
3D-ALMOND-QSAR Models to Predict the Antidepressant Effect of Some Natural Compounds

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Natural Compounds Structures
2.2. Prediction of Compounds Drug- and Lead-Likeness Features
2.3. Computational Pharmacokinetics and Pharmacogenomics Profiles of Natural Compounds
2.4. Building 3D-ALMOND-QSAR to Predict Natural Compounds Effects
2.5. Molecular Docking Protocol
3. Results
3.1. Drug-Likeness, Pharmacokinetics, and Pharmacogenomics Profiles of Compounds
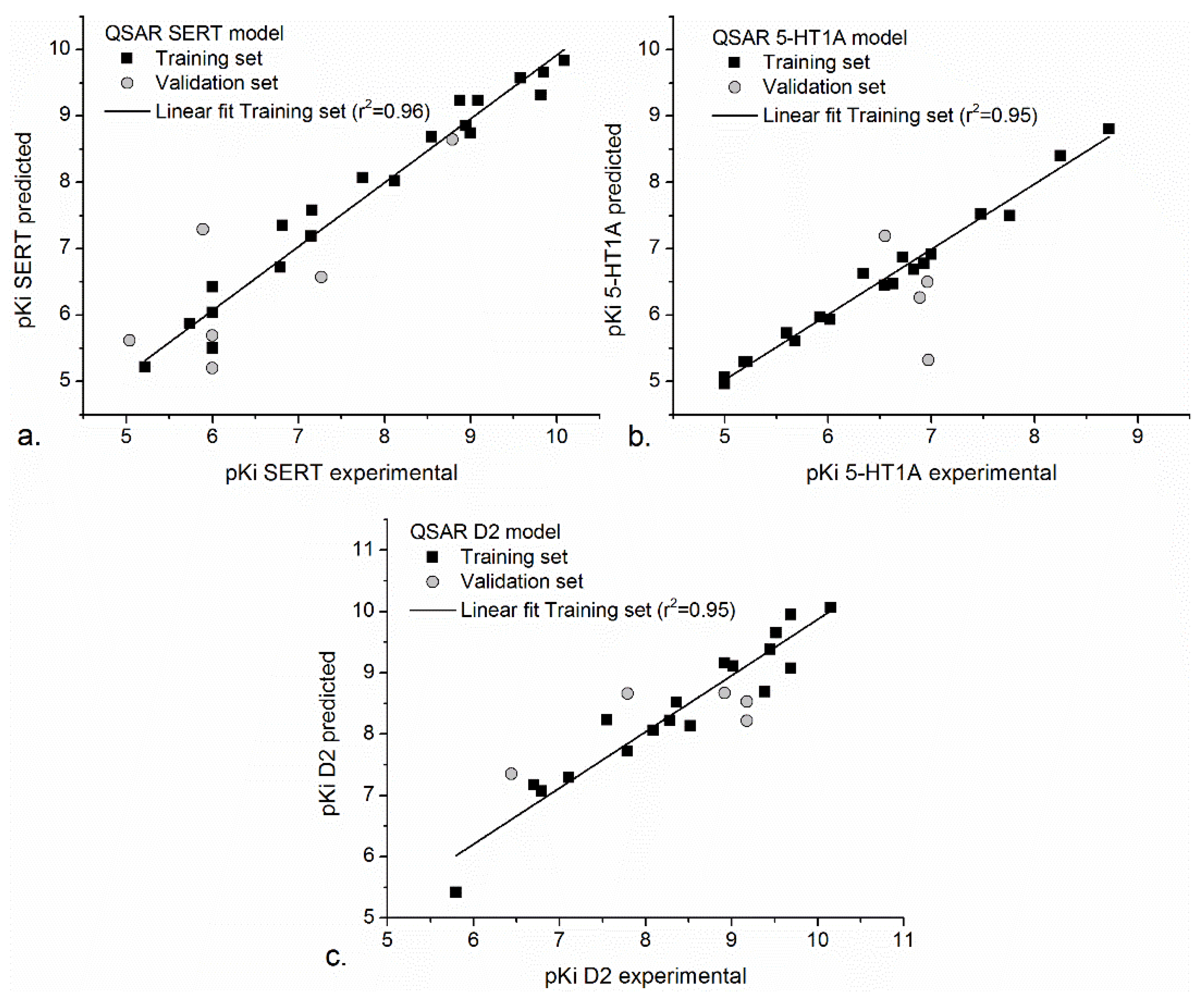
3.2. Natural Compounds’ Antidepressant Activities Predicted by 3D-ALMOND-QSAR
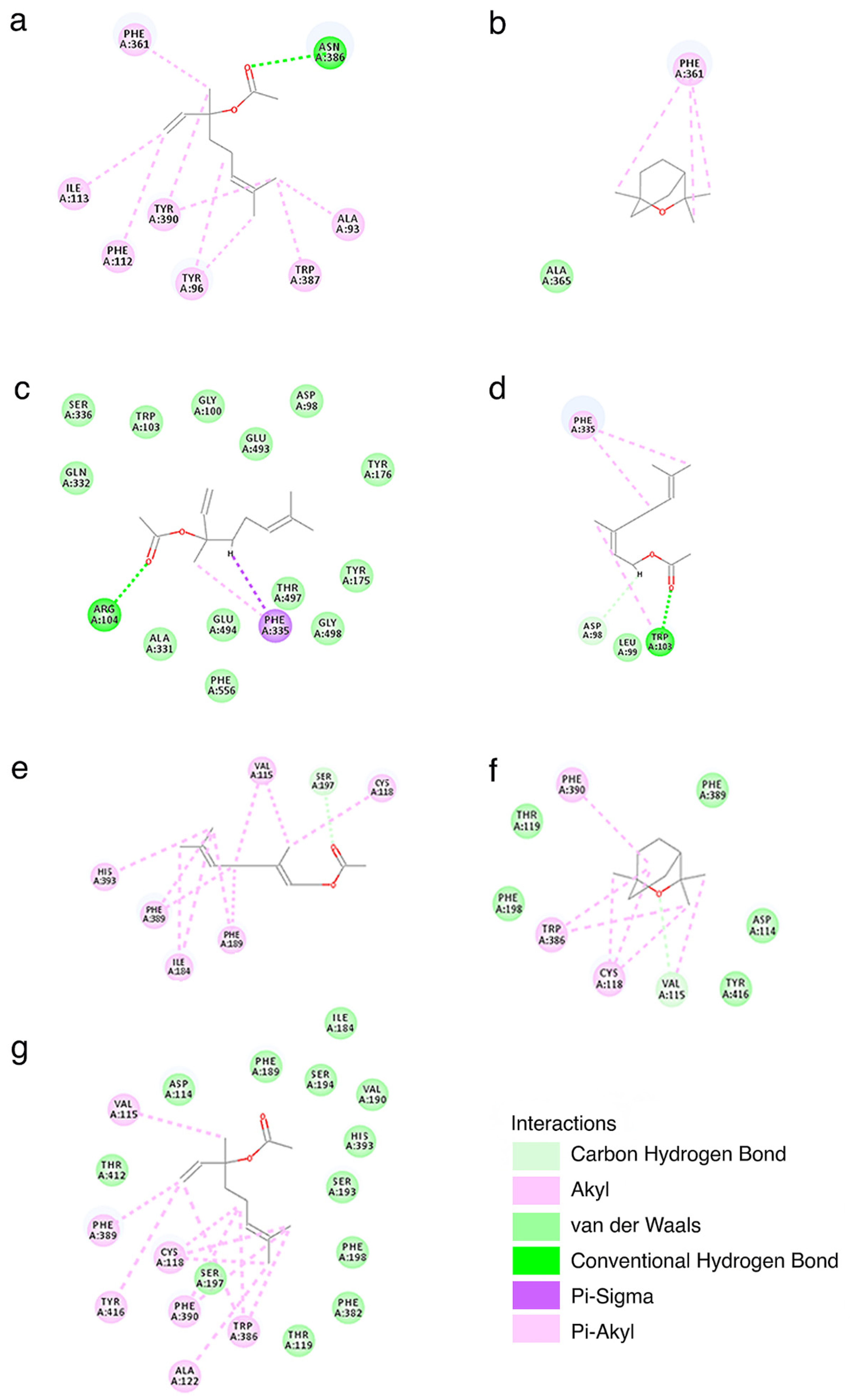
3.3. Molecular Docking
4. Discussion
4.1. Assessment of Compounds Drug-Likeness Features
4.2. Computational Pharmacokinetics and Pharmacogenomics Profiles of Natural Compounds
4.3. Predicted Pharmacodynamic Profiles of Natural Compounds on SERT, 5H-T1A, and D2 Active Sites by 3D-ALMOND-QSAR
4.4. Molecular Basis of the Interaction between Lead Compounds and Targets
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Health Organization (WHO) Depression. Available online: https://www.who.int/news-room/fact-sheets/detail/depression (accessed on 24 April 2021).
- Carvalho, A.F.; Sharma, M.S.; Brunoni, A.R.; Vieta, E.; Fava, G.A. The Safety, Tolerability and Risks Associated with the Use of Newer Generation Antidepressant Drugs: A Critical Review of the Literature. Psychother. Psychosom. 2016, 85, 270–288. [Google Scholar] [CrossRef]
- Yeung, K.S.; Hernandez, M.; Mao, J.J.; Haviland, I.; Gubili, J. Herbal medicine for depression and anxiety: A systematic review with assessment of potential psycho-oncologic relevance. Phytother. Res. 2018, 32, 865–891. [Google Scholar] [CrossRef]
- Udrea, A.-M.; Puia, A.; Shaposhnikov, S.; Avram, S. Computational approaches of new perspectives in the treatment of depression during pregnancy. Target 2018, 3, 680–687. [Google Scholar] [CrossRef]
- Yu, Y.-C.; Li, J.; Zhang, M.; Pan, J.-C.; Yu, Y.; Zhang, J.-B.; Zheng, L.; Si, J.-M.; Xu, Y. Resveratrol improves brain-gut axis by regulation of 5-HT-dependent signaling in the rat model of irritable bowel syndrome. Front. Cell. Neurosci. 2019, 13, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, A.; Beidler, J.; Hong, M.Y. Resveratrol and depression in animal models: A systematic review of the biological mechanisms. Molecules 2018, 23, 2197. [Google Scholar] [CrossRef] [Green Version]
- Anjaneyulu, M.; Chopra, K.; Kaur, I. Antidepressant Activity of Quercetin, a Bioflavonoid, in Streptozotocin-Induced Diabetic Mice. J. Med. Food 2003, 6, 391–395. [Google Scholar] [CrossRef]
- Samad, N.; Saleem, A.; Yasmin, F.; Shehzad, M. Quercetin protects against stress-induced anxiety-and depression-like behavior and improves memory in male mice. Physiol. Res. 2018, 67, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Bhutada, P.; Mundhada, Y.; Bansod, K.; Ubgade, A.; Quazi, M.; Umathe, S.; Mundhada, D. Reversal by quercetin of corticotrophin releasing factor induced anxiety- and depression-like effect in mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, Y.M.; Adu-Frimpong, M.; Xu, X.; Yu, J. Biochemical significance of limonene and its metabolites: Future prospects for designing and developing highly potent anticancer drugs. Biosci. Rep. 2018, 38, BSR20181253. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.; Liu, Q.F.; Choi, B.; Shin, C.; Lee, B.; Yuan, C.; Song, Y.J.; Yun, H.S.; Lee, I.-S.; Koo, B.-S. Neuroprotective effects of limonene (+) against Aβ42-induced neurotoxicity in a Drosophila model of Alzheimer’s disease. Biol. Pharm. Bull. 2020, 43, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Lorigooini, Z.; Boroujeni, S.N.; Sayyadi-Shahraki, M.; Rahimi-Madiseh, M.; Bijad, E.; Amini-Khoei, H. Limonene through Attenuation of Neuroinflammation and Nitrite Level Exerts Antidepressant-Like Effect on Mouse Model of Maternal Separation Stress. Behav. Neurol. 2021, 2021, 8817309. [Google Scholar] [CrossRef] [PubMed]
- Yun, J. Limonene inhibits methamphetamine-induced locomotor activity via regulation of 5-HT neuronal function and dopamine release. Phytomedicine 2014, 21, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Long, Y.; Yu, S.; Li, D.; Yang, M.; Guan, Y.; Zhang, D.; Wan, J.; Liu, S.; Shi, A. Natural volatile oils derived from herbal medicines: A promising therapy way for treating depressive disorder. Pharmacol. Res. 2020, 164, 105376. [Google Scholar] [CrossRef]
- Caputo, L.; Nazzaro, F.; Souza, L.F.; Aliberti, L.; De Martino, L.; Fratianni, F.; Coppola, R.; De Feo, V. Laurus nobilis: Composition of essential oil and its biological activities. Molecules 2017, 22, 930. [Google Scholar] [CrossRef] [PubMed]
- Fidan, H.; Stefanova, G.; Kostova, I.; Stankov, S.; Damyanova, S.; Stoyanova, A.; Zheljazkov, V.D. Chemical composition and antimicrobial activity of Laurus nobilis L. essential oils from Bulgaria. Molecules 2019, 24, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiresmaeili, A.; Roohollahi, S.; Mostafavi, A.; Askari, N. Effects of oregano essential oil on brain TLR4 and TLR2 gene expression and depressive-like behavior in a rat model. Res. Pharm. Sci. 2018, 13, 130. [Google Scholar] [PubMed]
- Juergens, U. Anti-inflammatory properties of the monoterpene 1.8-cineole: Current evidence for co-medication in inflammatory airway diseases. Drug Res. 2014, 64, 638–646. [Google Scholar] [CrossRef]
- Kim, K.Y.; Seo, H.J.; Min, S.S.; Park, M.; Seol, G.H. The effect of 1, 8-cineole inhalation on preoperative anxiety: A randomized clinical trial. Evid.-Based Complementary Altern. Med. 2014, 2014, 820126. [Google Scholar] [CrossRef] [Green Version]
- Dougnon, G.; Ito, M. Inhalation administration of the bicyclic ethers 1, 8-and 1, 4-cineole prevent anxiety and depressive-like behaviours in mice. Molecules 2020, 25, 1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, A.L.; González-Trujano, M.E.; Pellicer, F.; López-Muñoz, F.J.; Navarrete, A. Antinociceptive effect and GC/MS analysis of Rosmarinus officinalis L. essential oil from its aerial parts. Planta Med. 2009, 75, 508–511. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Dong, K.; Ma, Y.; Jin, Q.; Yin, S.; Wang, S. Hepatoprotective effects of chamazulene against alcohol-induced liver damage by alleviation of oxidative stress in rat models. Open Life Sci. 2020, 15, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.J.; Xie, S.X.; Keefe, J.R.; Soeller, I.; Li, Q.S.; Amsterdam, J.D. Long-term chamomile (Matricaria chamomilla L.) treatment for generalized anxiety disorder: A randomized clinical trial. Phytomedicine 2016, 23, 1735–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malcolm, B.J.; Tallian, K. Essential oil of lavender in anxiety disorders: Ready for prime time? Ment. Health Clin. 2017, 7, 147–155. [Google Scholar] [CrossRef]
- Donelli, D.; Antonelli, M.; Bellinazzi, C.; Gensini, G.F.; Firenzuoli, F. Effects of lavender on anxiety: A systematic review and meta-analysis. Phytomedicine 2019, 65, 153099. [Google Scholar] [CrossRef]
- López, V.; Nielsen, B.; Solas, M.; Ramírez, M.J.; Jäger, A.K. Exploring pharmacological mechanisms of lavender (Lavandula angustifolia) essential oil on central nervous system targets. Front. Pharmacol. 2017, 8, 280. [Google Scholar] [CrossRef]
- Saki, K.; Bahmani, M.; Rafieian-Kopaei, M. The effect of most important medicinal plants on two importnt psychiatric disorders (anxiety and depression)-a review. Asian Pac. J. Trop. Med. 2014, 7, S34–S42. [Google Scholar] [CrossRef] [Green Version]
- Nazıroğlu, M.; Kozlu, S.; Yorgancıgil, E.; Uğuz, A.C.; Karakuş, K. Rose oil (from Rosa × damascena Mill.) vapor attenuates depression-induced oxidative toxicity in rat brain. J. Nat. Med. 2013, 67, 152–158. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, L.; Feng, L.; Yao, L. The anxiolytic effect of essential oil of Cananga odorata exposure on mice and determination of its major active constituents. Phytomedicine 2016, 23, 1727–1734. [Google Scholar] [CrossRef]
- FooDB, Version 1.0. Available online: www.foodb.ca (accessed on 24 June 2021).
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Avram, S.; Mernea, M.; Bagci, E.; Hritcu, L.; Borcan, L.C.; Mihailescu, D.F. Advanced Structure-activity Relationships Applied to Mentha spicata L. Subsp. spicata Essential Oil Compounds as AChE and NMDA Ligands, in Comparison with Donepezil, Galantamine and Memantine—New Approach in Brain Disorders Pharmacology. CNS Neurol. Disord.-Drug Targets 2017, 16, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Duda-Seiman, D.; Borcan, F.; Wolschann, P. QSAR-CoMSIA applied to antipsychotic drugs with their dopamine D2 and serotonine 5HT2A membrane receptors. J. Serb. Chem. Soc. 2011, 76, 263–281. [Google Scholar] [CrossRef]
- Avram, S.; Buiu, C.; Duda-Seiman, D.M.; Duda-Seiman, C.; Mihailescu, D. 3D-QSAR design of new escitalopram derivatives for the treatment of major depressive disorders. Sci. Pharm. 2010, 78, 233–248. [Google Scholar] [CrossRef] [Green Version]
- Silverman, R.B. Chapter 2-Drug Discovery, Design, and Development. In The Organic Chemistry of Drug Design and Drug Action, 2nd ed.; Silverman, R.B., Ed.; Academic Press: San Diego, CA, USA, 2004; pp. 7–120. [Google Scholar]
- Floresta, G.; Patamia, V.; Gentile, D.; Molteni, F.; Santamato, A.; Rescifina, A.; Vecchio, M. Repurposing of FDA-Approved Drugs for Treating Iatrogenic Botulism: A Paired 3D-QSAR/Docking Approach. Chem. Med. Chem. 2020, 15, 256–262. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Y.; He, Y.; Zhu, H.; Gao, Y.; Li, Z.; Zhu, J.; Sun, X.; Fang, F.; Wen, H.; et al. Combined pharmacophore modeling, 3D-QSAR and docking studies to identify novel HDAC inhibitors using drug repurposing. J. Biomol. Struct. Dyn. 2020, 38, 533–547. [Google Scholar] [CrossRef]
- Tejera, E.; Munteanu, C.R.; López-Cortés, A.; Cabrera-Andrade, A.; Pérez-Castillo, Y. Drugs repurposing using QSAR, docking and molecular dynamics for possible inhibitors of the SARS-CoV-2 Mpro protease. Molecules 2020, 25, 5172. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Dubey, A.; Kamboj, N.K.; Sahoo, A.K.; Kang, S.G.; Yadava, U. Drug repurposing for ligand-induced rearrangement of Sirt2 active site-based inhibitors via molecular modeling and quantum mechanics calculations. Sci. Rep. 2021, 11, 10169. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Douguet, D. Data Sets Representative of the Structures and Experimental Properties of FDA-Approved Drugs. ACS Med. Chem. Lett. 2018, 9, 204–209. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2021.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Durán, Á.; Zamora, I.; Pastor, M. Suitability of GRIND-Based Principal Properties for the Description of Molecular Similarity and Ligand-Based Virtual Screening. J. Chem. Inf. Modeling 2009, 49, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- PDSP (Psychoactive Drug Screening Program) Ki Database. Available online: https://pdsp.unc.edu/databases/kidb.php (accessed on 24 June 2021).
- Coleman, J.A.; Navratna, V.; Antermite, D.; Yang, D.; Bull, J.A.; Gouaux, E. Chemical and structural investigation of the paroxetine-human serotonin transporter complex. Elife 2020, 9, e56427. [Google Scholar] [CrossRef]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Gagnon, J.K.; Law, S.M.; Brooks, C.L., 3rd. Flexible CDOCKER: Development and application of a pseudo-explicit structure-based docking method within CHARMM. J. Comput. Chem. 2016, 37, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Rao, P.P.; Pham, A.T.; Shakeri, A.; El Shatshat, A.; Zhao, Y.; Karuturi, R.C.; Hefny, A.A. Drug repurposing: Dipeptidyl peptidase IV (DPP4) inhibitors as potential agents to treat SARS-CoV-2 (2019-nCov) infection. Pharmaceuticals 2021, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2020, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Höfer, P.; Schosser, A.; Calati, R.; Serretti, A.; Massat, I.; Kocabas, N.A.; Konstantinidis, A.; Linotte, S.; Mendlewicz, J.; Souery, D.; et al. The impact of Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes on suicide attempt and suicide risk-a European multicentre study on treatment-resistant major depressive disorder. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Cañizares-Carmenate, Y.; Mena-Ulecia, K.; MacLeod Carey, D.; Perera-Sardiña, Y.; Hernández-Rodríguez, E.W.; Marrero-Ponce, Y.; Torrens, F.; Castillo-Garit, J.A. Machine learning approach to discovery of small molecules with potential inhibitory action against vasoactive metalloproteases. Mol. Divers. 2021, 1–15. [Google Scholar] [CrossRef]
- Udrescu, M.; Udrescu, L. A Drug Repurposing Method Based on Drug-Drug Interaction Networks and Using Energy Model Layouts. Methods Mol. Biol. 2019, 1903, 185–201. [Google Scholar]
- Udrescu, L.; Sbârcea, L.; Topîrceanu, A.; Iovanovici, A.; Kurunczi, L.; Bogdan, P.; Udrescu, M. Clustering drug-drug interaction networks with energy model layouts: Community analysis and drug repurposing. Sci. Rep. 2016, 6, 32745. [Google Scholar] [CrossRef] [Green Version]
- Udrescu, L.; Bogdan, P.; Chiş, A.; Sîrbu, I.O.; Topîrceanu, A.; Văruţ, >R.-M.; Udrescu, M. Uncovering New Drug Properties in Target-Based Drug–Drug Similarity Networks. Pharmaceutics 2020, 12, 879. [Google Scholar] [CrossRef]
- Sánchez-Martínez, J.D.; Bueno, M.; Alvarez-Rivera, G.; Tudela, J.; Ibañez, E.; Cifuentes, A. In Vitro neuroprotective potential of terpenes from industrial orange juice by-products. Food Funct. 2021, 12, 302–314. [Google Scholar] [CrossRef]
- Zhan, Y.Y.; Liang, B.Q.; Li, X.Y.; Gu, E.M.; Dai, D.P.; Cai, J.P.; Hu, G.X. The effect of resveratrol on pharmacokinetics of aripiprazole in vivo and in vitro. Xenobiotica 2016, 46, 439–444. [Google Scholar] [CrossRef]
- Yim, S.K.; Kim, K.; Chun, S.; Oh, T.; Jung, W.; Jung, K.; Yun, C.H. Screening of Human CYP1A2 and CYP3A4 Inhibitors from Seaweed In Silico and In Vitro. Mar. Drugs 2020, 18, 603. [Google Scholar] [CrossRef]
- Elbarbry, F.; Ung, A.; Abdelkawy, K. Studying the Inhibitory Effect of Quercetin and Thymoquinone on Human Cytochrome P450 Enzyme Activities. Pharmacogn. Mag. 2018, 13, S895–S899. [Google Scholar]
- Ganzera, M.; Schneider, P.; Stuppner, H. Inhibitory effects of the essential oil of chamomile (Matricaria recutita L.) and its major constituents on human cytochrome P450 enzymes. Life Sci. 2006, 78, 856–861. [Google Scholar] [CrossRef]
- Koyama, S.; Heinbockel, T. The Effects of Essential Oils and Terpenes in Relation to Their Routes of Intake and Application. Int. J. Mol. Sci. 2020, 21, 1558. [Google Scholar] [CrossRef] [Green Version]
- Saiyudthong, S.; Mekseepralard, C. Effect of Inhaling Bergamot Oil on Depression-Related Behaviors in Chronic Stressed Rats. J. Med Assoc. Thail. 2015, 98 (Suppl. 9), S152–S159. [Google Scholar]
- Garzoli, S.; Turchetti, G.; Giacomello, P.; Tiezzi, A.; Laghezza Masci, V.; Ovidi, E. Liquid and Vapour Phase of Lavandin (Lavandula × intermedia) Essential Oil: Chemical Composition and Antimicrobial Activity. Molecules 2019, 24, 2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal, M.; Ameno, K.; Ameno, S.; Morishita, J.; Wang, W.; Kumihashi, M.; Ikuo, U.; Miki, T.; Ijiri, I. Changes in cholinergic function in the frontal cortex and hippocampus of rat exposed to ethanol and acetaldehyde. Neuroscience 2007, 144, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Tozar, T.; Santos Costa, S.; Udrea, A.-M.; Nastasa, V.; Couto, I.; Viveiros, M.; Pascu, M.L.; Romanitan, M.O. Anti-staphylococcal activity and mode of action of thioridazine photoproducts. Sci. Rep. 2020, 10, 18043. [Google Scholar] [CrossRef] [PubMed]
- Udrea, A.-M.; Avram, S.; Nistorescu, S.; Pascu, M.-L.; Romanitan, M.O. Laser irradiated phenothiazines: New potential treatment for COVID-19 explored by molecular docking. J. Photochem. Photobiol. B Biol. 2020, 211, 111997. [Google Scholar] [CrossRef]
- Avram, S.; Puia, A.; Udrea, A.M.; Mihailescu, D.; Mernea, M.; Dinischiotu, A.; Oancea, F.; Stiens, J. Natural Compounds Therapeutic Features in Brain Disorders by Experimental, Bioinformatics and Cheminformatics Methods. Curr. Med. Chem. 2020, 27, 78–98. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Compound | DrugBank Accession Number | Medical Applications | Foodb Accession Number | Medical Applications |
|---|---|---|---|---|
| resveratrol | DB02709 | anti-inflammatory, antioxidant, anticancer effects | FDB031212 | suppresses NF-kappa B activation in HSV infected cells |
| quercetin | DB04216 | specific quinone reductase 2 (QR2) inhibitor, may contribute to killing the malaria causing parasites | FDB011904 | non-specific protein kinase enzyme inhibitor, agonist of the G protein-coupled estrogen receptor in human breast cancer cell lines |
| limonene | DB08921 | common in cosmetic products, a flavoring to mask the bitter taste of alkaloids, a fragrance in perfumery | FDB013567 | antimicrobial, expectorant |
| sabinene | not available | not available | FDB001456 | not available |
| 1,8-cineole | DB03852 | controls airway mucus hypersecretion and asthma via anti-inflammatory cytokine inhibition, eucalyptol reduces inflammation and pain | FDB014616 | antibronchitis, antiallergic |
| chamazulene | DB15931 | not available | FDB015363 | analgesic, antioxidant |
| linalyl acetate | not available | not available | FDB019133 | antimicrobial, antioxidant, flavor |
| germacrene D | DB11276 | as component of pine needle oil is used as disinfectant, lubricant, sanitizer, antimicrobial, insecticide | FDB003856 | pesticide |
| nerol | not available | not available | FDB014945 | antimicrobial, flavor, perfumery |
| neryl acetate | not available | not available | FDB013794 | antimicrobial, flavor, perfumery |
| Compound | Smiles | Natural Source/FooDB Id | Lipinski | Veber | Ghose | Egan | |
|---|---|---|---|---|---|---|---|
| 1,8-cineole [PubChem ID = 2758] |  | CC1(C2CCC(O1)(CC2)C)C | eucalyptus, sage [FDB014616] | YES | YES | No; 1 violation: MW < 160 | YES |
| limonene [PubChem ID = 22311] |  | CC1=CCC(CC1)C(=C)C | peppermint, spearmint [FDB013567] | YES | YES | No; 1 violation: MW < 160 | YES |
| sabinene [PubChem ID = 18818] |  | CC(C)C12CCC(=C)C1C2 | lemon, mint [FDB001454] | YES | YES | No; 1 violation: MW < 160 | YES |
| resveratrol [PubChem ID = 445154] |  | C1=CC(=CC=C1C=CC2=CC(=CC(=C2)O)O)O | skin of grapes [FDB031212] | YES | YES | YES | YES |
| chamazulene [PubChem ID = 10719] |  | CCC1=CC2=C(C=CC2=C(C=C1)C)C | german chamomile, roman chamomile [FDB015363] | YES | YES | YES | YES |
| germacrene D [PubChem ID = 5317570] |  | CC1=CCCC(=C)C=CC(CC1)C(C)C | Peppermint [FDB003856] | YES | YES | YES | YES |
| linalyl acetate [PubChem ID = 8294] |  | CC(=CCCC(C)(C=C)OC(=O)C)C | sage [FDB019133] | YES | YES | YES | YES |
| nerol [PubChem ID = 643820] |  | CC(=CCCC(=CCO)C)C | common grapes [FDB014945] | YES | YES | YES | YES |
| neryl acetate PubChem [ID = 1549025] |  | CC(=CCC/C(=C\COC(=O)C)/C)C | lemon balm, peppermint [FDB014946] | YES | YES | YES | YES |
| quercetin [PubChem ID = 5280343] |  | C1=CC(=C(C=C1C2=C(C(=O)C3=C(C=C(C=C3O2)O)O)O)O)O | Grape [FDB011904] | YES | YES | YES | YES |
| Compound | HIA | Log BBB | CNS Permeability | HFU | Max. Tolerated Dose (Human) | LD50 |
|---|---|---|---|---|---|---|
| 1,8-cineole | 96.50 | 0.36 | −2.97 | 0.55 | 0.55 | 2.01 |
| limonene | 95.89 | 0.72 | −2.37 | 0.48 | 0.77 | 1.88 |
| sabinene | 95.35 | 0.83 | −1.46 | 0.29 | 0.36 | 1.54 |
| resveratrol | 89.05 | −0.04 | −2.09 | 0.18 | 0.48 | 1.79 |
| chamazulene | 94.50 | 0.79 | −1.82 | 0.24 | 0.05 | 1.45 |
| germacrene D | 95.59 | 0.72 | −2.13 | 0.26 | 0.49 | 1.63 |
| linalyl acetate | 95.27 | 0.51 | −2.37 | 0.42 | 0.54 | 1.72 |
| nerol | 93.46 | 0.62 | −2.17 | 0.44 | 0.85 | 1.71 |
| neryl acetate | 96.06 | 0.56 | −2.19 | 0.37 | 0.74 | 1.95 |
| quercetin | 77.20 | −1.09 | −3.06 | 0.20 | 0.49 | 2.47 |
| Compound | CYP2D6 Substrate/ Inhibitor | CYP3A4 Substrate/ Inhibitor | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor |
|---|---|---|---|---|---|
| 1,8-cineole | no/no | no/no | no | no | no |
| limonene | no/no | no/no | no | no | no |
| sabinene | no/no | no/no | no | no | no |
| resveratrol | no/yes | no/yes | yes | yes | yes |
| chamazulene | no/no | no/no | no | no | no |
| germacrene d | no/no | no/no | no | no | no |
| linalyl acetate | no/no | no/no | no | no | no |
| nerol | no/no | no/no | no | no | no |
| neryl acetate | no/no | no/no | no | no | no |
| quercetin | no/no | no/no | yes | no | no |
| Statistical Parameters | QSAR-SERT | QSAR-5HT-1A | QSAR-D2 |
|---|---|---|---|
| No. of molecules in a training set | 21 | 19 | 19 |
| q2 | 0.80 | 0.90 | 0.83 |
| r2 | 0.96 | 0.95 | 0.95 |
| SDEP | 0.50 | 0.29 | 0.40 |
| Compounds | pKiSERTexp/ pKiSERTpredicted | pKi 5-HT1Aexp/ pKi 5-HT1Apredicted | pKi D2exp/ pKi D2predicted |
|---|---|---|---|
| amitriptyline | 8.55/8.68 | 6.34/6.62 | 6.70/7.17 |
| citalopram | 9.00/8.74 | - | - |
| clomipramine | 9.85/9.66 | - | 7.11/7.29 |
| desipramine | 7.75/8.07 | 5.19/5.29 | 5.80/5.42 |
| doxepin | 7.16/7.58 | 6.55/6.45 | 6.44/7.35 |
| escitalopram | 8.95/8.85 | 5.00/5.06 | - |
| fluoxetine | 9.09/9.23 | 5.00/4.96 | - |
| imipramine | 9.82/9.31 | - | - |
| lofepramine | 7.15/7.19 | - | - |
| paroxetine | 10.09/9.83 | - | - |
| sertraline | 9.58/9.57 | - | - |
| trazodone | 6.79/6.72 | 7.00/6.91 | - |
| venlafaxine | 8.12/8.02 | - | - |
| aripiprazole | 5.74/5.87 | 8.25/8.40 | 9.18/8.53 |
| chlorpromazine | 8.88/9.23 | 6.93/6.78 | 9.18/8.22 |
| clozapine | 6.00/5.49 | 6.97/5.32 | 7.55/8.23 |
| fluphenazine | 5.22/5.22 | 6.83/6.68 | 9.69/9.95 |
| haloperidol | 6.00/5.51 | 5.92/5.97 | 9.45/9.38 |
| risperidone | 6.00/6.04 | 6.72/6.87 | 9.52/9.65 |
| sertindole | 6.00/6.42 | 6.55/7.19 | 9.02/9.11 |
| zotepine | 6.82/7.35 | 6.89/6.26 | 8.09/8.06 |
| bupropion | 5.04/5.62 | - | - |
| olanzapine | 6.00/5.69 | 5.68/5.61 | 8.52/8.13 |
| quetiapine, | 6.00/5.20 | 6.63/6.47 | 7.79/8.66 |
| thioridazine | 5.89/7.29 | 6.96/6.50 | 9.39/8.69 |
| ziprasidone | 7.27/6.57 | 8.72/8.80 | 8.92/8.67 |
| fluvoxamine | 8.79/8.64 | - | - |
| iloperidone | - | 7.48/7.52 | 9.45/9.38 |
| loxapine | - | 5.60/5.73 | 8.28/8.22 |
| prochlorperazine | - | 5.22/5.30 | 9.69/9.07 |
| spiperone | - | 7.76/7.50 | 10.15/10.06 |
| trifluoperidine | - | 6.02/5.93 | - |
| mesoridazine | - | - | 8.36/8.52 |
| promazine | - | - | 6.79/7.07 |
| remoxipride | - | - | 7.79/7.72 |
| trifluoperazine | - | - | 8.92/9.16 |
| Natural compounds | |||
| 1,8-cineole | 8.57 (−1.52) | 7.09 (−1.63) | 8.11 (−2.04) |
| limonene | 9.45 (−0.64) | 6.73 (−1.99) | 7.99 (−2.16) |
| sabinene | 9.37 (−0.72) | 6.68 (−2.04) | 7.98 (−2.17) |
| resveratrol | 8.68 (−1.41) | 5.23 (−3.49) | 6.72 (−3.43) |
| chamazulene | 9.50 (−0.59) | 6.51 (−2.21) | 7.96 (−2.19) |
| germacrene D | 9.52 (−0.57) | 6.49 (−2.23) | 7.90 (−2.25) |
| linalyl acetate | 9.40 (−0.69) | 6.89 (−1.83) | 8.11 (−2.04) |
| nerol | 9.74 (−0.35) | 6.17 (−2.55) | 8.05 (−2.10) |
| neryl acetate | 10.61 (0.52) | 6.06 (−2.66) | 8.14 (−2.01) |
| quercetin | 6.42 (−3.67) | 5.87 (−2.85) | 8.39 (−1.76) |
| Compound | Target | -CDOCKER_ENERGY | -CDOCKER_INTERACTION_ENERGY |
|---|---|---|---|
| linalyl acetate | SERT | 4.65 | 31.46 |
| linalyl acetate | D2 | −3.94 | 30.82 |
| linalyl acetate | 5-HT1A | −1.96 | 27.17 |
| neryl acetate | SERT | −11.64 | 31.29 |
| neryl acetate | 5-HT1A | −9.79 | 33.85 |
| 1,8-cineole | D2 | −14.66 | 17.25 |
| 1,8-cineole | 5-HT1A | −8.22 | 19.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avram, S.; Stan, M.S.; Udrea, A.M.; Buiu, C.; Boboc, A.A.; Mernea, M. 3D-ALMOND-QSAR Models to Predict the Antidepressant Effect of Some Natural Compounds. Pharmaceutics 2021, 13, 1449. https://doi.org/10.3390/pharmaceutics13091449
Avram S, Stan MS, Udrea AM, Buiu C, Boboc AA, Mernea M. 3D-ALMOND-QSAR Models to Predict the Antidepressant Effect of Some Natural Compounds. Pharmaceutics. 2021; 13(9):1449. https://doi.org/10.3390/pharmaceutics13091449
Chicago/Turabian StyleAvram, Speranta, Miruna Silvia Stan, Ana Maria Udrea, Cătălin Buiu, Anca Andreea Boboc, and Maria Mernea. 2021. "3D-ALMOND-QSAR Models to Predict the Antidepressant Effect of Some Natural Compounds" Pharmaceutics 13, no. 9: 1449. https://doi.org/10.3390/pharmaceutics13091449