
Physiologically Based Pharmacokinetic (PBPK) Modeling for Predicting Brain Levels of Drug in Rat



Abstract
:
1. Introduction
- Brain-specific properties, such as the properties of the brain vascular network and the different brain barriers, the characteristics of the brain tissue and the CSF, the fluid movements within the brain or the presence of metabolic enzymes in the CNS;
- Drug-specific properties, such as molecular (molecular weight, polar surface area, shape or number of hydrogen bond donors and acceptors), physicochemical (pKa, solubility or lipophilicity) and pharmacokinetic properties;
- Processes affecting drug distribution within the brain, e.g., the drug transport through the brain vascular system, the brain barriers or within the brain fluids, the drug extra-/intracellular exchange, the drug binding or the drug metabolism.
2. Materials and Methods
2.1. Drugs and Products
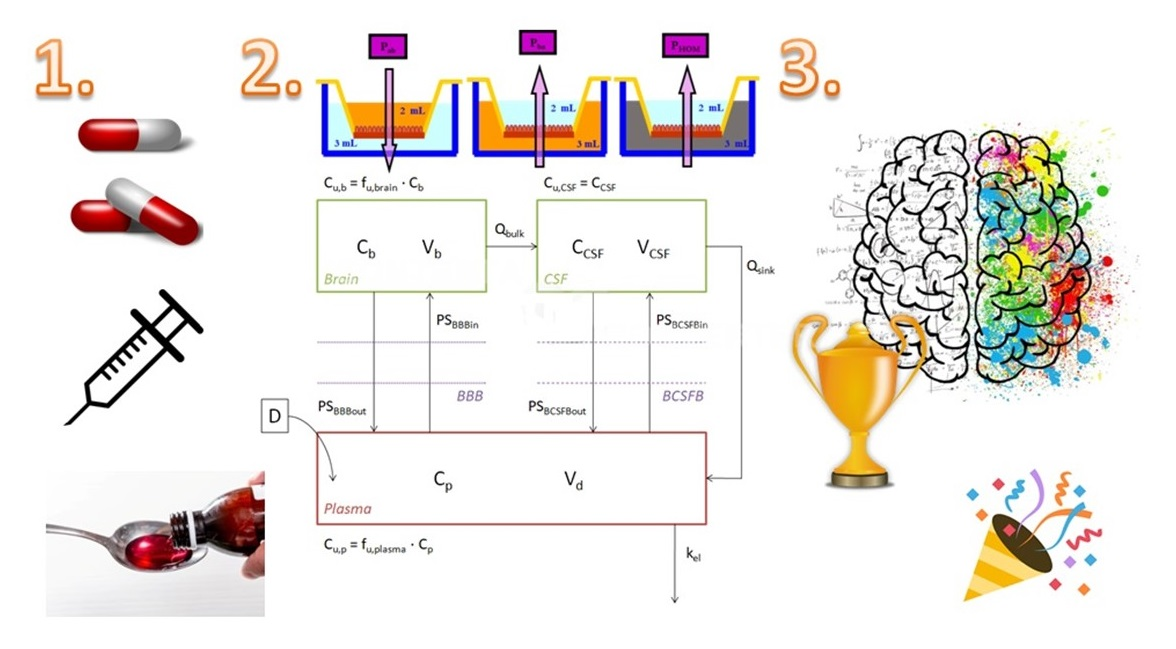
2.2. Cell Culture and Permeability Studies
- Standard AB. This experiment was designed for obtaining the apparent influx permeability (Papp A→B). The drug was dissolved in HBSS; this solution was put into the apical chamber of the transwell and the basolateral chamber was filled with cleaned HBSS. Four samples were taken from the basolateral chamber at pre-established times (15, 30, 60 and 90 min) [11,12]. Three replicates were carried out for each drug;
- Standard BA. In this case, the montage was the opposite to the first condition and the basolateral chamber was filled with a drug solution in HBSS, while the apical chamber was filled with cleaned HBSS. Four samples were taken from the apical chamber at pre-established times (15, 30, 60 and 90 min) [11,12]. With this test, the apparent efflux permeability (Papp B→A) was obtained. Three replicates were carried out for each drug;
- Brain homogenate BA. This last condition gives the free drug apparent efflux permeability (Papp HOM), as the drug is added to the basolateral chamber after being dissolved in a 1:3 pig brain homogenate:phosphate buffer (180 mM, pH 7.4) solution, and only the free fraction of drug will be able to cross to the apical chamber, where 4 samples were taken at pre-established times (15, 30, 60 and 90 min) [11,12]. Three replicates were carried out for each drug.
2.3. HPLC Analysis of the Samples
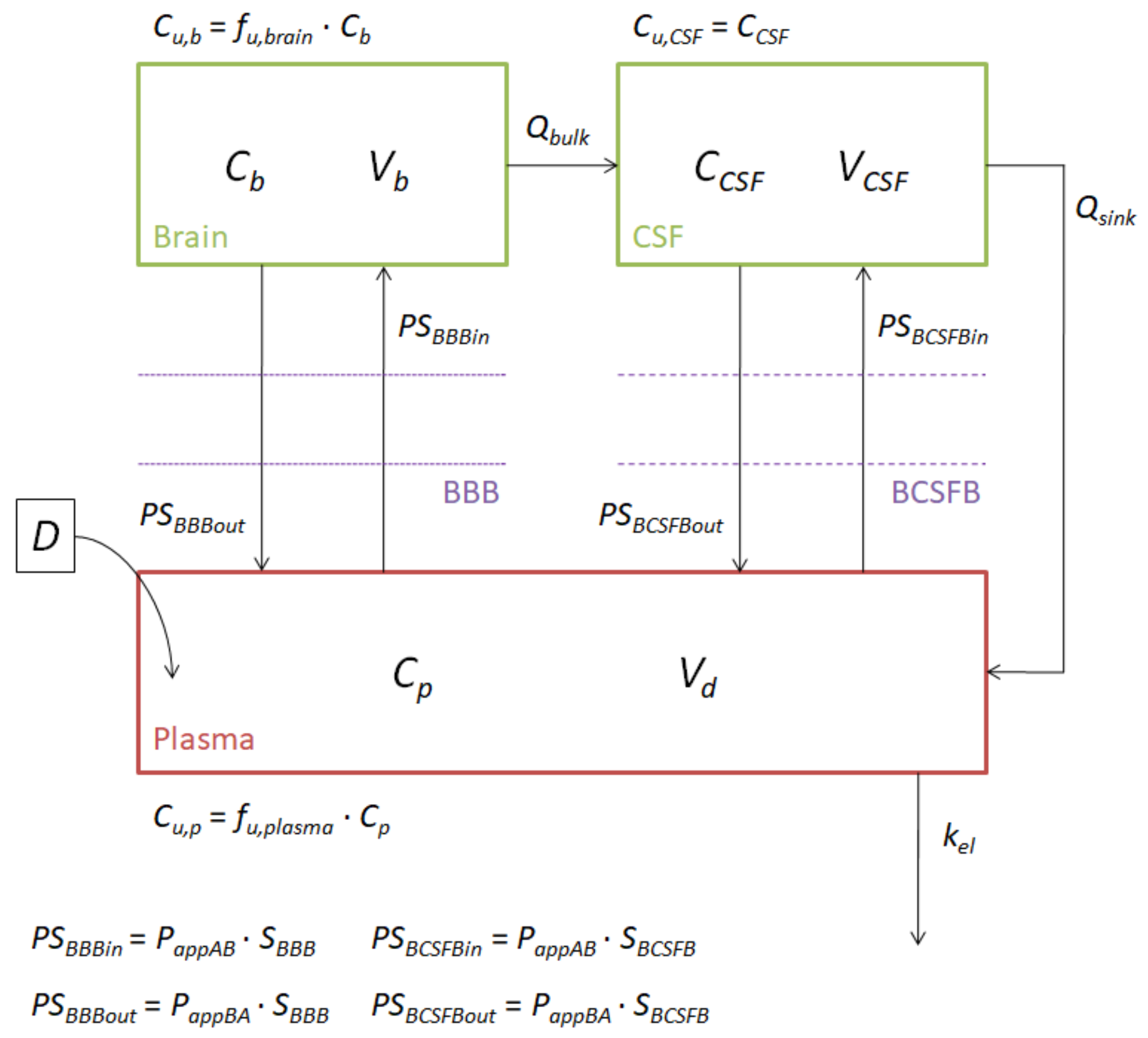
2.4. Model Construction
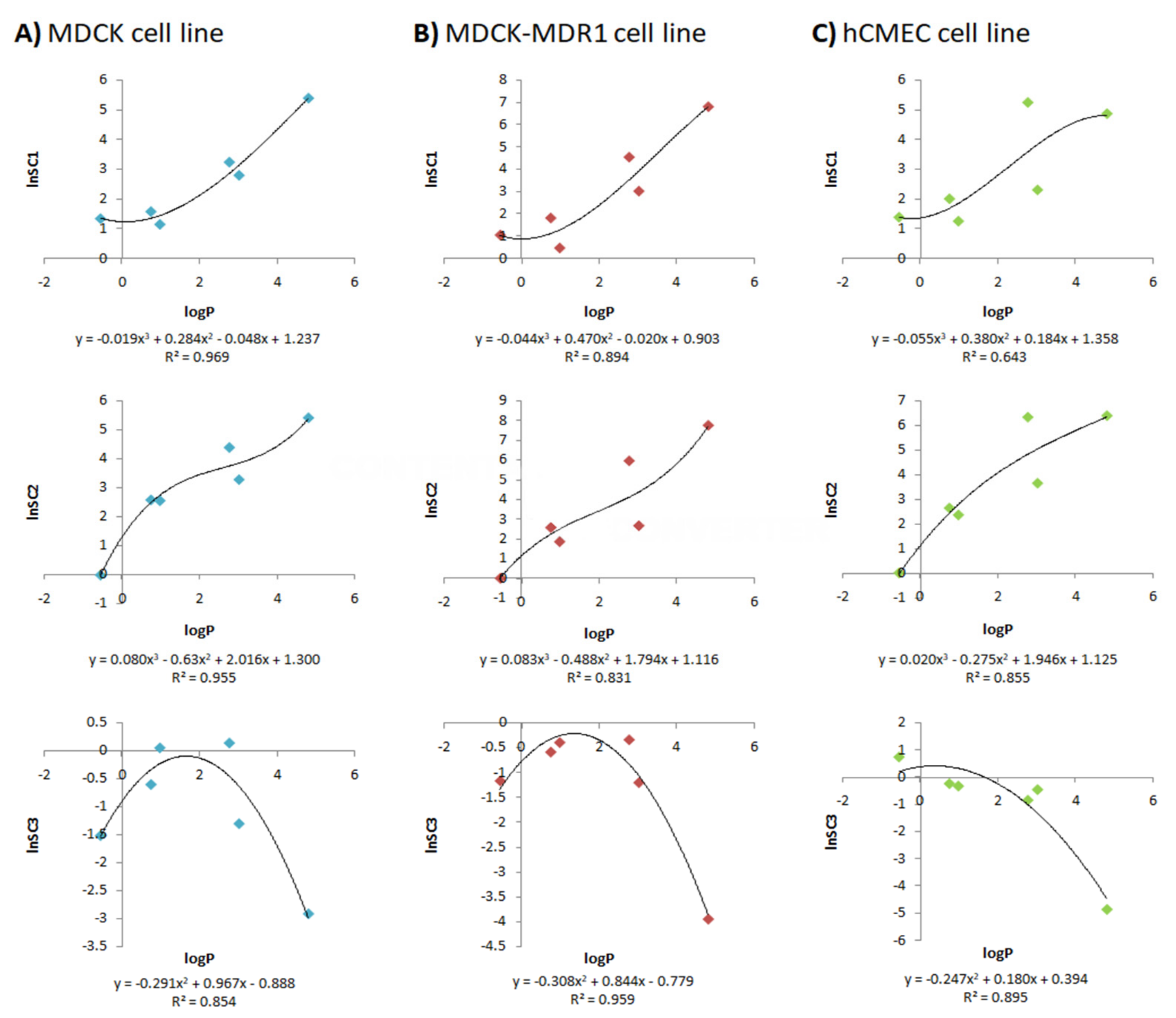
2.5. Quantitative Structure–Property Relationships (QSPRs)
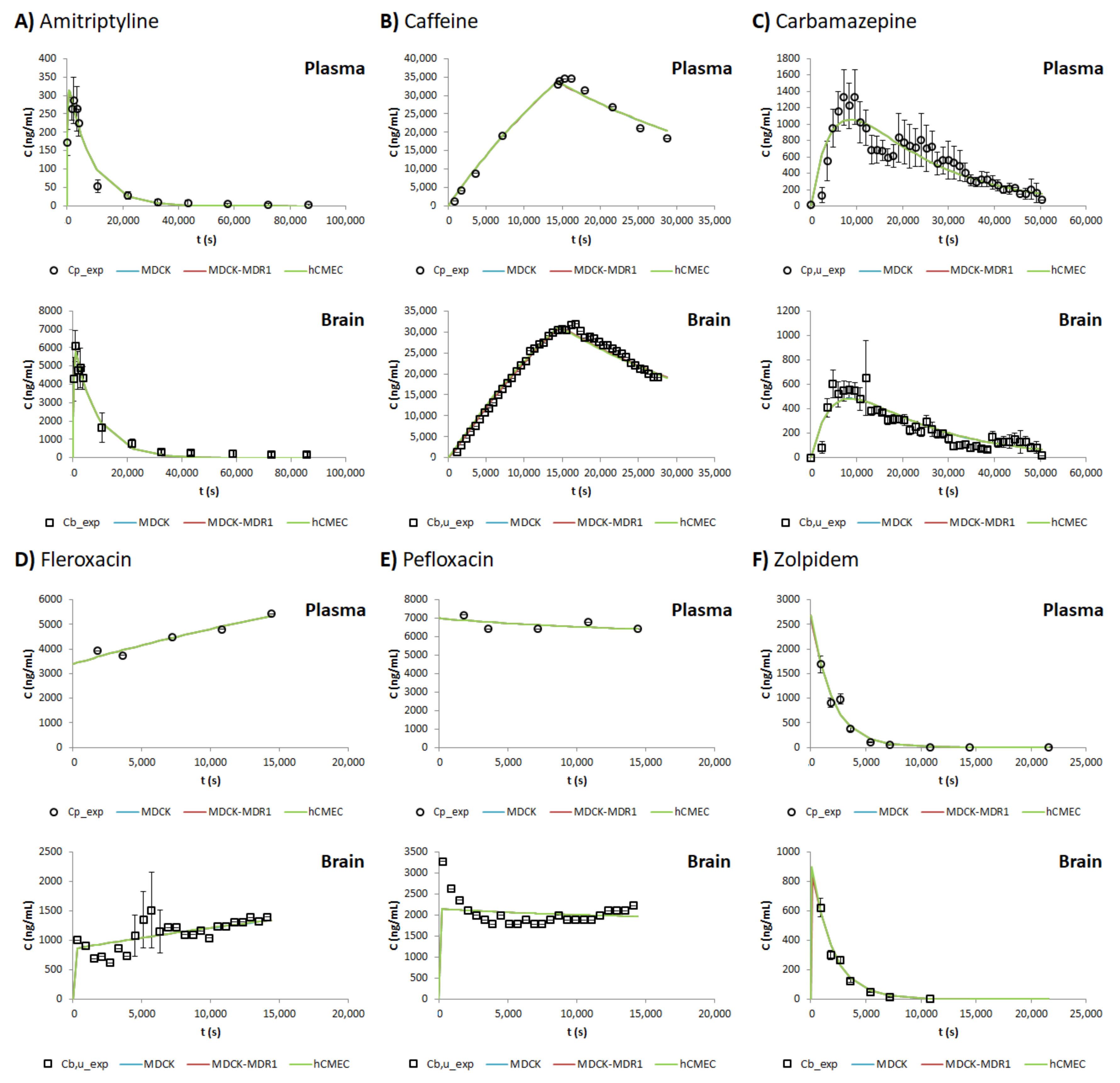
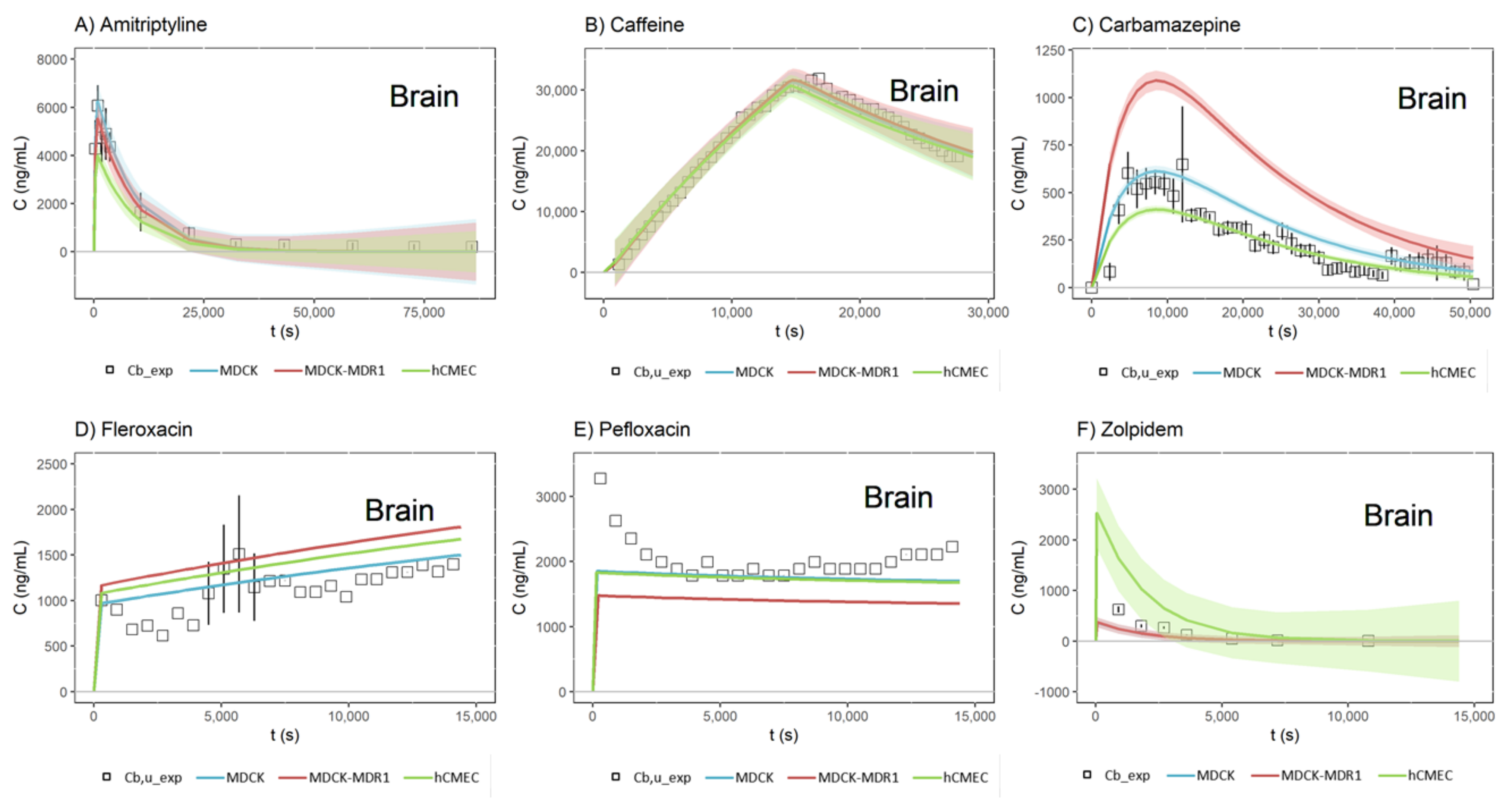
3. Results
4. Discussion
- SC1 and SC2 were equivalent to those defined by Ball et al., as they transform the apparent permeabilities of the MDCK, MDCK-MDR1 and hCMEC/D3 cell lines into influx and efflux clearances through the BBB. The main justification for introducing these scaling factors is the difference between the rat BBB and the in vitro monolayers. For instance, in terms of tight junctions, it has been proven that primary rat brain endothelial cell cultures have high levels of occludin, endothelial cell-specific adhesion molecule (ESAM) and claudin-5, while in MDCK and MDCK-MDR1 cells, the most abundant proteins are claudin-1 and claudin-2, and in hCMEC/D3, claudin-11 [34]. Furthermore, there are differences in the morphology of the different cell lines and in transporter expressions [34];
- The reasons for adding SC3, which re-scale the in vitro fu,brain, were, on the one hand, to bypass the inter-species differences, as the brain homogenate used in the in vitro studies came from pigs and the model was to be used to predict rat brain profiles, and, on the other hand, to correct the possible error present in the parameter due to the homogenization process, as brain structures are broken and membrane and internal proteins get mixed.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Telano, L.N.; Baker, S. Physiology, Cerebral Spinal Fluid (CSF); StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Vendel, E.; Rottschäfer, V.; de Lange, E.C.M. The need for mathematical modelling of spatial drug distribution within the brain. Fluids Barriers CNS 2019, 16, 12. [Google Scholar] [CrossRef]
- Abbot, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Sánchez-Dengra, B.; González-Álvarez, I.; Bermejo, M.; González-Álvarez, M. Nanomedicine in the Treatment of Pathologies of the Central Nervous System Advances in Nanomedicine. In Advances in Nanomedicine; Open Access Ebooks: Las Vegas, NV, USA, 2020. [Google Scholar]
- Hammarlund-Udenaes, M.; Fridén, M.; Syvänen, S.; Gupta, A. On the rate and extent of drug delivery to the brain. Pharm. Res. 2008, 25, 1737–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangas-Sanjuan, V.; González-Alvarez, M.; Gonzalez-Alvarez, I.; Bermejo, M. Drug penetration across the blood-brain barrier: An overview. Ther. Deliv. 2010, 1, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, C.; Graff, C.L.; Pollack, G.M. Variable modulation of opioid brain uptake by P-glycoprotein in mice. Biochem. Pharmacol. 2004, 67, 269–276. [Google Scholar] [CrossRef]
- Loryan, I.; Sinha, V.; Mackie, C.; van Peer, A.; Drinkenburg, W.; Vermeulen, A.; Morrison, D.; Monshouwer, M.; Heald, D.; Hammarlund-Udenaes, M. Mechanistic understanding of brain drug disposition to optimize the selection of potential neurotherapeutics in drug discovery. Pharm. Res. 2014, 31, 2203–2219. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A Method to Predict Blood-Brain Barrier Permeability of Drug-Like Compounds Using Molecular Dynamics Simulations. Biophys. J. 2014, 107, 630–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geldenhuys, W.J.; Mohammad, A.S.; Adkins, C.E.; Lockman, P.R. Molecular determinants of blood-brain barrier permeation. Ther. Deliv. 2015, 6, 961–971. [Google Scholar] [CrossRef]
- Mangas-Sanjuan, V.; González-Álvarez, I.; González-Álvarez, M.; Casabó, V.G.; Bermejo, M. Innovative in vitro method to predict rate and extent of drug delivery to the brain across the blood-brain barrier. Mol. Pharm. 2013, 10, 3822–3831. [Google Scholar] [CrossRef]
- Sánchez-Dengra, B.; González-Álvarez, I.; Sousa, F.; Bermejo, M.; González-Álvarez, M.; Sarmento, B. In vitro model for predicting the access and distribution of drugs in the brain using hCMEC/D3 cells. Eur. J. Pharm. Biopharm. 2021, 163, 120–126. [Google Scholar] [CrossRef]
- Mangas-Sanjuan, V.; Gonzalez-Alvarez, M.; Gonzalez-Alvarez, I.; Bermejo, M. In vitro methods for assessing drug access to the brain. In Advances in Non-Invasive Drug Delivery to the Brain; Future Science Ltd.: London, UK, 2015; pp. 44–61. ISBN 9781909453937. [Google Scholar]
- Ball, K.; Bouzom, F.; Scherrmann, J.M.; Walther, B.; Declèves, X. A physiologically based modeling strategy during preclinical CNS drug development. Mol. Pharm. 2014, 11, 836–848. [Google Scholar] [CrossRef]
- Engelhard, H.H.; Arnone, G.D.; Mehta, A.I.; Nicholas, M.K. Biology of the Blood-Brain and Blood-Brain Tumor Barriers. In Handbook of Brain Tumor Chemotherapy, Molecular Therapeutics, and Immunotherapy: Second Edition; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 113–125. ISBN 9780128121009. [Google Scholar]
- Coudore, F.; Besson, A.; Eschalier, A.; Lavarenne, J.; Fialip, J. Plasma and brain pharmacokinetics of amitriptyline and its demethylated and hydroxylated metabolites after one and six half-life repeated administrations to rats. Gen. Pharmacol. 1996, 27, 215–219. [Google Scholar] [CrossRef]
- Hansen, D.K.; Scott, D.O.; Otis, K.W.; Lunte, S.M. Comparison of in vitro BBMEC permeability and in vivo CNS uptake by microdialysis sampling. J. Pharm. Biomed. Anal. 2002, 27, 945–958. [Google Scholar] [CrossRef]
- Graumlich, J.F.; McLaughlin, R.G.; Birkhahn, D.; Shah, N.; Burk, A.; Jobe, P.C.; Dailey, J.W. Subcutaneous microdialysis in rats correlates with carbamazepine concentrations in plasma and brain. Epilepsy Res. 2000, 40, 25–32. [Google Scholar] [CrossRef]
- Ooie, T.; Terasaki, T.; Suzuki, H.; Sugiyama, Y. Quantitative brain microdialysis study on the mechanism of quinolones distribution in the central nervous system. Drug Metab. Dispos. 1997, 25, 784–789. [Google Scholar] [PubMed]
- Garrigou-Gadenne, D.; Burke, J.T.; Durand, A.; Depoortere, H.; Thénot, J.P.; Morselli, P.L. Pharmacokinetics, brain distribution and pharmaco-electrocorticographic profile of zolpidem, a new hypnotic, in the rat. J. Pharmacol. Exp. Ther. 1989, 248, 1283–1288. [Google Scholar] [PubMed]
- González-Alvarez, I.; Fernández-Teruel, C.; Casabó-Alós, V.G.; Garrigues, T.M.; Polli, J.E.; Ruiz-García, A.; Bermejo, M. In situ kinetic modelling of intestinal efflux in rats: Functional characterization of segmental differences and correlatlon with in vitro results. Biopharm. Drug Dispos. 2007, 28, 229–239. [Google Scholar] [CrossRef]
- Fernandez-Teruel, C.; Gonzalez-Alvarez, I.; Casabó, V.; Ruiz-Garcia, A.; Bermejo, B. Kinetic modelling of the intestinal transport of sarafloxacin. Studies in situ in rat and in vitro in Caco-2 cells. J. Drug Target. 2005, 13, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Fridén, M.; Gupta, A.; Antonsson, M.; Bredberg, U.; Hammarlund-Udenaes, M. In vitro methods for estimating unbound drug concentrations in the brain interstitial and intracellular fluids. Drug Metab. Dispos. 2007, 35, 1711–1719. [Google Scholar] [CrossRef] [Green Version]
- Kodaira, H.; Kusuhara, H.; Fujita, T.; Ushiki, J.; Fuse, E.; Sugiyama, Y. Quantitative evaluation of the impact of active efflux by P-glycoprotein and breast cancer resistance protein at the blood-brain barrier on the predictability of the unbound concentrations of drugs in the brain using cerebrospinal fluid concentration as a. J. Pharmacol. Exp. Ther. 2011, 339, 935–944. [Google Scholar] [CrossRef] [Green Version]
- DrugBank DrugBank. Available online: https://www.drugbank.ca/ (accessed on 14 May 2020).
- Remmel, R.P.; Sinz, M.W.; Cloyd, J.C. Dose-Dependent Pharmacokinetics of Carbamazepine in Rats: Determination of the Formation Clearance of Carbamazepine-10,11-epoxide. Pharm. Res. An Off. J. Am. Assoc. Pharm. Sci. 1990, 7, 513–517. [Google Scholar] [CrossRef]
- Weidekamm, E.; Portmann, R.; Suter, K.; Partos, C.; Dell, D.; Lucker, P.W. Single- and multiple-dose pharmacokinetics of fleroxacin, a trifluorinated quinolone, in humans. Antimicrob. Agents Chemother. 1987, 31, 1909–1914. [Google Scholar] [CrossRef] [Green Version]
- Bulitta, J.B.; Jiao, Y.; Landersdorfer, C.B.; Sutaria, D.S.; Tao, X.; Shin, E.; Höhl, R.; Holzgrabe, U.; Stephan, U.; Sörgel, F. Comparable bioavailability and disposition of pefloxacin in patients with cystic fibrosis and healthy volunteers assessed via population pharmacokinetics. Pharmaceutics 2019, 11, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chartrand, S.A. Quinolones in Animal Models of Infection. In The New Generation of Quinolones; Siporin, C., Heifetz, C., Domagala, J., Eds.; Marcel Dekker: New York, NY, USA, 1990; ISBN 0-8247-8224-0. [Google Scholar]
- Bransford, P.; Cook, J.; Gupta, M.; Haertter, S.; He, H.; Ju, R.; Kanodia, J.; Lennernäs, H.; Lindley, D.; Polli, J.E.; et al. ICH M9 Guideline in Development on Biopharmaceutics Classification System-Based Biowaivers: An Industrial Perspective from the IQ Consortium. Mol. Pharm. 2020, 17, 361–372. [Google Scholar] [CrossRef] [PubMed]
- EMA-Committee for Medicinal Products for Human Use (CHMP). Guideline on the Pharmacokinetic and Clinical Evaluation of Modified Release Dosage Forms (EMA/CPMP/EWP/280/96 Corr1); European Medicines Agency: London, UK, 2014. [Google Scholar]
- Ruiz Picazo, A.; Martinez-Martinez, M.T.; Colón-Useche, S.; Iriarte, R.; Sánchez-Dengra, B.; González-Álvarez, M.; García-Arieta, A.; González-Álvarez, I.; Bermejo, M. In Vitro Dissolution as a Tool for Formulation Selection: Telmisartan Two-Step IVIVC. Mol. Pharm. 2018, 15, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Campos, A.; Sánchez-Dengra, B.; Merino, V.; Dahan, A.; González-Álvarez, I.; García-Arieta, A.; González-Álvarez, M.; Bermejo, M. Candesartan Cilexetil In Vitro-In Vivo Correlation: Predictive Dissolution as a Development Tool. Pharmaceutics 2020, 12, 633. [Google Scholar] [CrossRef]
- Veszelka, S.; Tóth, A.; Walter, F.R.; Tóth, A.E.; Gróf, I.; Mészáros, M.; Bocsik, A.; Hellinger, É.; Vastag, M.; Rákhely, G.; et al. Comparison of a rat primary cell-based blood-brain barrier model with epithelial and brain endothelial cell lines: Gene expression and drug transport. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garberg, P.; Ball, M.; Borg, N.; Cecchelli, R.; Fenart, L.; Hurst, R.D.; Lindmark, T.; Mabondzo, A.; Nilsson, J.E.; Raub, T.J.; et al. In vitro models for the blood-brain barrier. Toxicol. Vitr. 2005, 19, 299–334. [Google Scholar] [CrossRef]
- Molino, Y.; Jabès, F.; Lacassagne, E.; Gaudin, N.; Khrestchatisky, M. Setting-up an In Vitro Model of Rat Blood-brain Barrier (BBB): A Focus on BBB Impermeability and Receptor-mediated Transport. J. Vis. Exp. 2014, 88, 51278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-vitro blood-brain barrier models for drug screening and permeation studies: An overview. Drug Des. Devel. Ther. 2019, 13, 3591–3605. [Google Scholar] [CrossRef] [Green Version]
- Bermejo, M.; Avdeef, A.; Ruiz, A.; Nalda, R.; Ruell, J.A.; Tsinman, O.; González, I.; Fernández, C.; Sánchez, G.; Garrigues, T.M.; et al. PAMPA-A drug absorption in vitro model: 7. Comparing rat in situ, Caco-2, and PAMPA permeability of fluoroquinolones. Eur. J. Pharm. Sci. 2004, 21, 429–441. [Google Scholar] [CrossRef]
- Garrigues, T.M.; Pérez-Varona, A.T.; Climent, E.; Bermejo, M.V.; Martín-Villodre, A.; Plá-Delfina, J.M. Gastric absorption of acidic xenobiotics in the rat: Biophysical interpretation of an apparently atypical behaviour. Int. J. Pharm. 1990, 64, 127–138. [Google Scholar] [CrossRef]
- Sánchez-Castaño, G.; Ruíz-García, A.; Bañón, N.; Bermejo, M.; Merino, V.; Freixas, J.; Garriguesx, T.M.; Plá-Delfina, J.M. Intrinsic absolute bioavailability prediction in rats based on in situ absorption rate constants and/or in vitro partition coefficients: 6-Fluoroquinolones. J. Pharm. Sci. 2000, 89, 1395–1403. [Google Scholar] [CrossRef]
- Lanevskij, K.; Dapkunas, J.; Juska, L.; Japertas, P.; Didziapetris, R. QSAR analysis of blood-brain distribution: The influence of plasma and brain tissue binding. J. Pharm. Sci. 2011, 100, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Esaki, T.; Kawashima, H.; Natsume-Kitatani, Y.; Nagao, C.; Ohashi, R.; Mizuguchi, K. Predicting Fraction Unbound in Human Plasma from Chemical Structure: Improved Accuracy in the Low Value Ranges. Mol. Pharm. 2018, 15, 5302–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Horie, K.; Borchardt, R.T. Are MDCK cells transfected with the human MRP2 gene a good model of the human intestinal mucosa? Pharm. Res. 2002, 19, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.J.; Mendes, B.; Martins, S.; Sarmento, B. Cell-based in vitro models for studying blood-brain barrier (BBB) permeability. In Concepts and Models for Drug Permeability Studies: Cell and Tissue Based In Vitro Culture Models; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 169–188. ISBN 9780081001141. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | fu,plasma | Vd (cm3) * | kel (s−1) * | ka (s−1) * | D (ng) | k0 (ng/s) | Rat Weight (g) |
|---|---|---|---|---|---|---|---|
| Amitriptyline | 0.090 a | 4000 c,+ | 7.70 × 10−5 h | 1.54·10−4 # | 5,000,000 h | 250 h | |
| Caffeine | 0.917 b | 180 c,+ | 3.55 × 10−5 i,$ | 833.333 i | 300 i | ||
| Carbamazepine | 0.385 b | 1490.5 d | 4.50 × 10−5 j | 8.99·10−5 # | 3,600,000 j | 300 j | |
| Fleroxacin | 0.793 b | 427.5 e,+ | 7.13 × 10−5 k | 1,114,350 l | 83.125 l | 285 l | |
| Pefloxacin | 0.860 b | 361.1f,+ | 5.83 × 10−5 k | 3,676,500 l | 214.542 l | 285 l | |
| Zolpidem | 0.267 b | 304 g | 1.56 × 10−4 g | 499,700 g | 190 g |
| Drug | MDCK Cell Line | MDCK-MDR1 Cell Line | hCMEC Cell Line | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Papp A→B (×10−6 cm/s) | Papp B→A (×10−6 cm/s) | fu,brain | Papp A→B (×10−6 cm/s) | Papp B→A (×10−6 cm/s) | fu,brain | Papp A→B (×10−6 cm/s) | Papp B→A (×10−6 cm/s) | fu,brain | |
| Amitriptyline | 74.77 a | 178.48 a | 0.037 a | 17.95 a | 16.91 a | 0.104 a | 124.24 b | 66.21 b | 0.252 b |
| Caffeine | 26.10 | 35.31 | 0.857 | 33.57 | 30.59 | 0.613 | 63.93 | 194.70 | 0.095 |
| Carbamazepine | 114.64 | 78.66 | 0.673 | 142.96 | 75.64 | 0.238 | 70.14 b | 51.93 b | 0.386 b |
| Fleroxacin | 88.48 | 63.44 | 0.471 | 67.40 | 42.57 | 0.813 | 29.96 b | 25.73 b | 0.743 b |
| Pefloxacin | 41.21 | 37.49 c | 0.910 c | 30.82 | 35.39 c | 0.931 c | 24.95 b | 33.14 b | 0.642 b |
| Zolpidem | 21.32 | 36.48 c | 0.971 c | 8.92 | 33.43 c | 0.881 c | 106.16 b | 80.76 b | 0.408 b |
| Drug | Vd (cm3) | kel (s−1) | ka (s−1) | MDCK Cell Line | MDCK-MDR1 Cell Line | hCMEC Cell Line | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SC1 | SC2 | SC3 | SC1 | SC2 | SC3 | SC1 | SC2 | SC3 | ||||
| Amitriptyline | 14,632.6 | 1.17 × 10−4 | 5.86 × 10−3 | 220.59 | 224.82 | 0.05 | 920.39 | 2377.06 | 0.02 | 132.89 | 606.69 | 0.01 |
| Caffeine | 273.6 | 3.55 × 10−5 | 3.85 | 1.00 | 0.22 | 2.85 | 1.00 | 0.31 | 4.09 | 1.00 | 2.15 | |
| Carbamazepine | 827.0 | 5.39 × 10−5 | 2.15 × 10−4 | 25.71 | 81.01 | 1.16 | 95.00 | 391.22 | 0.71 | 193.66 | 569.98 | 0.44 |
| Fleroxacin | 327.3 | 2.69 × 10−5 | 3.18 | 12.96 | 1.07 | 1.59 | 6.43 | 0.68 | 3.58 | 10.65 | 0.74 | |
| Pefloxacin | 524.3 | 6.75 × 10−5 | 4.88 | 13.32 | 0.55 | 6.15 | 13.19 | 0.56 | 7.59 | 14.09 | 0.81 | |
| Zolpidem | 185.9 | 5.12 × 10−4 | 16.53 | 26.79 | 0.27 | 20.59 | 14.51 | 0.30 | 10.26 | 38.70 | 0.65 | |
| Profile | MDCK Cell Line | MDCK-MDR1 Cell Line | hCMEC Cell Line | |||
|---|---|---|---|---|---|---|
| %PE Cmax | %PE AUC | %PE Cmax | %PE AUC | %PE Cmax | %PE AUC | |
| Fitted | 14.34 | 5.28 | 15.05 | 5.45 | 13.56 | 5.16 |
| Simulated | 19.23 | 22.34 | 35.71 | 48.21 | 49.77 | 46.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Dengra, B.; Gonzalez-Alvarez, I.; Bermejo, M.; Gonzalez-Alvarez, M. Physiologically Based Pharmacokinetic (PBPK) Modeling for Predicting Brain Levels of Drug in Rat. Pharmaceutics 2021, 13, 1402. https://doi.org/10.3390/pharmaceutics13091402
Sánchez-Dengra B, Gonzalez-Alvarez I, Bermejo M, Gonzalez-Alvarez M. Physiologically Based Pharmacokinetic (PBPK) Modeling for Predicting Brain Levels of Drug in Rat. Pharmaceutics. 2021; 13(9):1402. https://doi.org/10.3390/pharmaceutics13091402
Chicago/Turabian StyleSánchez-Dengra, Bárbara, Isabel Gonzalez-Alvarez, Marival Bermejo, and Marta Gonzalez-Alvarez. 2021. "Physiologically Based Pharmacokinetic (PBPK) Modeling for Predicting Brain Levels of Drug in Rat" Pharmaceutics 13, no. 9: 1402. https://doi.org/10.3390/pharmaceutics13091402