
Investigating In Vitro and Ex Vivo Properties of Artemether/Lumefantrine Double-Fixed Dose Combination Lipid Matrix Tablets Prepared by Hot Fusion


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Solid Lipid Dispersions Utilizing Hot Fusion
2.3. Full Factorial Experimental Design
2.4. Preparation of Directly Compressed Tablet Formulations
2.5. Pharmacotechnical Characterization
2.6. Assay
2.7. Dissolution Behavior
2.8. Analysis of Drug Release Mechanism
2.9. In Vitro Permeability Studies
2.10. Statistical Data Analysis
3. Results and Discussions
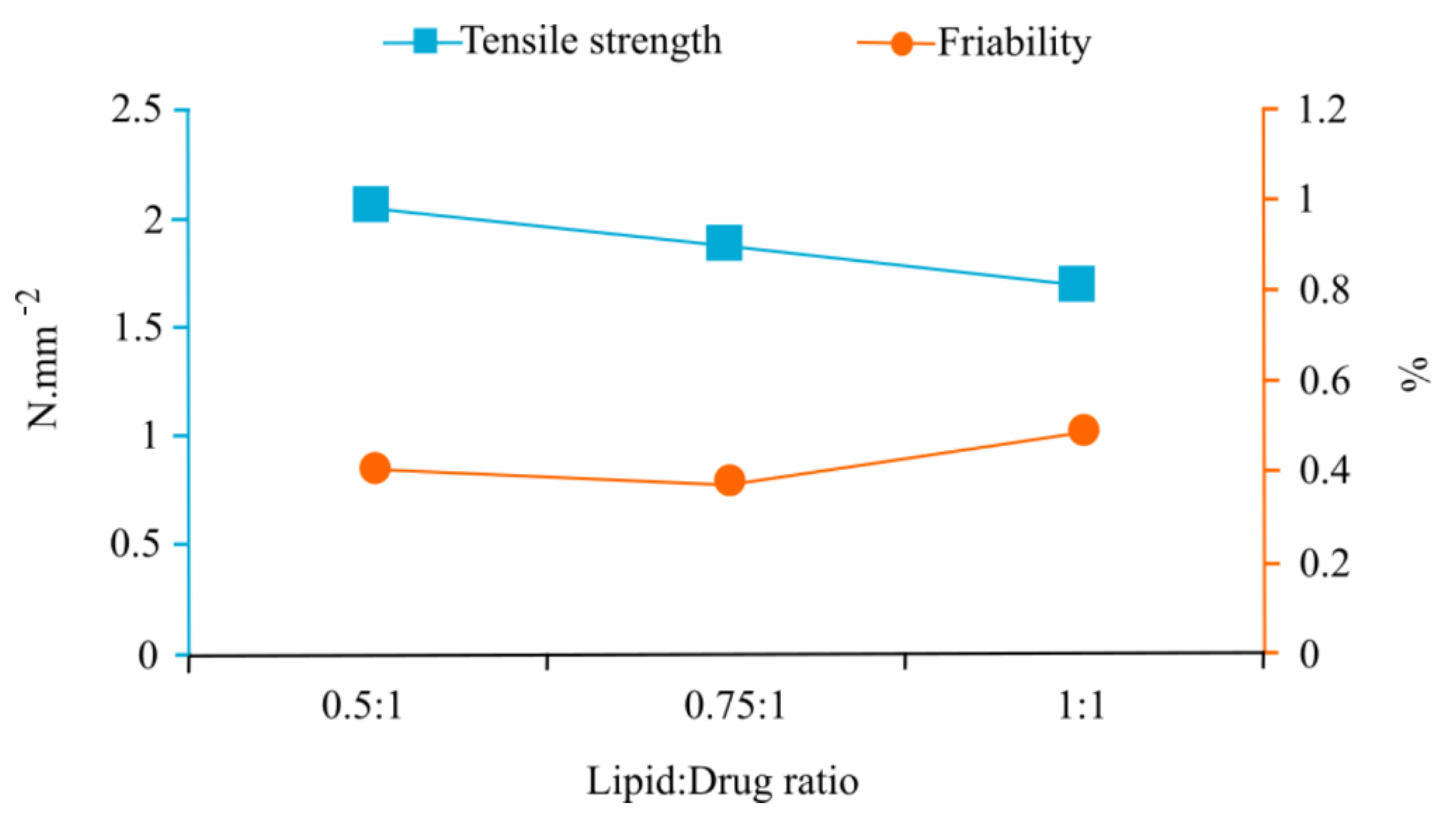
3.1. Analysis of Physical Tablet Properties
3.2. Assay
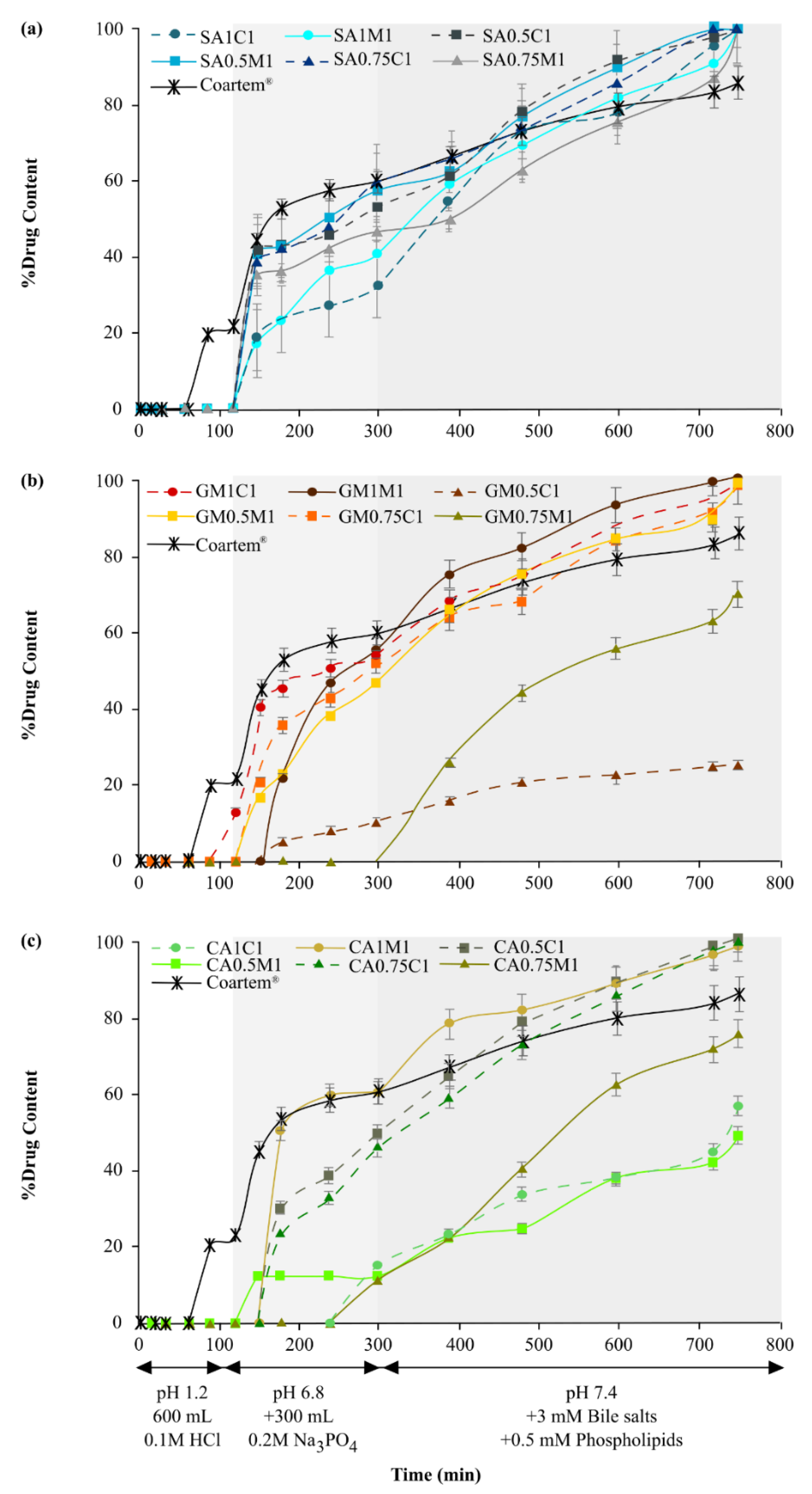
3.3. Dissolution Behavior
3.4. In Vitro Permeability Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The World Health Organization. World Malaria Report. Available online: https://www.who.int/news-room/feature-stories/detail/world-malaria-report-2019 (accessed on 14 December 2019).
- Khuda, F.; Iqbal, Z.; Shah, Y.; Ahmmad, L.; Nasir, F.; Khan, A.Z.; Shahbaz, N. Method development and validation for simultaneous determination of lumefantrine and its major metabolite, desbutyl lumefantrine in human plasma using RP-HPLC/UV detection. J. Chromatogr. B 2014, 944, 114–122. [Google Scholar] [CrossRef]
- Lindenberg, M.; Kopp, S.; Dressmann, J.B. Classification of orally administered drugs on the World Health Organization model list of essential medicines according to the biopharmaceutical classification system. Eur. J. Pharm. Biopharm. 2004, 58, 265–278. [Google Scholar] [CrossRef]
- Van, V.M.; Brockman, A.; Gemperli, B. Randomized comparison of artemether benflumetol and artesunate-mefloquine in treatment of multidrug-resistant falciparum malaria. Antimicrob. Agents Chemother. 1998, 42, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Ambike, A.A.; Mahadik, K.R.; Paradkar, A. Stability study of amorphous valdecoxib. Int. J. Pharm. 2004, 282, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergström, C.A.S.; Porter, C.J.S. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv. Drug Deliv. Rev. 2016, 101, 167–194. [Google Scholar] [CrossRef]
- Hofmann, A.F.; Borgström, B. The intraluminal phase of fat digestion in man: The lipid content of the micellar and oil phases of intestinal content obtained during fat digestion and absorption. J. Clin. Investig. 1964, 43, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Humberstone, A.J.; Porter, C.J.H.; Charman, W.N. A physiological basis for the effect of food on the absolute oral bioavailability of halofantrene. J. Pharm. Sci. 1996, 85, 525–529. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Pouton, C.W.; Cuine, J.F.; Charman, W.N. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 673–691. [Google Scholar] [CrossRef]
- Wilkins, C.A.; du Plessis, L.H.; Viljoen, J.M. Characterization of solid lipid dispersions prepared by hot fusion containing a double-fixed dose combination of artemether and lumefantrine. Drug Dev. Ind. Pharm. 2020, 46, 1289–1297. [Google Scholar] [CrossRef]
- Mu, H.; Holm, R.; Müllertz, A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int. J. Pharm. 2013, 453, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–633. [Google Scholar] [CrossRef]
- Corrigan, O.I. Mechanisms of dissolution of fast release solid dispersions. Drug Dev. Ind. Pharm. 1985, 11, 697–724. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef]
- Janssens, S.; van den Mooter, G. Review: Physical chemistry of solid dispersions. J. Pharm. Pharmacol. 2009, 61, 1571–1586. [Google Scholar] [CrossRef]
- Edueng, K.; Mahlin, D.; Bergström, C.A.S. The need of restructuring the disordered science of amorphous drug formulations. Pharm. Res. 2017, 34, 1754–1772. [Google Scholar] [CrossRef] [Green Version]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1675. [Google Scholar] [CrossRef]
- Kleberg, K.; Jacobsen, J.; Müllertz, A. Characterising the behaviour of poorly water soluble drugs in the intestine: Application of biorelevant media for solubility, dissolution and transport studies. J. Pharm. Pharmacol. 2010, 62, 1656–1668. [Google Scholar] [CrossRef]
- Zoeller, T.; Klein, S. Simplified biorelevant media for screening dissolution performance of poorly soluble drugs. Dissolut. Technol. 2007, 14, 8–13. [Google Scholar] [CrossRef]
- Sunesen, V.H.; Pedersen, B.L.; Kristensen, H.G.; Müllertz, A. In vivo in vitro correlations for a poorly soluble drug, danazol, using the flow-through dissolution method with biorelevant dissolution media. Eur. J. Pharm. Sci. 2005, 24, 305–313. [Google Scholar] [CrossRef]
- Wang, Q.; Fotaki, N.; Mao, Y. Biorelevant dissolution: Methodology and application in drug development. Dissolut. Technol. 2009, 16, 6–12. [Google Scholar] [CrossRef]
- Obaidat, A.A.; Obaidat, R.M. Release of tramadol hydrochloride from matrices prepared using glyceryl behenate. Eur. J. Pharm. Biopharm. 2001, 52, 231–235. [Google Scholar] [CrossRef]
- Özyazici, M.; Gökҫe, E.H.; Ertan, G. Release and diffusional modelling of metronidazole lipid matrices. Eur. J. Pharm. Biopharm. 2006, 63, 331–339. [Google Scholar] [CrossRef] [PubMed]
- British Pharmacopoeia. Available online: https://www.pharmacopoeia.com/ (accessed on 23 February 2021).
- Shang, C.; Sinka, I.C.; Jayaraman, B.; Pan, J. Break force and tensile strength relationships for curved faced tablets subject to diametrical compression. Int. J. Pharm. 2013, 442, 57–64. [Google Scholar] [CrossRef]
- Costa, C.; Nogueira, F.H.A.; Pianetti, G.A. Simultaneous determination of artemether and lumefantrine in fixed dose combination tablets by HPLC with UV detection. J. Pharm. Biomed. Anal. 2008, 48, 951–954. [Google Scholar] [CrossRef]
- Obitte, N.C.; Chukwu, A.; Onyishi, I.V. The use of a pH-dependent and non pH-dependent natural hydrophobic biopolymer (Landolphia owariensis latex) as capsule coating agents in in vitro controlled release of metronidazole for possible colon targeted delivery. Int. J. Appl. Res. Nat. Prod. 2010, 3, 1–17. [Google Scholar] [CrossRef]
- Vertzoni, M.; Fotaki, N.; Kostewicz, E.; Stippler, E.; Leuner, C.; Nicolaides, E.; Dressman, J.; Reppas, C. Dissolution media simulating the intraluminal composition of the small intestine: Physiological issues and practical aspects. J. Pharm. Pharmacol. 2004, 56, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modelling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Costa, F.O.; Sousa, J.J.S.; Pais, A.A.C.C.; Formosinho, S.J. Comparison of dissolution profiles of Ibuprofen pellets. J. Control. Release 2003, 89, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.W.; Flanner, H.H. Mathematical comparison of dissolution profiles. Pharm. Technol. 1996, 20, 64–74. [Google Scholar] [CrossRef]
- Costa, P.; Lobo, J.M.S. Modelling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Hansen, T.S.; Nilsen, O.G. Echinacea purpurea and P-glycoprotein drug transport in Caco-2 cells. Phytother. Res. 2009, 23, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Hellum, B.H.; Nilsen, O.G. In vitro inhibition of CYP3A4 metabolism and P-Glycoprotein-mediated transport by trade herbal products. Basic Clin. Pharmacol. Toxicol. 2008, 102, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Adeleye, O.A.; Femi-Oyewo, M.N.; Odeniyi, M.A. Effect of compression pressure on mechanical and release properties of tramadol matrix tablets. Curr. Issues Pharm. Med. Sci. 2015, 28, 120–125. [Google Scholar] [CrossRef] [Green Version]
- Sheskey, P.J.; Robb, R.T.; Moore, R.D.; Boyce, B.M. Effects of lubricant level, method of mixing, and duration of mixing on a controlled-release matrix tablet containing hydroxypropyl methylcellulose. Drug Dev. Ind. Pharm. 1995, 21, 2151–2165. [Google Scholar] [CrossRef]
- Podczeck, F.; Miah, Y. The influence of particle size and shape on the angle of internal friction and the flow factor of unlubricated and lubricated powders. Int. J. Pharm. 1994, 144, 187–194. [Google Scholar] [CrossRef]
- Sabir, A.; Evans, B.; Jain, S. Formulation and process optimization to eliminate picking from market image tablets. Int. J. Pharm. 2001, 215, 123–135. [Google Scholar] [CrossRef]
- Yamamura, T.; Ohta, T.; Taira, T.; Ogawa, Y.; Sakai, Y.; Moribe, K.; Yamamoto, K. Effects of automated external lubrication on tablet properties and the stability of eprazinone hydrochloride. Int. J. Pharm. 2009, 370, 1–7. [Google Scholar] [CrossRef]
- Bossert, J.; Stamm, A. Effect of mixing on the lubrication of crystalline lactose by magnesium stearate. Drug Dev. Ind. Pharm. 1980, 6, 573–589. [Google Scholar] [CrossRef]
- Dansereau, R.; Peck, G.E. The effect of the variability in the physical and chemical properties of magnesium stearate on the properties of compressed tablets. Drug Dev. Ind. Pharm. 1987, 64, 51–65. [Google Scholar] [CrossRef]
- Bolhuis, G.K.; Hollzer, A.W. Lubricant sensitivity. In Pharmaceutical Powder Compaction Technology; Alderborn, G., Nystrom, C., Eds.; Marcel Dekker: New York, NY, USA, 1996; pp. 517–560. [Google Scholar]
- Kikuta, J.; Kitamori, N. Effect of mixing time on the lubrication properties of magnesium stearate and the final characteristics of the compressed tablets. Drug Dev. Ind. Pharm. 1994, 20, 343–355. [Google Scholar] [CrossRef]
- Schlagenhauf-Lawlor, P. Travellers’ Malaria, 2nd ed.; BC Decker Inc.: Ontario, CA, USA, 2008; pp. 1–388. [Google Scholar]
- Gray, V.A. Power of the dissolution test in distinguishing a change in dosage form critical quality attributes. AAPS PharmSciTech 2018, 19, 3328–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, R.; Silva, C.; Chaves, L. Tissue-Based In Vitro and Ex Vivo Models for Intestinal Permeability Studies. 2016. Available online: http://dx.doi.org/10.1016/B978-0-08-100094-6.00013-4 (accessed on 24 November 2019).
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridging the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Exp. | X1 | X2 | X3 | X4 | Code | Exp. | X1 | X2 | X3 | X4 | Code | Exp. | X1 | X2 | X3 | X4 | Code |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −1 | −1 | 0 | 0 | SA0.5C1 | 13 | 0 | −1 | 0 | 0 | GM0.5C1 | 25 | 1 | −1 | 0 | 0 | CA0.5C1 |
| 2 | −1 | 0 | 0 | 0 | SA0.75C1 | 14 | 0 | 0 | 0 | 0 | GM0.75C1 | 26 | 1 | 0 | 0 | 0 | CA0.75C1 |
| 3 | −1 | 1 | 0 | 0 | SA1C1 | 15 | 0 | 1 | 0 | 0 | GM1C1 | 27 | 1 | 1 | 0 | 0 | CA1C1 |
| 4 | −1 | −1 | 1 | 0 | SA0.5M1 | 16 | 0 | −1 | 1 | 0 | GM0.5M1 | 28 | 1 | −1 | 1 | 0 | CA0.5M1 |
| 5 | −1 | 0 | 1 | 0 | SA0.75M1 | 17 | 0 | 0 | 1 | 0 | GM0.75M1 | 29 | 1 | 0 | 1 | 0 | CA0.75M1 |
| 6 | −1 | 1 | 1 | 0 | SA1M1 | 18 | 0 | 1 | 1 | 0 | GM1M1 | 30 | 1 | 1 | 1 | 0 | CA1M1 |
| 7 | −1 | −1 | 0 | 1 | SA0.5C1.25 | 19 | 0 | −1 | 0 | 1 | GM0.5C1.25 | 31 | 1 | −1 | 0 | 1 | CA0.5C1.25 |
| 8 | −1 | 0 | 0 | 1 | SA0.75C1.25 | 20 | 0 | 0 | 0 | 1 | GM0.75C1.25 | 32 | 1 | 0 | 0 | 1 | CA0.75C1.25 |
| 9 | −1 | 1 | 0 | 1 | SA1C1.25 | 21 | 0 | 1 | 0 | 1 | GM1C1.25 | 33 | 1 | 1 | 0 | 1 | CA1C1.25 |
| 10 | −1 | −1 | 1 | 1 | SA0.5M1.25 | 22 | 0 | −1 | 1 | 1 | GM0.5M1.25 | 34 | 1 | −1 | 1 | 1 | CA0.5M1.25 |
| 11 | −1 | 0 | 1 | 1 | SA0.75M1.25 | 23 | 0 | 0 | 1 | 1 | GM0.75M1.25 | 35 | 1 | 0 | 1 | 1 | CA0.75M1.25 |
| 12 | −1 | 1 | 1 | 1 | SA1M1.25 | 24 | 0 | 1 | 1 | 1 | GM1M1.25 | 36 | 1 | 1 | 1 | 1 | CA1M1.25 |
| Factor | Mass Variation | Friability (%) | Crushing Strength | Tensile Strength | |||
|---|---|---|---|---|---|---|---|
| Mass (mg) | %RSD | (N) | %RSD | (N·mm−2) | %RSD | ||
| MicroceLac®100 | 494.939 | 1.390 | 0.429 | 165.41 | 19.611 | 1.850 | 0.148 |
| CombiLac® | 494.533 | 1.710 | 0.519 | 170.56 | 20.400 | 1.978 | 0.216 |
| Stearic Acid | 499.783 | 1.027 | 0.217 | 185.13 | 24.490 | 2.031 | 0.249 |
| Glycerol Monostearate | 498.592 | 1.912 | 0.209 | 149.58 | 25.886 | 1.613 | 0.086 |
| Cetyl Alcohol | 485.833 | 1.719 | 0.997 | 169.23 | 12.528 | 2.097 | 0.211 |
| 0.5:1 | 495.775 | 2.088 | 0.459 | 182.25 | 11.078 | 2.084 | 0.142 |
| 0.75:1 | 496.500 | 1.411 | 0.430 | 165.18 | 27.746 | 1.923 | 0.169 |
| 1:1 | 491.930 | 1.158 | 0.534 | 156.51 | 21.192 | 1.735 | 0.235 |
| MgSt 1% | 497.130 | 1.411 | 0.460 | 167.99 | 20.497 | 1.914 | 0.216 |
| MgSt 1.25% | 492.339 | 1.694 | 0.489 | 167.97 | 19.514 | 1.914 | 0.148 |
| Factor | Variable | API Content (%) | |
|---|---|---|---|
| Artemether | Lumefantrine | ||
| Lipid type | Stearic acid | 85.56 | 111.94 |
| Glycerol monostearate | 83.62 | 92.52 | |
| Cetyl alcohol | 87.26 | 96.67 | |
| Filler type | MicroceLac®100 | 90.42 | 104.34 |
| CombiLac® | 80.55 | 96.41 | |
| Lipid:drug ratio | 0.5:1 | 97.58 | 107.20 |
| 0.75:1 | 79.72 | 106.22 | |
| 1:1 | 79.14 | 87.71 | |
| Formulation | MDT | Fit Factors | Goodness of Fit | Best-Fit Values | ||||
|---|---|---|---|---|---|---|---|---|
| f1 | f2 | Rsqr adj | AIC | MSC | Tlag | Model Variable | ||
| SA 0.5M1 a | 310.500 | 30.674 | 43.502 | 0.9899 | 97.517 | 3.803 | 120.000 | kKP: 8.98; n: 0.37 |
| SA0.5C1 a | 315.000 | 20.247 | 50.338 | 0.9847 | 104.317 | 3.402 | 120.000 | kKP: 8.114; n: 0.385 |
| SA 0.75M1 b | 382.400 | 39.821 | 38.111 | 0.9712 | 111.122 | 2.743 | 120.000 | k1: 4.398; k2: 2.216; m: 0.261 |
| SA 0.75C1 b | 315.500 | 19.180 | 49.743 | 0.9927 | 92.358 | 4.091 | 120.000 | k1: 8.131; k2: 2.486; m: 0.244 |
| SA 1M1 c | 374.000 | 29.238 | 42.157 | 0.9978 | 69.193 | 5.464 | 120.000 | k1: 2.704; k2: 0.047 |
| SA 1C1 c | 386.500 | 43.479 | 35.107 | 0.9900 | 94.958 | 3.986 | 120.000 | k1: 1.956; k2: 0.08 |
| GM0.5M1 a | 355.800 | 50.274 | 32.880 | 0.9968 | 76.064 | 5.094 | 130.442 | kKP: 3.264; n: 0.528 |
| GM0.5C1 a | 323.100 | 80.552 | 22.248 | 0.9951 | 39.170 | 4.685 | 152.871 | kKP: 0.677; n: 0.573 |
| GM0.75M1 c | 499.400 | 66.135 | 25.610 | 0.9981 | 54.831 | 5.756 | 349.090 | k1: 4.403; k2: −0.051 |
| GM0.75C1 a | 350.800 | 28.360 | 43.340 | 0.9956 | 81.279 | 4.718 | 120.000 | kKP: 4.305; n: 0.481 |
| GM1M1 c | 313.000 | 45.026 | 32.888 | 0.9990 | 59.719 | 6.239 | 165.510 | k1: 5.997; k2: −0.075 |
| GM1C1 b | 310.900 | 42.262 | 34.766 | 0.9920 | 93.219 | 4.026 | 117.890 | k1: 10.179; k2: 0.823; m: 0.285 |
| CA 0.5M1 a | 407.200 | 65.595 | 26.848 | 0.9719 | 85.176 | 2.971 | 45.934 | kKP: 0.035; n: 1.098 |
| CA0.5C1 b | 341.000 | 30.807 | 40.008 | 0.9978 | 72.708 | 5.387 | 150.000 | k1: 4.199; k2: 2.153; m: 0.279 |
| CA 0.75M1 a | 454.100 | 73.660 | 24.011 | 0.9957 | 71.334 | 4.935 | 265.246 | kKP: 0.570; n: 0.793 |
| CA 0.75C1 c | 358.000 | 31.522 | 39.295 | 0.9988 | 60.730 | 6.066 | 150.000 | k1: 3.456; k2: 0.026 |
| CA 1M1 a | 285.300 | 23.989 | 42.616 | 0.9961 | 83.323 | 4.7571 | 150.000 | kKP: 18.868; n: 0.256 |
| CA 1C1 c | 472.900 | 72.880 | 24.405 | 0.9885 | 75.041 | 3.913 | 240.000 | k1: 1.469; k2: 0.034 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilkins, C.A.; du Plessis, L.H.; Viljoen, J.M. Investigating In Vitro and Ex Vivo Properties of Artemether/Lumefantrine Double-Fixed Dose Combination Lipid Matrix Tablets Prepared by Hot Fusion. Pharmaceutics 2021, 13, 922. https://doi.org/10.3390/pharmaceutics13070922
Wilkins CA, du Plessis LH, Viljoen JM. Investigating In Vitro and Ex Vivo Properties of Artemether/Lumefantrine Double-Fixed Dose Combination Lipid Matrix Tablets Prepared by Hot Fusion. Pharmaceutics. 2021; 13(7):922. https://doi.org/10.3390/pharmaceutics13070922
Chicago/Turabian StyleWilkins, Christi A., Lissinda H. du Plessis, and Joe M. Viljoen. 2021. "Investigating In Vitro and Ex Vivo Properties of Artemether/Lumefantrine Double-Fixed Dose Combination Lipid Matrix Tablets Prepared by Hot Fusion" Pharmaceutics 13, no. 7: 922. https://doi.org/10.3390/pharmaceutics13070922