. 2008, 13, 196–212. [Google Scholar] [CrossRef]
- Gallo, M.; Moreno, E.; Defaus, S.; Ortega-Alvaro, A.; Gonzalez, A.; Robledo, P.; Cavaco, M.; Neves, V.; Castanho, M.A.R.B.; Casadó, V.; et al. Orally Active Peptide Vector Allows Using Cannabis to Fight Pain While Avoiding Side Effects. J. Med. Chem. 2021, 64, 6937–6948. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Navarro, G.; Franco, R.; Andreu, D. A2A Receptor Homodimer-Disrupting Sequence Efficiently Delivered by a Protease-Resistant, Cyclic CPP Vector. Int. J. Mol. Sci. 2019, 20, 4937. [Google Scholar] [CrossRef] [Green Version]
- Ferre, S.; Ciruela, F.; Borycz, J.; Solinas, M.; Quarta, D.; Antoniou, K.; Quiroz, C.; Justinova, Z.; Lluis, C.; Franco, R.; et al. Adenosine A1-A2A receptor heteromers: New targets for caffeine in the brain. Front. Biosci. 2008, 13, 2391–2399. [Google Scholar] [CrossRef] [Green Version]
- Ferré, S.; Quiroz, C.; Woods, A.S.; Cunha, R.; Popoli, P.; Ciruela, F.; Lluis, C.; Franco, R.; Azdad, K.; Schiffmann, S.N. An update on adenosine A2A-dopamine D2 receptor interactions: Implications for the function of G protein-coupled receptors. Curr. Pharm. Des. 2008, 14, 1468–1474. [Google Scholar] [CrossRef] [Green Version]
- Casadó-Anguera, V.; Bonaventura, J.; Moreno, E.; Navarro, G.; Cortés, A.; Ferré, S.; Casadó, V. Evidence for the heterotetrameric structure of the adenosine A2A-dopamine D2 receptor complex. Biochem. Soc. Trans. 2016, 44, 595–600. [Google Scholar] [CrossRef]
- Torvinen, M.; Marcellino, D.; Canals, M.; Agnati, L.F.; Lluis, C.; Franco, R.; Fuxe, K. Adenosine A2A receptor and dopamine D3 receptor interactions: Evidence of functional A2A/D3 heteromeric complexes. Mol. Pharmacol. 2005, 67, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Fernández-Dueñas, V.; López-Cano, M.; Taura, J.; Watanabe, M.; Ferrer, I.; Luján, R.; Ciruela, F. Adenosine A(2A)-Cannabinoid CB(1) Receptor Heteromers in the Hippocampus: Cannabidiol Blunts Δ(9)-Tetrahydrocannabinol-Induced Cognitive Impairment. Mol. Neurobiol. 2019, 56, 5382–5391. [Google Scholar] [CrossRef] [Green Version]
- Moreno, E.; Chiarlone, A.; Medrano, M.; Puigdellívol, M.; Bibic, L.; Howell, L.A.; Resel, E.; Puente, N.; Casarejos, M.J.; Perucho, J.; et al. Singular Location and Signaling Profile of Adenosine A(2A)-Cannabinoid CB(1) Receptor Heteromers in the Dorsal Striatum. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 964–977. [Google Scholar] [CrossRef] [Green Version]
- Márquez-Gómez, R.; Robins, M.T.; Gutiérrez-Rodelo, C.; Arias, J.-M.; Olivares-Reyes, J.-A.; van Rijn, R.M.; Arias-Montaño, J.-A. Functional histamine H(3) and adenosine A(2A) receptor heteromers in recombinant cells and rat striatum. Pharmacol. Res. 2018, 129, 515–525. [Google Scholar] [CrossRef]
- Łukasiewicz, S.; Błasiak, E.; Faron-Górecka, A.; Polit, A.; Tworzydło, M.; Górecki, A.; Wasylewski, Z.; Dziedzicka-Wasylewska, M. Fluorescence studies of homooligomerization of adenosine A2A and serotonin 5-HT1A receptors reveal the specificity of receptor interactions in the plasma membrane. Pharmacol. Rep. 2007, 59, 379–392. [Google Scholar] [PubMed]
- Ferre, S.; Ciruela, F.; Woods, A.S.; Canals, M.; Burgueno, J.; Marcellino, D.; Karcz-Kubicha, M.; Hope, B.T.; Morales, M.; Popoli, P.; et al. Glutamate mGlu5-Adenosine A2A-Dopamine D2 Receptor Interactions in the Striatum. Implications for Drug Therapy in Neuro-psychiatric Disorders and Drug Abuse. Curr. Med. Chem.-Cent. Nerv. Syst. Agents 2003, 3, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Ferré, S.; Goldberg, S.R.; Lluis, C.; Franco, R. Looking for the role of cannabinoid receptor heteromers in striatal function. Neuropharmacology 2009, 56 (Suppl. S1), 226–234. [Google Scholar] [CrossRef] [Green Version]
- Borroto-Escuela, D.O.; Hinz, S.; Navarro, G.; Franco, R.; Müller, C.E.; Fuxe, K. Understanding the Role of Adenosine A2AR Heteroreceptor Complexes in Neurodegeneration and Neuroinflammation. Front. Neurosci. 2018, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Atangcho, L.; Navaratna, T.; Thurber, G.M. Hitting Undruggable Targets: Viewing Stabilized Peptide Development through the Lens of Quantitative Systems Pharmacology. Trends Biochem. Sci. 2019, 44, 241–257. [Google Scholar] [CrossRef]
- Nischan, N.; Herce, H.D.; Natale, F.; Bohlke, N.; Budisa, N.; Cardoso, M.C.; Hackenberger, C.P.R. Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem. Int. Ed. Engl. 2015, 54, 1950–1953. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPCR Complex | TMs Involved in Dimerization | Synthetic TM Disruptor Peptide | In Vitro/In Vivo Assays Performed | Patho-Physiological Implication | Ref. |
|---|---|---|---|---|---|
| A2AR-D2R | TM4/5 interface | A2AR TM5 |
| Cocaine use | [53] |
| APJR-OX1R | TM4/5 interface | APJ TM4, TM5 |
| - | [60] |
| APJR homodimer | TM1, TM2, TM3, TM4 | TM1, TM2, TM3, TM4 |
| - | [61] |
| A2AR-CB1R | TM 5/6 interface | CB1R TM5 TM6 A2AR TM5 TM6 |
| Glutamate release | [59] |
| A1R-A2AR | TM 5/6 interface | A2AR TM4, TM5, TM6 A1R TM5 and TM6 |
| Neurodegeneration Neuroinflammation | [62] |
| CB1R-5HT2AR | TM 5/6 interface | CB1R TM5, TM6 |
| Cognitive impairment | [40] |
| M3R homodimer | TM1, TM5, TM7 | TM1-TM5-TM7 |
| - | [63] |
| CCKR homodimer | TM6 | TM6 |
| - | [64] |
| CCR5 homodimer | TM1, TM2, TM4 | TM1, TM4 |
| - | [65] |
| RhoR homodimer | TM1,TM2, TM4, TM5, H8 | TM1, TM2, TM4, TM5 |
| Phototransduction | [66] |
| β2AR homodimer | TM1, TM5, TM6, H8 | TM6 |
| - | [17] |
| SCTR | TM4 | TM4 |
| Liver diseases | [55] |
| AT1aR-SCTR | TM1/2 interface TM4/4 interface | AT1aR TM1, TM4 SCTR TM2, TM4 |
| Hyperosmolality-induced drinking | [54] |
| FZD6 homodimer | TM4, TM5 | TM4, TM5 |
| Cancer and neurologic disorders | [67] |
| MOR-DOR | MOR TM1 | MOR TM1 |
| Morphine tolerance | [68] |
| Heteromer | Ligand | Implication | Ref. |
|---|---|---|---|
| A1R-A2AR | Caffein (A1R, A2R antagonist) | Drug tolerance | [98] |
| A2AR-D2R | A2AR antagonists, D2R agonists | Parkinson’s disease, schizophrenia, drug addiction | [99,100] |
| D3R-A2AR | CGS-21680 (A2AR agonist) | Schizophrenia | [101] |
| CB1R-A2AR | CBD (CB1R agonist) | Cognitive impairment | [102,103] |
| A2AR-mGlu5R | CHPG (mGluR5 agonist) | Parkinson’s disease | [23] |
| A2AR-H3R | RAMH (H3R agonist) | Autism, obsessive and compulsive disorder | [104] |
| A2AR-5HT1AR | CGS 21,680 (A2AR agonist), 8-OH-DPAT (5HT1AR agonist), SCH 58,216 (A2AR antagonist), methysergide (5HT1AR antagonist) | Dyskinesia | [105] |
| A2AR-D2R-mGlu5R | A2AR agonists, A2AR antagonists, D2R agonists, D2R antagonists, mGlu5R agonists | Psychosis, Parkinson’s disease, drug abuse | [106] |
| CB1R-A2AR-D2R | TBD | Endocannabinoid modulation | [107] |
| A2AR-D2R-NMDAR | α-synuclein | Neurodegeneration, neuroinflammation | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo, M.; Defaus, S.; Andreu, D. Disrupting GPCR Complexes with Smart Drug-like Peptides. Pharmaceutics 2022, 14, 161. https://doi.org/10.3390/pharmaceutics14010161
Gallo M, Defaus S, Andreu D. Disrupting GPCR Complexes with Smart Drug-like Peptides. Pharmaceutics. 2022; 14(1):161. https://doi.org/10.3390/pharmaceutics14010161
Chicago/Turabian StyleGallo, Maria, Sira Defaus, and David Andreu. 2022. "Disrupting GPCR Complexes with Smart Drug-like Peptides" Pharmaceutics 14, no. 1: 161. https://doi.org/10.3390/pharmaceutics14010161