
Antioxidant and Antiproliferative Activity of Finasteride against Glioblastoma Cells

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drug
2.3. Transfection and Reporter Assays
2.4. Protein Preparation and Immunoblot Analysis
2.5. Immunofluorescence
2.6. ROS Detection
2.7. Monitoring of the Mitochondrial Membrane Potential
2.8. Quantitative Real-Time RT-PCR
2.9. Statistical Analysis
3. Results
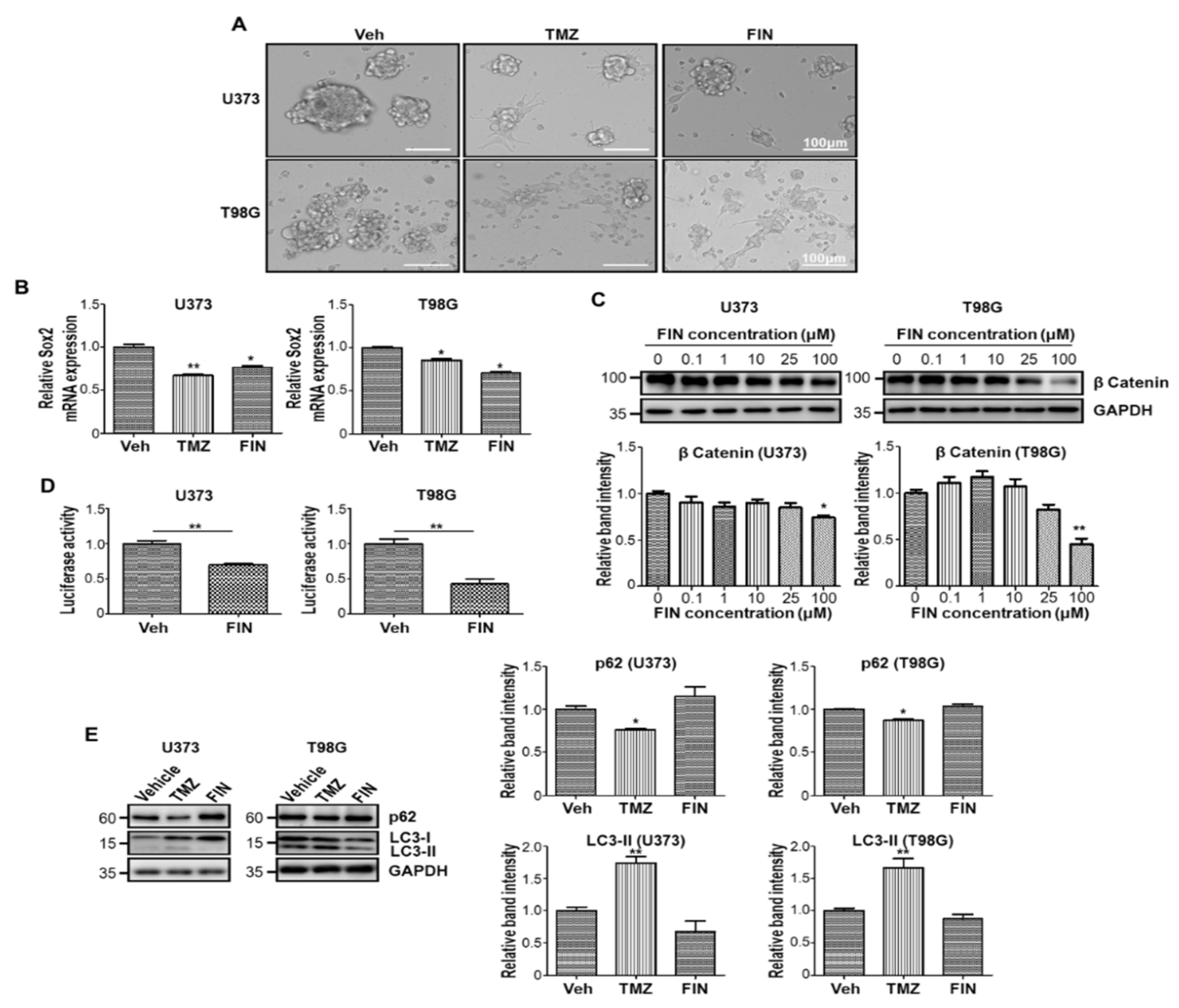
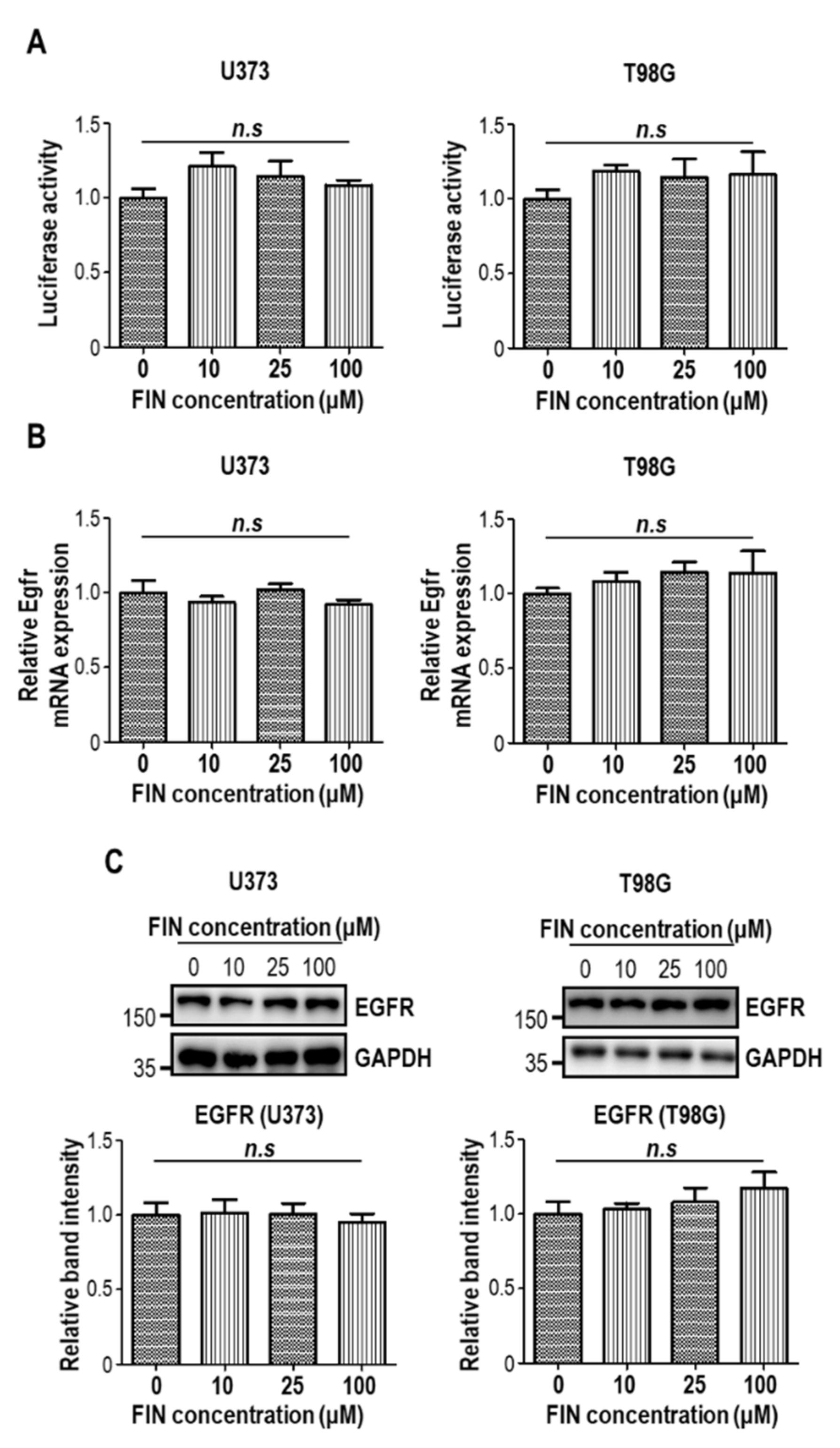
3.1. High-Dose Finasteride Downregulates β-Catenin Protein Level
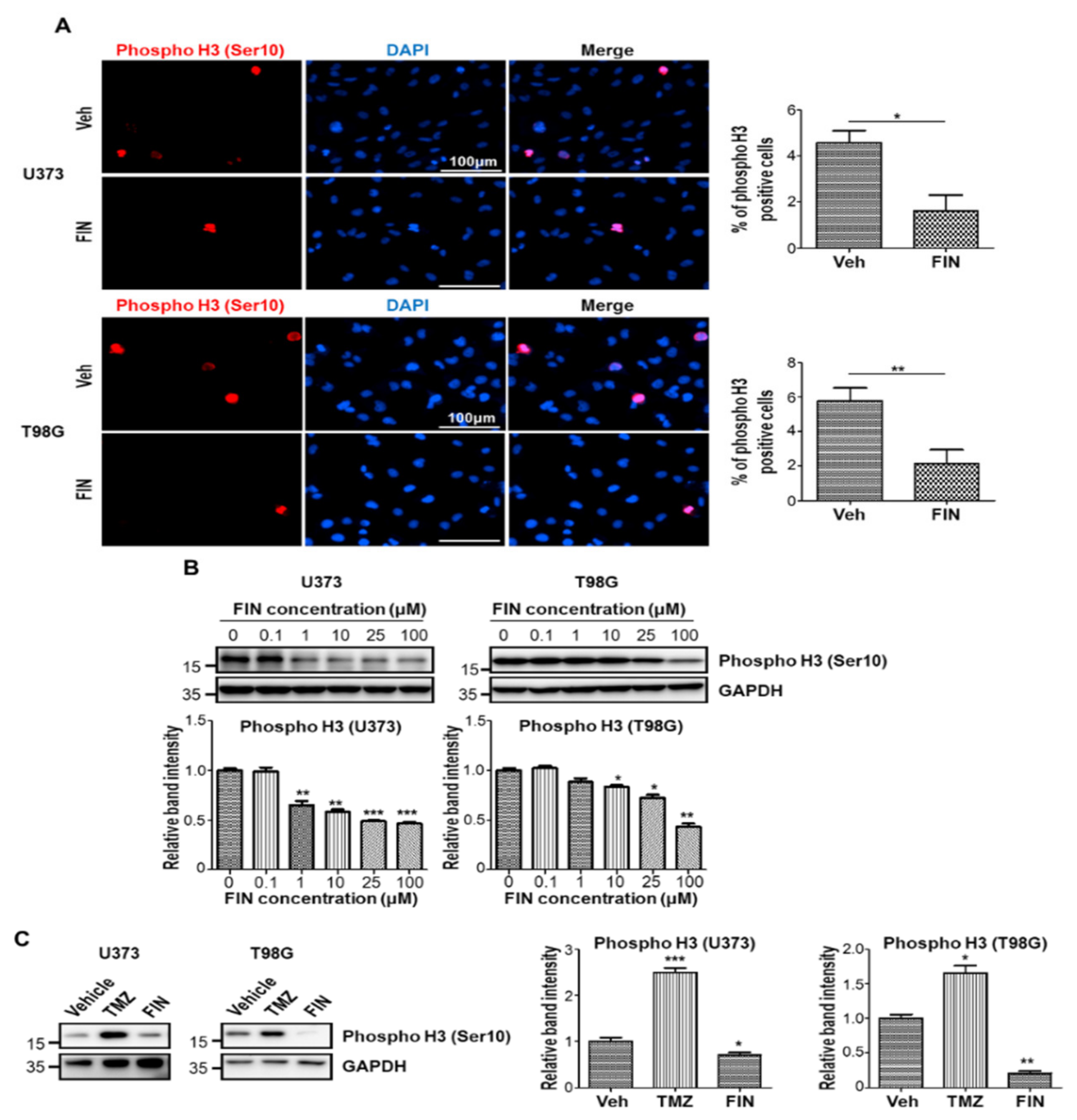
3.2. High-Dose Finasteride Shows Antiproliferative Function against Glioblastoma
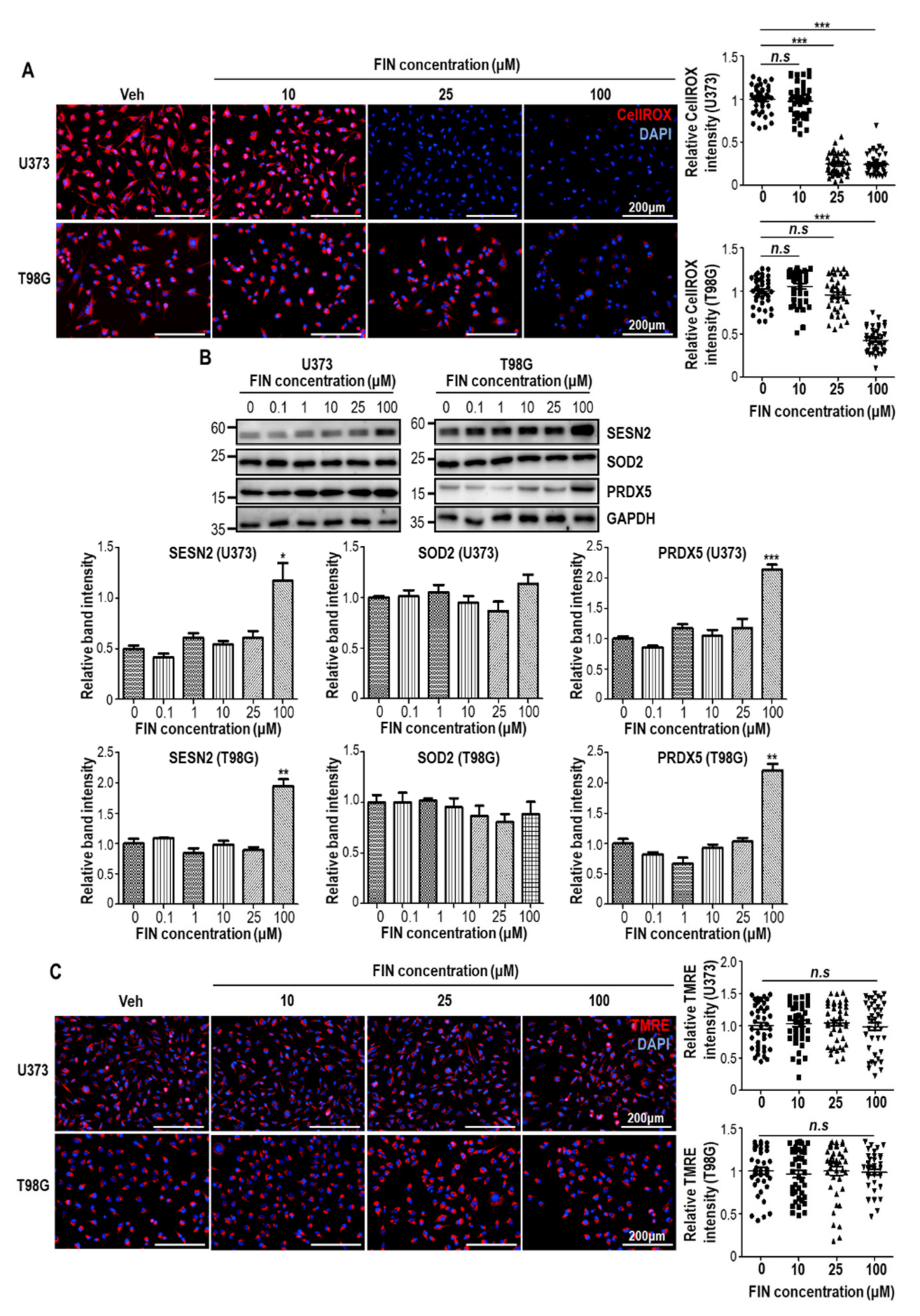
3.3. Finasteride Diminishes Intracellular Reactive Oxygen Species (ROS) Level through Inducing the Expression of Antioxidant Genes
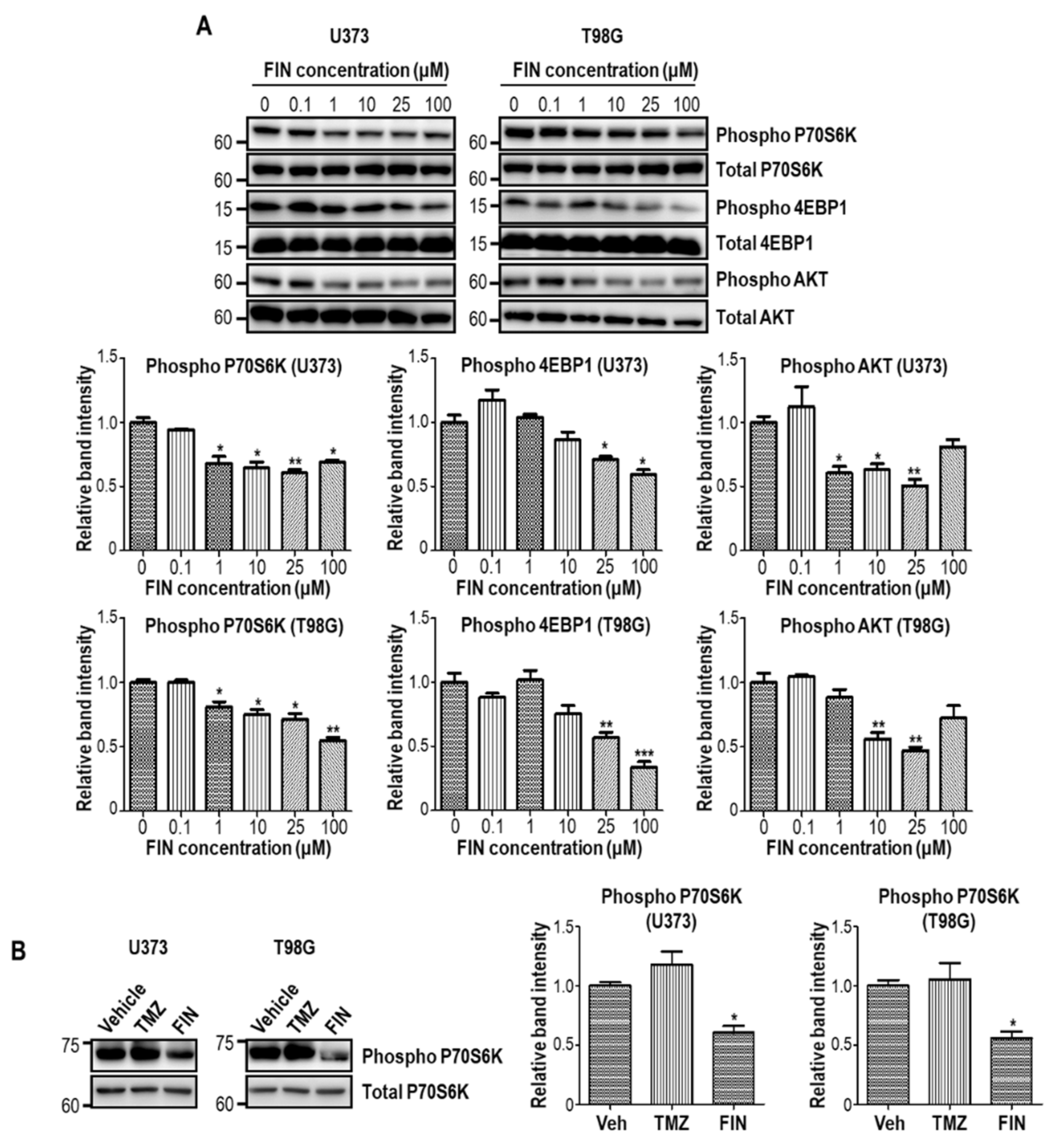
3.4. High-Dose Finasteride Suppresses AKT/mTOR Signaling in Glioblastoma Cells
3.5. Finasteride Seems Less Effective in Inducing DNA Damage than Temozolomide
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, H.J.; Kim, D.Y. Present and Future of Anti-Glioblastoma Therapies: A Deep Look into Molecular Dependencies/Features. Molecules 2020, 25, 4641. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Lara-Velazquez, M.; Al-Kharboosh, R.; Jeanneret, S.; Vazquez-Ramos, C.; Mahato, D.; Tavanaiepour, D.; Rahmathulla, G.; Quinones-Hinojosa, A. Advances in Brain Tumor Surgery for Glioblastoma in Adults. Brain Sci. 2017, 7, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Baker, S.D.; Wirth, M.; Statkevich, P.; Reidenberg, P.; Alton, K.; Sartorius, S.E.; Dugan, M.; Cutler, D.; Batra, V.; Grochow, L.B.; et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin. Cancer Res. 1999, 5, 309–317. [Google Scholar] [PubMed]
- Rabe, M.; Dumont, S.; Alvarez-Arenas, A.; Janati, H.; Belmonte-Beitia, J.; Calvo, G.F.; Thibault-Carpentier, C.; Sery, Q.; Chauvin, C.; Joalland, N.; et al. Identification of a transient state during the acquisition of temozolomide resistance in glioblastoma. Cell Death Dis. 2020, 11, 19. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.Z.; Wang, Z.F.; Lan, T.; Huang, W.H.; Zhao, Y.H.; Ma, C.; Li, Z.Q. Carmustine as a Supplementary Therapeutic Option for Glioblastoma: A Systematic Review and Meta-Analysis. Front. Neurol. 2020, 11, 1036. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef] [Green Version]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.J.; Kwon, H.S.; Kang, M.; Leem, H.H.; Lee, K.H.; Kim, D.Y. The Antitumor Natural Compound Falcarindiol Disrupts Neural Stem Cell Homeostasis by Suppressing Notch Pathway. Int. J. Mol. Sci. 2018, 19, 3432. [Google Scholar] [CrossRef] [Green Version]
- Beier, D.; Rohrl, S.; Pillai, D.R.; Schwarz, S.; Kunz-Schughart, L.A.; Leukel, P.; Proescholdt, M.; Brawanski, A.; Bogdahn, U.; Trampe-Kieslich, A.; et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008, 68, 5706–5715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, H.W.; Leung, C.O.; Lau, E.Y.; Chung, K.P.S.; Mok, E.H.; Lei, M.M.L.; Leung, R.W.H.; Tong, M.; Keng, V.W.; Ma, C.; et al. EPHB2 activates beta-catenin to enhance cancer stem cell properties and drive sorafenib resistance in hepatocellular carcinoma. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Agathocleous, M.; Iordanova, I.; Willardsen, M.I.; Xue, X.Y.; Vetter, M.L.; Harris, W.A.; Moore, K.B. A directional Wnt/beta-catenin-Sox2-proneural pathway regulates the transition from proliferation to differentiation in the Xenopus retina. Development 2009, 136, 3289–3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeman, M.T.; Slusarski, D.C.; Kaykas, A.; Louie, S.H.; Moon, R.T. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol. 2003, 13, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Yun, E.J.; Kim, S.; Hsieh, J.T.; Baek, S.T. Wnt/beta-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 2020, 11, 771. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- The Gene Ontology, C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Fielden, M.R.; Brennan, R.; Gollub, J. A gene expression biomarker provides early prediction and mechanistic assessment of hepatic tumor induction by nongenotoxic chemicals. Toxicol. Sci. 2007, 99, 90–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmeda, D.; Castel, S.; Vilaro, S.; Cano, A. Beta-catenin regulation during the cell cycle: Implications in G2/M and apoptosis. Mol. Biol. Cell 2003, 14, 2844–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambright, H.G.; Meng, P.; Kumar, A.P.; Ghosh, R. Inhibition of PI3K/AKT/mTOR axis disrupts oxidative stress-mediated survival of melanoma cells. Oncotarget 2015, 6, 7195–7208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funato, Y.; Michiue, T.; Asashima, M.; Miki, H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat. Cell Biol. 2006, 8, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, S.; Marinkovic, D.; Mungamuri, S.K.; Zhang, X.; Tong, W.; Sellers, R.; Ghaffari, S. ROS-mediated amplification of AKT/mTOR signalling pathway leads to myeloproliferative syndrome in Foxo3(−/−) mice. EMBO J. 2010, 29, 4118–4131. [Google Scholar] [CrossRef] [Green Version]
- Saadeh, F.S.; Mahfouz, R.; Assi, H.I. EGFR as a clinical marker in glioblastomas and other gliomas. Int. J. Biol. Markers 2018, 33, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Ronellenfitsch, M.W.; Brucker, D.P.; Burger, M.C.; Wolking, S.; Tritschler, F.; Rieger, J.; Wick, W.; Weller, M.; Steinbach, J.P. Antagonism of the mammalian target of rapamycin selectively mediates metabolic effects of epidermal growth factor receptor inhibition and protects human malignant glioma cells from hypoxia-induced cell death. Brain 2009, 132, 1509–1522. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Lozano, D.C.; Velazquez-Vazquez, D.E.; Del Moral-Morales, A.; Camacho-Arroyo, I. Dihydrotestosterone Induces Proliferation, Migration, and Invasion of Human Glioblastoma Cell Lines. OncoTargets Ther. 2020, 13, 8813–8823. [Google Scholar] [CrossRef] [PubMed]
- Pinacho-Garcia, L.M.; Valdez, R.A.; Navarrete, A.; Cabeza, M.; Segovia, J.; Romano, M.C. The effect of finasteride and dutasteride on the synthesis of neurosteroids by glioblastoma cells. Steroids 2020, 155, 108556. [Google Scholar] [CrossRef]
- Patil, V.; Pal, J.; Somasundaram, K. Elucidating the cancer-specific genetic alteration spectrum of glioblastoma derived cell lines from whole exome and RNA sequencing. Oncotarget 2015, 6, 43452–43471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, H.K.; Seo, S.Y.; Kim, J.H.; Kim, H.J.; Chie, E.K.; Kim, S.K.; Kim, I.H. Disulfiram, a Re-positioned Aldehyde Dehydrogenase Inhibitor, Enhances Radiosensitivity of Human Glioblastoma Cells In Vitro. Cancer Res. Treat. 2019, 51, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthurs, A.L.; Keating, D.J.; Stringer, B.W.; Conn, S.J. The Suitability of Glioblastoma Cell Lines as Models for Primary Glioblastoma Cell Metabolism. Cancers 2020, 12, 3722. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Byun, J.S.; Kwon, H.S.; Kim, D.Y. Cellular toxicity driven by high-dose vitamin C on normal and cancer stem cells. Biochem. Biophys. Res. Commun. 2018, 497, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Tsuchihashi, K.; Okazaki, S.; Ohmura, M.; Ishikawa, M.; Sampetrean, O.; Onishi, N.; Wakimoto, H.; Yoshikawa, M.; Seishima, R.; Iwasaki, Y.; et al. The EGF Receptor Promotes the Malignant Potential of Glioma by Regulating Amino Acid Transport System xc(-). Cancer Res. 2016, 76, 2954–2963. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequence (5’ to 3’) |
|---|---|
| hRPL32 | GAAGTTCCTGGTCCACAACG GCGATCTCGGCACAGTAAG |
| hSox2 | GAGCTTTGCAGGAAGTTTGC GCAAGAAGCCTCTCCTTGAA |
| hEGFR | TCCCCGTAATTATGTGGTGAC AGGCCCTTCGCACTTCTTAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.J.; Kim, T.-J.; Kim, Y.G.; Seong, C.; Cho, J.-H.; Kim, W.; Lee, K.-H.; Kim, D.-Y. Antioxidant and Antiproliferative Activity of Finasteride against Glioblastoma Cells. Pharmaceutics 2021, 13, 1410. https://doi.org/10.3390/pharmaceutics13091410
Kim HJ, Kim T-J, Kim YG, Seong C, Cho J-H, Kim W, Lee K-H, Kim D-Y. Antioxidant and Antiproliferative Activity of Finasteride against Glioblastoma Cells. Pharmaceutics. 2021; 13(9):1410. https://doi.org/10.3390/pharmaceutics13091410
Chicago/Turabian StyleKim, Hyeon Ji, Tae-Jun Kim, Yu Gyung Kim, Chaeeun Seong, Jin-Hwa Cho, Wanil Kim, Kyung-Ha Lee, and Do-Yeon Kim. 2021. "Antioxidant and Antiproliferative Activity of Finasteride against Glioblastoma Cells" Pharmaceutics 13, no. 9: 1410. https://doi.org/10.3390/pharmaceutics13091410