
Formulation Development of Sublingual Cyclobenzaprine Tablets Empowered by Standardized and Physiologically Relevant Ex Vivo Permeation Studies


Abstract
:1. Introduction
2. Materials and Methods
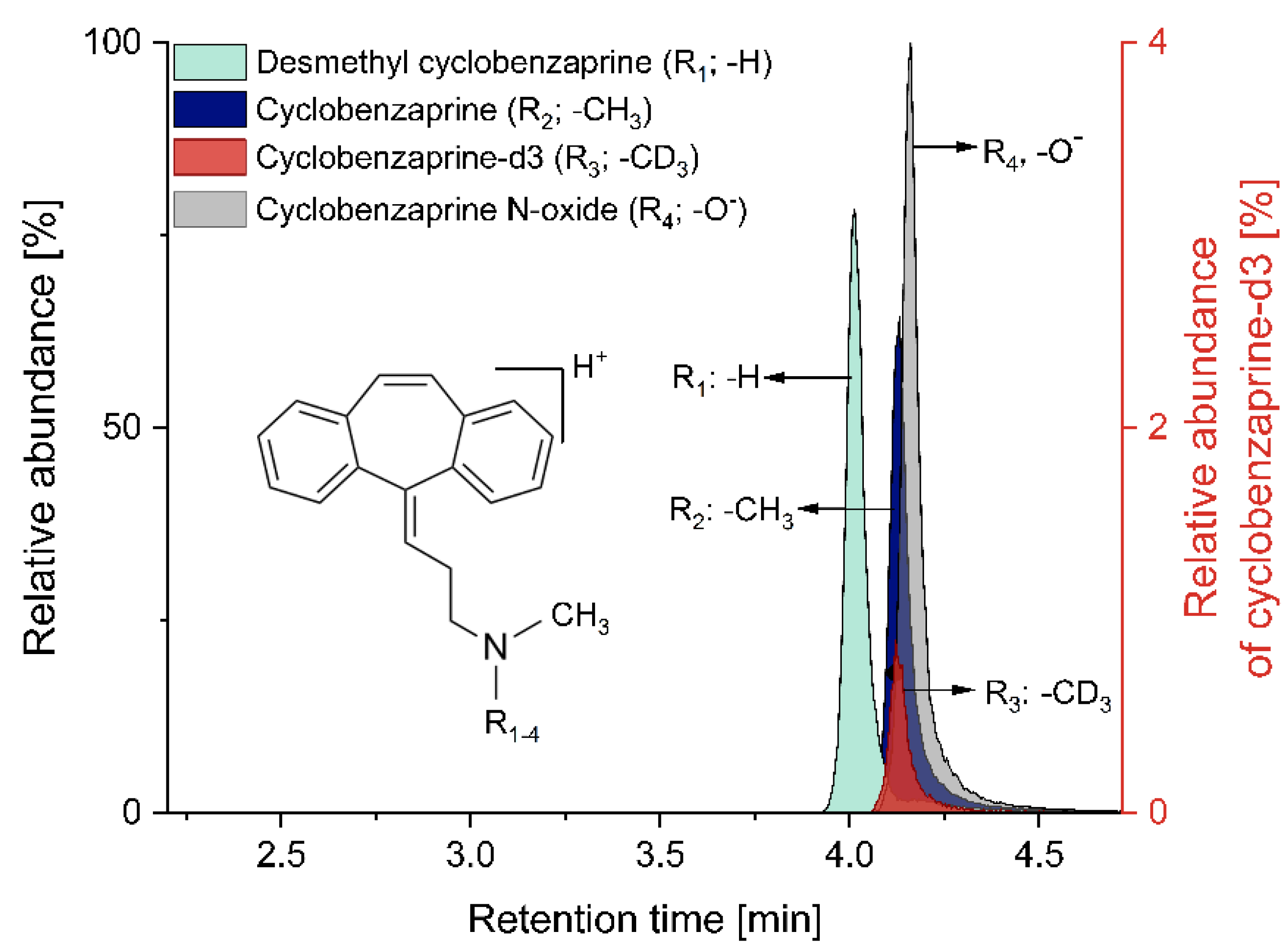
2.1. Simultaneous Quantification of Cyclobenzaprine and Its Related Compounds
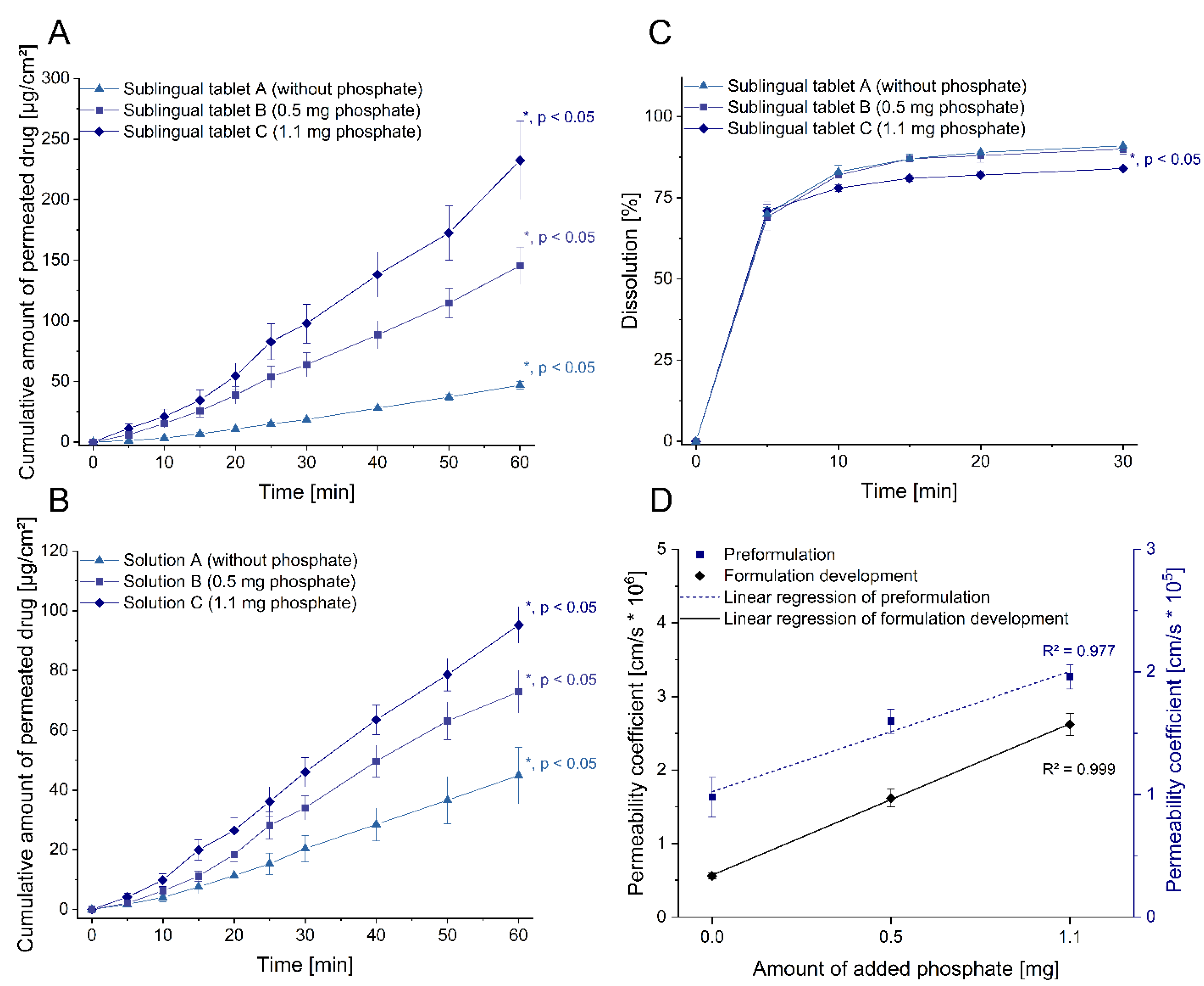
2.2. Sublingual Formulation Development Guided by Permeation Studies
2.3. Standardized and Physiologically Relevant Permeation Model
2.3.1. Model Set-up
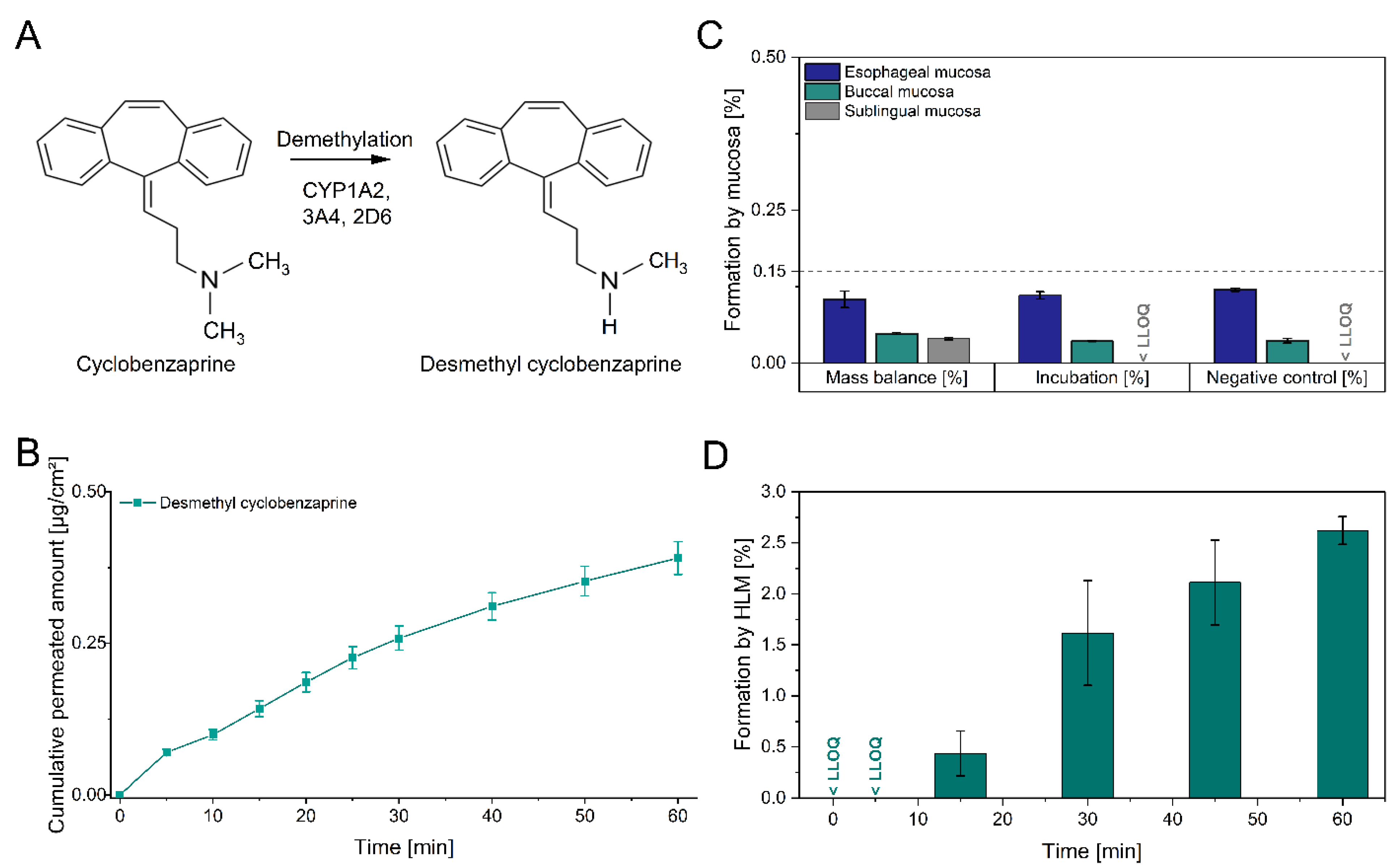
2.3.2. Metabolization of Cyclobenzaprine during Mucosal Permeation
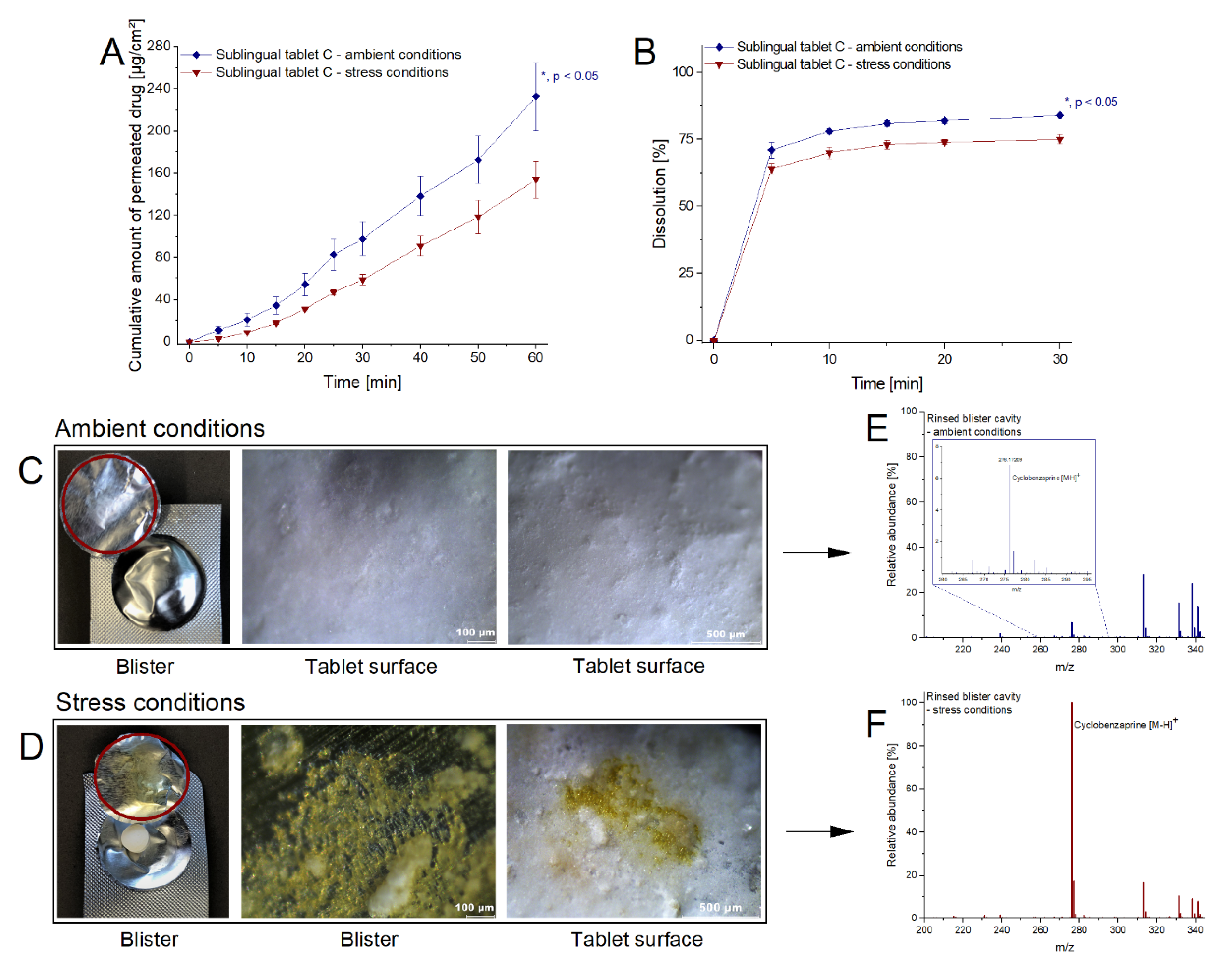
2.3.3. Impact of Alteration in Dosage Forms on Drug Liberation and Absorption
2.4. Dissolution Studies
3. Results and Discussion
3.1. Simultaneous Quantification of Cyclobenzaprine and Its Related Compounds
3.2. Sublingual Formulation Development Guided by Permeation Studies
3.3. Metabolism of Cyclobenzaprine during Mucosal Administration
3.4. Impact of Alteration in Dosage Forms on Drug Liberation and Absorption
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Schiele, J.T.; Quinzler, R.; Klimm, H.-D.; Pruszydlo, M.G.; Haefeli, W.E. Difficulties swallowing solid oral dosage forms in a general practice population: Prevalence, causes, and relationship to dosage forms. Eur. J. Clin. Pharmacol. 2013, 69, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.; Pintado, M.E.; Sarmento, B. In vivo, ex vivo and in vitro assessment of buccal permeation of drugs from delivery systems. Expert Opin. Drug Deliv. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Gilhotra, R.M.; Ikram, M.; Srivastava, S.; Gilhotra, N. A clinical perspective on mucoadhesive buccal drug delivery systems. J. Biomed. Res. 2014, 28, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Pérez, M.Á.; Sanz, M.B.; Sanjuan, V.M.; González-Álvarez, M.; Álvarez, I.G. Importance and applications of cell- and tissue-based in vitro models for drug permeability screening in early stages of drug development. In Concepts and Models for Drug Permeability Studies: Cell and Tissue Based In Vitro Culture Models; Sarmento, B., Ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2016; pp. 3–29. ISBN 9780081000946. [Google Scholar]
- Kalia, V.; Garg, T.; Rath, G.; Goyal, A.K. Development and evaluation of a sublingual film of the antiemetic granisetron hydrochloride. Artif. Cells Nanomed. Biotechnol. 2016, 44, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Kolli, C.S.; Pather, I. Characterization Methods for Oral Mucosal Drug Delivery. In Oral Mucosal Drug Delivery and Therapy; Rathbone, M.J., Şenel, S., Pather, I., Eds.; Springer: New York, NY, USA; Heidelberg, Germany; Dordrecht, The Netherlands; London, UK, 2015; pp. 125–148. ISBN 978-1-4899-7557-7. [Google Scholar]
- Patel, V.F.; Liu, F.; Brown, M.B. Modeling the oral cavity: In vitro and in vivo evaluations of buccal drug delivery systems. J. Control. Release 2012, 161, 746–756. [Google Scholar] [CrossRef]
- Castro, P.; Madureira, R.; Sarmento, B.; Pintado, M. Tissue-based in vitro and ex vivo models for buccal permeability studies. In Concepts and Models for Drug Permeability Studies: Cell and Tissue Based In Vitro Culture Models; Sarmento, B., Ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2016; pp. 189–202. ISBN 9780081000946. [Google Scholar]
- AlAli, A.S.; Aldawsari, M.F.; Alalaiwe, A.; Almutairy, B.K.; Al-Shdefat, R.; Walbi, I.A.; Fayed, M.H. Exploitation of Design-of-Experiment Approach for Design and Optimization of Fast-Disintegrating Tablets for Sublingual Delivery of Sildenafil Citrate with Enhanced Bioavailability Using Fluid-Bed Granulation Technique. Pharmaceutics 2021, 13, 870. [Google Scholar] [CrossRef] [PubMed]
- Pather, S.I.; Rathbone, M.J.; Senel, S. Current status and the future of buccal drug delivery systems. Expert Opin. Drug Deliv. 2008, 5, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Majid, H.; Bartel, A.; Burckhardt, B.B. Development, validation and standardization of oromucosal ex-vivo permeation studies for implementation in quality-controlled environments. J. Pharm. Biomed. Anal. 2021, 194, 113769. [Google Scholar] [CrossRef]
- Majid, H.; Bartel, A.; Burckhardt, B.B. Predictivity of Standardized and Controlled Permeation Studies: Ex vivo—In vitro—In vivo Correlation for Sublingual Absorption of Propranolol. Under Revision. Eur. J. Phar. Biophar. in press.
- Majid, H.; Puzik, A.; Maier, T.; Eberhard, D.; Bartel, A.; Mueller, H.-C.; Burckhardt, B.B. Exploring the transmucosal permeability of cyclobenzaprine: A comparative preformulation by standardized and controlled ex vivo and in vitro permeation studies. Int. J. Pharm. 2021, 601, 120574. [Google Scholar] [CrossRef]
- Chou, R.; Peterson, K.; Helfand, M. Comparative efficacy and safety of skeletal muscle relaxants for spasticity and musculoskeletal conditions: A systematic review. J. Pain Symptom Manag. 2004, 28, 140–175. [Google Scholar] [CrossRef]
- Moldofsky, H.; Harris, H.W.; Archambault, W.T.; Kwong, T.; Lederman, S. Effects of bedtime very low dose cyclobenzaprine on symptoms and sleep physiology in patients with fibromyalgia syndrome: A double-blind randomized placebo-controlled study. J. Rheumatol. 2011, 38, 2653–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, K. Sleep Dysfunction in Fibromyalgia and Therapeutic Approach Options. OBM Neurobiol. 2020, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Koenen, K.C.; Ratanatharathorn, A.; Ng, L.; McLaughlin, K.A.; Bromet, E.J.; Stein, D.J.; Karam, E.G.; Meron Ruscio, A.; Benjet, C.; Scott, K.; et al. Posttraumatic stress disorder in the World Mental Health Surveys. Psychol. Med. 2017, 47, 2260–2274. [Google Scholar] [CrossRef] [PubMed]
- Spoormaker, V.I.; Montgomery, P. Disturbed sleep in post-traumatic stress disorder: Secondary symptom or core feature? Sleep Med. Rev. 2008, 12, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Sartori, S.B.; Singewald, N. Novel pharmacological targets in drug development for the treatment of anxiety and anxiety-related disorders. Pharmacol. Ther. 2019, 204, 107402. [Google Scholar] [CrossRef]
- Sullivan, G.M.; Gendreau, R.M.; Gendreau, J.; Peters, P.; Peters, A.; Engels, J.; Daugherty, B.L.; Vaughn, B.; Weathers, F.W.; Lederman, S. Randomized clinical trial of bedtime sublingual cyclobenzaprine (TNX-102 SL) in military-related PTSD and the role of sleep quality in treatment response. Psychiatry Res. 2021, 301, 113974. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services Food and Drug Administration. Bioanalytical Method Validation Guidance for Industry; U.S. Department of Health and Human Services Food and Drug Administration: Silver Spring, MD, USA, 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 1 July 2021).
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline Validation of Analytical Procedures: Text and Methodology: Q2(R1); International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 2005; Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 10 July 2021).
- European Medicines Agency. Guideline Bioanalytical Method Validation: EMEA/CHMP/EWP/192217/2009; European Medicines Agency: Amsterdam, The Netherlands, 2011; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 1 July 2021).
- Telò, I.; Tratta, E.; Guasconi, B.; Nicoli, S.; Pescina, S.; Govoni, P.; Santi, P.; Padula, C. In-vitro characterization of buccal iontophoresis: The case of sumatriptan succinate. Int. J. Pharm. 2016, 506, 420–428. [Google Scholar] [CrossRef]
- Diaz del Consuelo, I.; Jacques, Y.; Pizzolato, G.-P.; Guy, R.H.; Falson, F. Comparison of the lipid composition of porcine buccal and esophageal permeability barriers. Arch. Oral Biol. 2005, 50, 981–987. [Google Scholar] [CrossRef]
- Diaz del Consuelo, I.; Pizzolato, G.-P.; Falson, F.; Guy, R.H.; Jacques, Y. Evaluation of pig esophageal mucosa as a permeability barrier model for buccal tissue. J. Pharm. Sci. 2005, 94, 2777–2788. [Google Scholar] [CrossRef]
- Diaz del Consuelo, I.; Falson, F.; Guy, R.H.; Jacques, Y. Transport of fentanyl through pig buccal and esophageal epithelia in vitro: Influence of concentration and vehicle pH. Pharm. Res. 2005, 22, 1525–1529. [Google Scholar] [CrossRef]
- Kerski, S.; Rathsack, W.; Stodt, G. DE202015004165U1—Permeation cell with isofill chamber. Deutsches Patent- und Markenamt DE202015004165U1, 15 October 2015. Available online: https://patents.google.com/patent/DE202015004165U1/en (accessed on 3 September 2021).
- Yu, J. Cyclobenzaprine. In Handbook of Metabolic Pathways of Xenobiotics; Lee, P.W., Ed.; Wiley: Chichester, UK, 2014; pp. 1–5. ISBN 9781118541203. [Google Scholar]
- Wang, R.W.; Liu, L.; Cheng, H. Identification of human liver cytochrome P450 isoforms involved in the in vitro metabolism of cyclobenzaprine. Drug Metab. Dispos. 1996, 24, 786–791. [Google Scholar]
- Nielsen, H.M.; Rassing, M.R. Nicotine permeability across the buccal TR146 cell culture model and porcine buccal mucosa in vitro: Effect of pH and concentration. Eur. J. Phar. Sci. 2002, 16, 151–157. [Google Scholar] [CrossRef]
- The United States Pharmacopeia. Revision Bulletin Cyclobenzaprine Hydrochloride. Chemical Medicines Monographs 4. Available online: https://www.uspnf.com/sites/default/files/usp_pdf/EN/USPNF/revisions/cyclobenzaprine-hcl-rb-notice-20191227.pdf (accessed on 8 July 2021).
- Cimolai, N. Cyclobenzaprine: A new look at an old pharmacological agent. Expert Rev. Clin. Pharmacol. 2009, 2, 255–263. [Google Scholar] [CrossRef]
- Hucker, H.B.; Stauffer, S.C.; Albert, K.S.; Lei, B.W. Plasma levels and bioavailability of cyclobenzaprine in human subjects. J. Clin. Pharmacol. 1977, 17, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, D.; Zhao, Z. Study of degradation mechanisms of cyclobenzaprine by LC-MS/MS. Anal. Methods 2014, 6, 2320–2330. [Google Scholar] [CrossRef]
- Stephenson, G.A.; Aburub, A.; Woods, T.A. Physical stability of salts of weak bases in the solid-state. J. Pharm. Sci. 2011, 100, 1607–1617. [Google Scholar] [CrossRef]
- Koranne, S.; Govindarajan, R.; Suryanarayanan, R. Investigation of Spatial Heterogeneity of Salt Disproportionation in Tablets by Synchrotron X-ray Diffractometry. Mol. Pharm. 2017, 14, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.A.; Luthra, S.; Shamblin, S.L.; Arora, K.K.; Krzyzaniak, J.F.; Taylor, L.S. Effect of excipient properties, water activity, and water content on the disproportionation of a pharmaceutical salt. Int. J. Pharm. 2018, 546, 226–234. [Google Scholar] [CrossRef]
- Tian, Y.; Orlu, M.; Woerdenbag, H.J.; Scarpa, M.; Kiefer, O.; Kottke, D.; Sjöholm, E.; Öblom, H.; Sandler, N.; Hinrichs, W.L.J.; et al. Oromucosal films: From patient centricity to production by printing techniques. Expert Opin. Drug Deliv. 2019, 16, 981–993. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte-Specific Parameters | Cyclobenzaprine | Cyclobenzaprine N-Oxide | Desmethyl Cyclobenzaprine | Cyclobenzaprine-d3 |
|---|---|---|---|---|
| Mass transition [m/z] | 276.2 → 215.0 | 292.4 → 231.2 | 262.4 → 231.2 | 279.2 → 215.0 |
| Declustering potential | 55 V | 55 V | 55 V | 55 V |
| Focusing potential | 380 V | 380 V | 380 V | 380 V |
| Entrance potential | 10 V | 9 V | 9 V | 10 V |
| Cell entrance potential | 21 V | 10 V | 10 V | 21 V |
| Collision energy | 61 V | 25 V | 25 V | 61 V |
| Cell exit potential | 10 V | 10 V | 10 V | 10 V |
| Mode | ESI (+) | |||
| Dwell time | 80 msec | |||
| Ingredients | Amount [%] | Amount [mg] |
|---|---|---|
| Cyclobenzaprine hydrochloride (API) (Hetero drugs Ltd., Hyderabad, India) | 3.7 | 2.80 |
| Crospovidone (Kollidon CL, BASF, Ludwigshafen, Germany) | 5.3 | 4.00 |
| Peppermint aroma (Symrise, Holzminden, Germany) | 3.7 | 2.80 |
| Sodium stearyl fumarate (Pruv, JRS Pharma, Rosenberg, Germany) | 2.6 | 2.00 |
|
Dipotassium hydrogen phosphate (Merck KGaA, Darmstadt, Germany) | 0.0 (SLT-A); 0.7 (SLT-B); 1.4 (SLT-C) | 0.00 (SLT-A); 0.53 (SLT-B); 1.05 (SLT-C) |
| Silicon dioxide (Syloid 244 FP, Grace, Worms, Germany) | 1.3 | 1.00 |
| Sucralose (Merck KGaA, Darmstadt, Germany) | 0.3 | 0.25 |
| Levomenthol (L-Menthol, BASF, Ludwigshafen, Germany) | 0.03 | 0.02 |
| Mannitol (Pearlitol 100 SD, Frankfurt, Germany) | ad 100 | ad 76.00 |
| Physical attributes | ||
| Shape | White round sublingual tablet | |
| Diameter | 0.6 cm | |
| Height | 0.27 cm | |
| Weight | 76.0 mg | |
| Analyte | Quality Control [ng/mL] | Relative Error [%] | CV [%] | |||||
|---|---|---|---|---|---|---|---|---|
| Within-Run | Between-Run | Within-Run | Between-Run | |||||
| Run 1 | Run 2 | Run 3 | ||||||
| Cyclobenzaprine | QC high | 476.19 | +4.50 | −0.24 | +1.97 | +2.08 | 3.46 | 3.87 |
| QC middle | 59.52 | +1.04 | +3.27 | +3.71 | +2.67 | 2.73 | 2.81 | |
| QC low | 3.72 | −6.06 | −6.74 | −5.03 | −5.94 | 4.08 | 4.08 | |
| QC LLOQ | 0.93 | +13.33 | +8.68 | +16.63 | +12.82 | 4.28 | 5.21 | |
| Desmethyl cyclobenzaprine | QC high | 476.19 | +4.15 | −3.98 | −5.18 | −0.66 | 3.57 | 6.07 |
| QC middle | 59.52 | +1.30 | +1.95 | +1.76 | +2.27 | 2.25 | 2.25 | |
| QC low | 3.72 | −2.21 | −5.04 | −4.70 | −3.41 | 4.25 | 4.25 | |
| QC LLOQ | 0.93 | +1.73 | +8.19 | +0.35 | +3.99 | 3.54 | 5.14 | |
| Cyclobenzaprine N-oxide | QC high | 476.19 | −0.74 | −8.36 | −2.30 | −3.21 | 3.77 | 5.37 |
| QC middle | 59.52 | −5.49 | −3.61 | −1.34 | −2.91 | 2.20 | 2.92 | |
| QC low | 3.72 | −0.47 | −3.99 | +0.57 | −0.71 | 4.92 | 5.02 | |
| QC LLOQ | 0.93 | +0.25 | −2.41 | −3.18 | −1.24 | 2.83 | 3.13 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majid, H.; Puzik, A.; Maier, T.; Merk, R.; Bartel, A.; Mueller, H.-C.; Burckhardt, B.B. Formulation Development of Sublingual Cyclobenzaprine Tablets Empowered by Standardized and Physiologically Relevant Ex Vivo Permeation Studies. Pharmaceutics 2021, 13, 1409. https://doi.org/10.3390/pharmaceutics13091409
Majid H, Puzik A, Maier T, Merk R, Bartel A, Mueller H-C, Burckhardt BB. Formulation Development of Sublingual Cyclobenzaprine Tablets Empowered by Standardized and Physiologically Relevant Ex Vivo Permeation Studies. Pharmaceutics. 2021; 13(9):1409. https://doi.org/10.3390/pharmaceutics13091409
Chicago/Turabian StyleMajid, Haidara, Andreas Puzik, Tanja Maier, Raphaela Merk, Anke Bartel, Hans-Christian Mueller, and Bjoern B. Burckhardt. 2021. "Formulation Development of Sublingual Cyclobenzaprine Tablets Empowered by Standardized and Physiologically Relevant Ex Vivo Permeation Studies" Pharmaceutics 13, no. 9: 1409. https://doi.org/10.3390/pharmaceutics13091409