
Scaling Approaches for Pediatric Dose Selection: The Fremanezumab (AJOVY®) Journey to Select a Phase 3 Dose Using Pharmacokinetic Data from a Phase 1 Study


Abstract
:1. Introduction
2. Materials and Methods
2.1. Overview of the Pharmacokinetic Model Development Process
2.2. Pediatric Population Pharmacokinetic Model Development
2.3. Evaluation of Different Revisions to the Pediatric Population Pharmacokinetic Model
- Fixed allometric exponents: applying conventional theoretical allometric exponents (0.75 for CL; 1.0 for Vc);
- Fixing the F1 and ALAG1 parameters to the estimates obtained in adults;
- Using less informative adult priors (25% RSE).
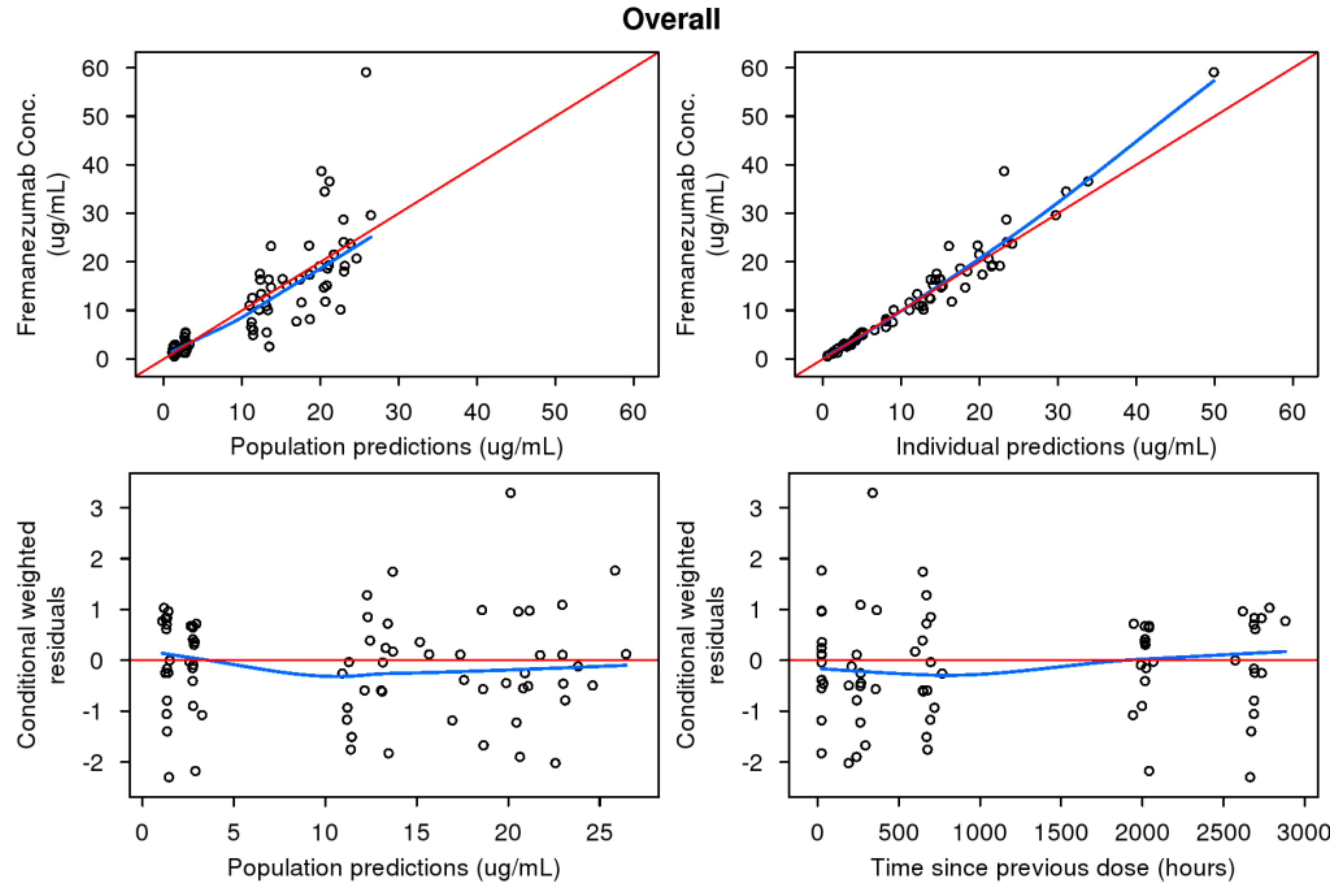
2.4. Model Evaluation
2.5. Simulations
3. Results
3.1. Previously Developed Adult Model Applied to Pediatric Data
3.2. Pediatric Population Pharmacokinetic Model Development
3.3. Evaluation of Different Revisions to the Pediatric Population Pharmacokinetic Model
3.3.1. Revised Pediatric Model with Fixed Allometric Exponents
3.3.2. Revised Pediatric Model with Bioavailability and Lag Time Fixed to Adult Values
3.3.3. Pediatric Model with Fixed Bioavailability and Lag Time from Adults and Less Informative Priors (25% RSE)
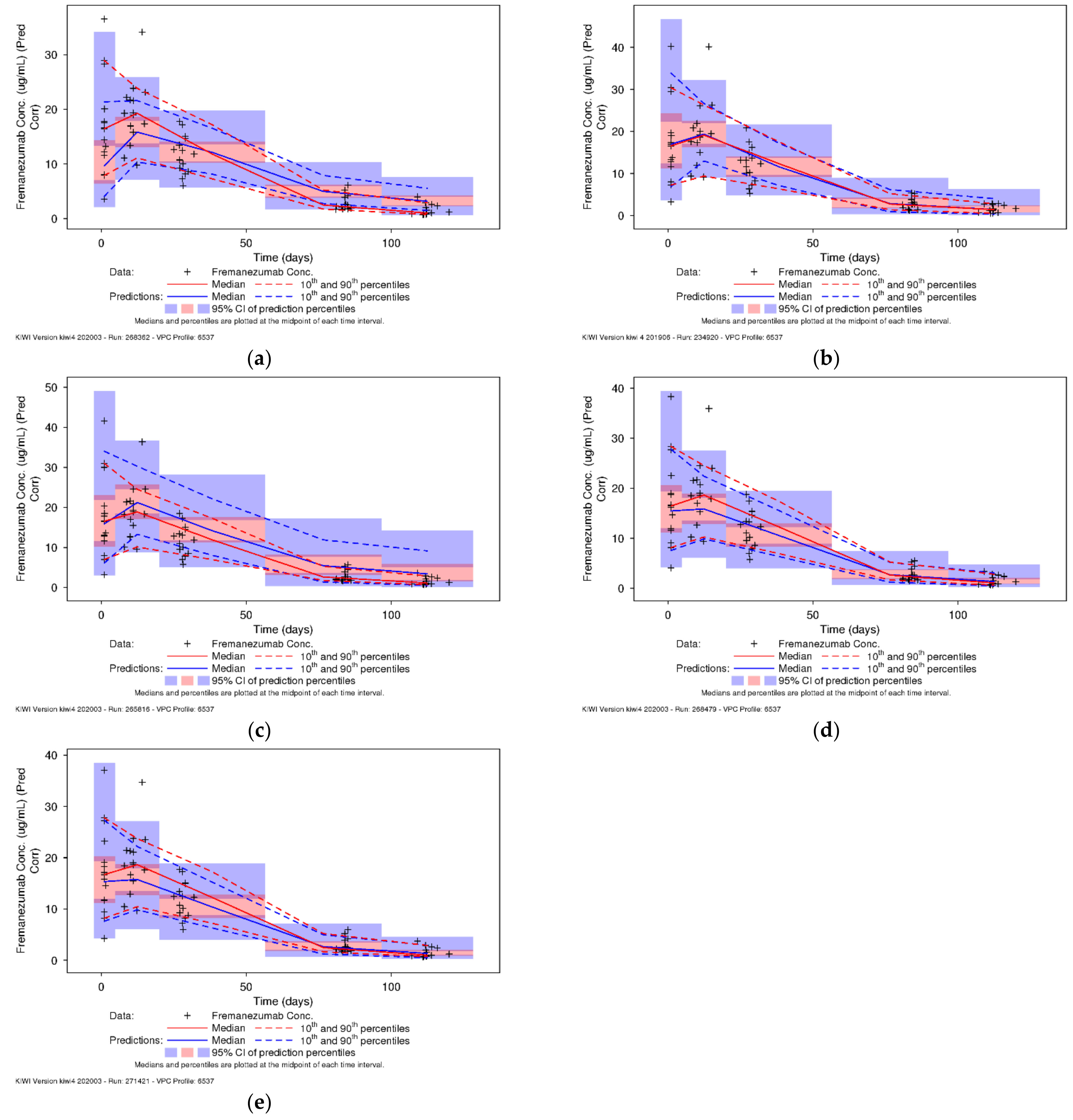
3.4. Predictive Performance of Pediatric Model to Support Phase 3 Development
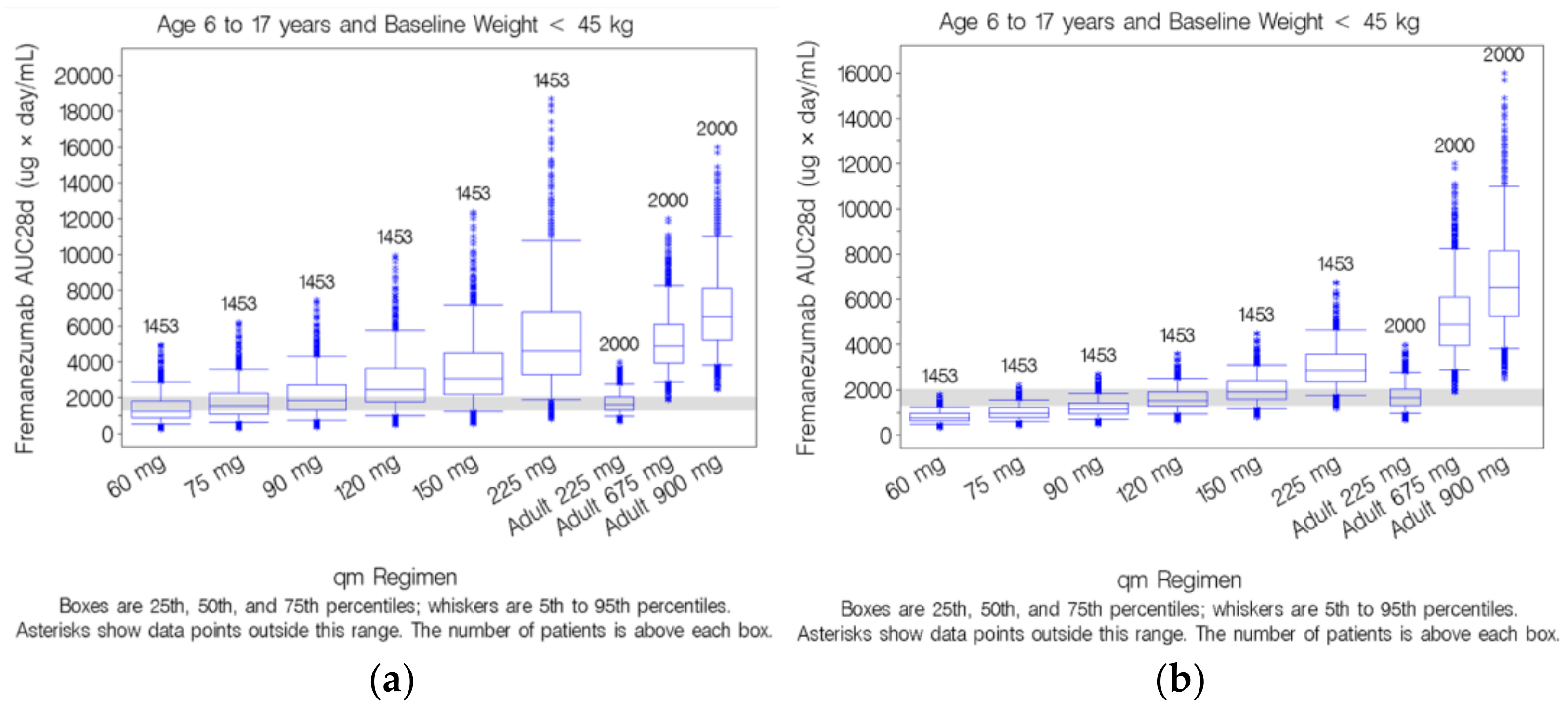
3.5. Simulation-Based Dose Selection for Pediatric Patients Weighing <45 kg
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abu-Arafeh, I.; Razak, S.; Sivaraman, B.; Graham, C. Prevalence of headache and migraine in children and adolescents: A systematic review of population-based studies. Dev. Med. Child Neurol. 2010, 52, 1088–1097. [Google Scholar] [CrossRef]
- Abu-Arefeh, I.; Russell, G. Prevalence of headache and migraine in schoolchildren. BMJ 1994, 309, 765–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurell, K.; Larsson, B.; Eeg-Olofsson, O. Prevalence of Headache in Swedish Schoolchildren, with a Focus on Tension-Type Headache. Cephalalgia 2004, 24, 380–388. [Google Scholar] [CrossRef]
- Özge, A.; Şaşmaz, T.; Buğdaycı, R.; Çakmak, S.E.; Kurt, A.Ö.; Kaleağası, S.H.; Siva, A. The prevalence of chronic and episodic migraine in children and adolescents. Eur. J. Neurol. 2012, 20, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Stovner, L.J.; Andree, C. Prevalence of headache in Europe: A review for the Eurolight project. J. Headache Pain 2010, 11, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.W. Pediatric Migraine. Neurol. Clin. 2009, 27, 481–501. [Google Scholar] [CrossRef] [PubMed]
- Fendrich, K.; Vennemann, M.; Pfaffenrath, V.; Evers, S.; May, A.; Berger, K.; Hoffmann, W. Headache Prevalence Among Adolescents—The German DMKG Headache Study. Cephalalgia 2007, 27, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Unalp, A.; Dirik, E.; Kurul, S. Prevalence and clinical findings of migraine and tension-type headache in adolescents. Pediatr. Int. 2007, 49, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Buse, D.C.; Loder, E.W.; Ms, J.A.G.; Stewart, W.F.; Reed, M.L.; Fanning, K.M.; Serrano, D.; Lipton, R.B. Sex Differences in the Prevalence, Symptoms, and Associated Features of Migraine, Probable Migraine and Other Severe Headache: Results of the American Migraine Prevalence and Prevention (AMPP) Study. Headache J. Head Face Pain 2013, 53, 1278–1299. [Google Scholar] [CrossRef]
- Burch, R.C.; Loder, S.; Loder, E.; Smitherman, T.A. The Prevalence and Burden of Migraine and Severe Headache in the United States: Updated Statistics from Government Health Surveillance Studies. Headache J. Head Face Pain 2015, 55, 21–34. [Google Scholar] [CrossRef]
- Victor, T.W.; Hu, X.; Campbell, J.C.; Buse, D.C.; Lipton, R.B. Migraine prevalence by age and sex in the United States: A life-span study. Cephalalgia 2010, 30, 1065–1072. [Google Scholar] [CrossRef]
- Eftekhari, S.; Edvinsson, L. Possible sites of action of the new calcitonin gene-related peptide receptor antagonists. Ther. Adv. Neurol. Disord. 2010, 3, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Olesen, J. CGRP in migraine. Cephalalgia 2011, 31, 638. [Google Scholar] [CrossRef] [PubMed]
- Ashina, M.; Bendtsen, L.; Jensen, R.; Schifter, S.; Jansen-Olesen, I.; Olesen, J. Plasma levels of calcitonin gene-related peptide in chronic tension-type headache. Neurology 2000, 55, 1335–1340. [Google Scholar] [CrossRef]
- Hansen, J.M.; Hauge, A.W.; Olesen, J.; Ashina, M. Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia 2010, 30, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Diener, H.-C.; Husstedt, I.W.; Goadsby, P.J.; Hall, D.; Meier, U.; Pollentier, S.; Lesko, L.M. Calcitonin Gene–Related Peptide Receptor Antagonist BIBN 4096 BS for the Acute Treatment of Migraine. N. Engl. J. Med. 2004, 350, 1104–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, T.W.; Ferrari, M.D.; Dodick, D.W.; Galet, V.; Kost, J.; Fan, X.; Leibensperger, H.; Froman, S.; Assaid, C.; Lines, C.; et al. Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: A randomised, placebo-controlled, parallel-treatment trial. Lancet 2008, 372, 2115–2123. [Google Scholar] [CrossRef]
- Hewitt, D.J.; Aurora, S.K.; Dodick, D.W.; Goadsby, P.J.; Ge, Y.J.; Bachman, R.; Taraborelli, D.; Fan, X.; Assaid, C.; Lines, C.; et al. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia 2011, 31, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Bigal, M.E.; Dodick, D.W.; Rapoport, A.M.; Silberstein, S.D.; Ma, Y.; Yang, R.; Loupe, P.S.; Burstein, R.; Newman, L.C.; Lipton, R.B. Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of high-frequency episodic migraine: A multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol. 2015, 14, 1081–1090. [Google Scholar] [CrossRef]
- Bigal, M.E.; Edvinsson, L.; Rapoport, A.M.; Lipton, R.B.; Spierings, E.L.H.; Diener, H.-C.; Burstein, R.; Loupe, P.S.; Ma, Y.; Yang, R.; et al. Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of chronic migraine: A multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol. 2015, 14, 1091–1100. [Google Scholar] [CrossRef]
- Bigal, M.E.; Rapoport, A.M.; Silberstein, S.D.; Walter, S.; Hargreaves, R.J.; Aycardi, E. From LBR-101 to Fremanezumab for Migraine. CNS Drugs 2018, 32, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Ajovy [Package Insert]; Teva Pharmaceuticals Inc.: North Wales, PA, USA, 2018; Revised Date: 9/2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761089s000lbl.pdf (accessed on 1 May 2021).
- Armour, K.L.; Clark, M.R.; Hadley, A.G.; Williamson, L.M. Recombinant human IgG molecules lacking Fcgamma receptor I binding and monocyte triggering activities. Eur. J. Immunol. 1999, 29, 2613–2624. [Google Scholar] [CrossRef]
- Cohen-Barak, O.; Weiss, S.; Rasamoelisolo, M.; Faulhaber, N.; Yeung, P.P.; Loupe, P.S.; Yoon, E.; Gandhi, M.D.; Spiegelstein, O.; Aycardi, E. A phase 1 study to assess the pharmacokinetics, safety, and tolerability of fremanezumab doses (225 mg, 675 mg and 900 mg) in Japanese and Caucasian healthy subjects. Cephalalgia 2018, 38, 1960–1971. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.M.; Dodick, D.W.; Yang, R.; Newman, L.C.; Li, T.; Aycardi, E.; Bigal, M.E. Fremanezumab as Add-On Treatment for Patients Treated with Other Migraine Preventive Medicines. Headache J. Head Face Pain 2017, 57, 1375–1384. [Google Scholar] [CrossRef]
- Dodick, D.W.; Silberstein, S.D.; Bigal, M.E.; Yeung, P.P.; Goadsby, P.J.; Blankenbiller, T.; Grozinski-Wolff, M.; Yang, R.; Ma, Y.; Aycardi, E. Effect of Fremanezumab Compared with Placebo for Prevention of Episodic Migraine. JAMA 2018, 319, 1999–2008. [Google Scholar] [CrossRef]
- Silberstein, S.D.; Dodick, D.W.; Bigal, M.E.; Yeung, P.P.; Goadsby, P.J.; Blankenbiller, T.; Grozinski-Wolff, M.; Yang, R.; Ma, Y.; Aycardi, E. Fremanezumab for the Preventive Treatment of Chronic Migraine. N. Engl. J. Med. 2017, 377, 2113–2122. [Google Scholar] [CrossRef]
- Fiedler-Kelly, J.B.; Cohen-Barak, O.; Morris, D.N.; Ludwig, E.; Rasamoelisolo, M.; Shen, H.; Levi, M. Population pharmacokinetic modelling and simulation of fremanezumab in healthy subjects and patients with migraine. Br. J. Clin. Pharmacol. 2019, 85, 2721–2733. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Barak, O.; Radivojevic, A.; Jones, A.; Fiedler-Kelly, J.; Gillespie, M.; Brennan, M.; Gutman, D.; Rasamoelisolo, M.; Hallak, H.; Loupe, P.; et al. Dose selection for fremanezumab (AJOVY) phase 3 pediatric migraine studies using pharmacokinetic data from a pediatric phase 1 study and a population pharmacokinetic modeling and simulation approach. Cephalalgia 2021, in press. [Google Scholar] [CrossRef]
- Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013, 33, 629–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherniakov, I.; Cohen-Barak, O.; Tiver, R.; Gillespie, M.; Kessler, Y.; Gutierrez, M.; Rasamoelisolo, M.; Li, S.; Shen, H.; Hallak, H.; et al. A pharmacokinetic bioequivalence study of fremanezumab administered subcutaneously using an autoinjector and a prefilled syringe. Clin. Pharmacol. Drug Dev. 2021. Available online: https://accp1.onlinelibrary.wiley.com/doi/full/10.1002/cpdd.902 (accessed on 26 April 2021). [CrossRef] [PubMed]
- SAS [Computer Program]; Version 9.4; SAS Institute: Cary, NC, USA, 2013.
- KIWI [Computer Program]; Version 4; Cognigen Corporation: Buffalo, NY, USA, 2019.
- Beal, S.L.; Sheiner, L.B.; Boeckmann, A.J.; Bauer, R.J. (Eds.) NONMEM 7.3.0 Users Guides; ICON Development Solutions LLC: Hanover, MD, USA, 2013. [Google Scholar]
- Bergstrand, M.; Hooker, A.C.; Wallin, J.E.; Karlsson, M.O. Prediction-Corrected Visual Predictive Checks for Diagnosing Nonlinear Mixed-Effects Models. AAPS J. 2011, 13, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holford, N. The Visual Predictive Check: Superiority to Standard Diagnostic (Rorschach) Plots. Poster Presented at Population Approach Group in Europe (PAGE); Pamplona, Spain, 16–17 June 2015. PAGE 14 (2005) Abstr 738 Poster. Available online: www.page-meeting.org/?abstract=738 (accessed on 26 April 2021).
- Centers for Disease Control and Prevention, National Center for Health Statistics. CDC Growth Charts: Data Tables: Selected Z-Score Values. Available online: https://www.cdc.gov/growthcharts/zscore.htm (accessed on 16 August 2019).
- United States Food and Drug Administration. Guidance for Industry: General Clinical Pharmacology Considerations for Pediatric Studies for Drugs and Biological Products; December 2014. Available online: https://www.fda.gov/media/90358/download (accessed on 26 April 2021).
- Frymoyer, A.; Hoekman, D.R.; Piester, T.L.; de Meij, T.G.; Hummel, T.Z.; Benninga, M.A.; Kindermann, A.; Park, K. Application of Population Pharmacokinetic Modeling for Individualized Infliximab Dosing Strategies in Crohn Disease. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Eckert, D.; Hyams, J.S.; Mensing, S.; Thakkar, R.B.; Robinson, A.M.; Rosh, J.R.; Ruemmele, F.M.; Awni, W.M. Pharmacokinetics and Exposure–Efficacy Relationship of Adalimumab in Pediatric Patients with Moderate to Severe Crohn’s Disease: Results from a randomized, multi-center, phase-3 study. Inflamm. Bowel Dis. 2015, 21, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Chen, J.; Yu, J.; Marathe, A.; Sahajwalla, C.; Borigini, M.; Maynard, J.; Burckart, G.J.; Wang, Y. Dosage Considerations for Canakinumab in Children with Periodic Fever Syndromes. Clin. Pharmacol. Ther. 2019, 106, 557–567. [Google Scholar] [CrossRef]
- Liu, X.I.; Dallmann, A.; Wang, Y.; Green, D.J.; Burnham, J.M.; Chiang, B.; Wu, P.; Sheng, M.; Lu, K.; Anker, J.N.V.D.; et al. Monoclonal Antibodies and Fc-Fusion Proteins for Pediatric Use: Dosing, Immunogenicity, and Modeling and Simulation in Data Submitted to the US Food and Drug Administration. J. Clin. Pharmacol. 2019, 59, 1130–1143. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Parameter | Previously Developed Adult Model Applied to Pediatric Data | Pediatric Model to Support Phase 3 Development 1 | Revised Pediatric Model with Fixed Allometric Exponents | Revised Pediatric Model with Bioavailability and Lag Time Fixed to Adult Values | Pediatric Model with Fixed Bioavailability and Lag Time from Adults and Less Informative Priors (25% RSE) |
|---|---|---|---|---|---|
| CL: central clearance (L/day) | 0.0902 (1.50) | 0.0905 (0.0937) | 0.0907 (0.213) | 0.0905 (0.305) | 0.106 (13.1) |
| CL: allometric exponent for weight (−) | 1.05 (4.33) | 0.245 (32.7) | 0.750 (FIXED) | 0.642 (10.6) | 0.833 (16.9) |
| Vc: central volume of distribution (L) | 1.88 (3.38) | 1.89 (0.213) | 1.90 (0.569) | 1.89 (0.512) | 2.18 (4.57) |
| Vc: allometric exponent for weight (−) | 1.53 (10.3) | 1.20 (32.7) | 1.00 (FIXED) | 2.56 (8.03) | 2.70 (6.66) |
| ka: absorption rate constant (1/day) | 0.180 (12.2) | 0.252 (14.8) | 0.262 (10.5) | 0.176 (2.72) | 0.178 (5.40) |
| Q: intercompartmental clearance (L/day) | 0.262 (FIXED) | 0.262 (FIXED) | 0.262 (FIXED) | 0.262 (FIXED) | 0.262 (FIXED) |
| Vp: peripheral volume of distribution (L) | 1.72 (FIXED) | 1.72 (FIXED) | 1.72 (FIXED) | 1.72 (FIXED) | 1.72 (FIXED) |
| F1: bioavailability | 0.658 (FIXED) | Apparent (F1 = 1) | Apparent (F1 = 1) | 0.658 (FIXED) | 0.658 (FIXED) |
| ALAG1: lag time (day) | 0.0803 (FIXED) | Removed | NE | 0.0803 (FIXED) | 0.0803 (FIXED) |
| Magnitude of Interindividual Variability (%RSE) | |||||
| Interindividual variability in CL | 23.4 %CV (4.60) | 34.2 %CV (53.2) | 53.8 %CV (31.2) | 24.9 %CV (68.3) | 23.6 %CV (63.8) |
| Interindividual variability in Vc | 35.1 %CV (19.9) | 79.9 %CV (49.6) | 80.8 %CV (51.4) | 58.9 %CV (133) | 56.1 %CV (138) |
| Interindividual variability in ka | 59.0 %CV (15.8) | NE | NE | 28.4 %CV (57.5) | 27.9 %CV (59.5) |
| Parameter Estimate (%RSE) | |||||
| Residual variability proportional component | 0.0531 (4.03) | 0.0338 (43.7) | 0.0333 (43.8) | 0.0795 (69.5) | 0.0793 (68.9) |
| Residual variability additive component | 0.204 (25.6) | NE | NE | NE | NE |
| VOF | NA | 201.396 | 214.782 | 219.479 | 218.022 |
| Body Weight | Pharmacokinetic Parameter | Pediatrics | ||
|---|---|---|---|---|
| <30 kg | >30 kg | All Patients | ||
| Number of Patients | 6 | 9 | 15 | |
| NCA (observed) [29] | Cmax (µg/mL) mean (SD) | 34.2 (13.5) | 16.3 (5.27) | 23.5 (12.8) |
| AUC28d (µg × h/mL) mean (SD) | 16,200 (5310) | 8840 (3080) | 11,800 (5410) | |
| NCA (predicted based on pediatric population pharmacokinetic model) | Cmax (μg/mL) mean (SD) | 31.8 (9.91) | 17.9 (5.02) | 23.5 (9.93) |
| AUC28d (μg × h/mL) mean (SD) | 14,200 (2120) | 9620 (3070) | 11,500 (3520) | |
| Number of Simulated Patients | 786 | 667 | 1453 | |
| Simulation NCA (based on pediatric population pharmacokinetic model) | Cmax (µg/mL) mean (SD) | 28.4 (9.11) | 22.6 (7.06) | 25.7 (8.73) |
| AUC28d (µg × h/mL) mean (SD) | 13,200 (2880) | 11,200 (2730) | 12,300 (3000) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, A.; Cohen-Barak, O.; Radivojevic, A.; Fiedler-Kelly, J. Scaling Approaches for Pediatric Dose Selection: The Fremanezumab (AJOVY®) Journey to Select a Phase 3 Dose Using Pharmacokinetic Data from a Phase 1 Study. Pharmaceutics 2021, 13, 785. https://doi.org/10.3390/pharmaceutics13060785
Jones A, Cohen-Barak O, Radivojevic A, Fiedler-Kelly J. Scaling Approaches for Pediatric Dose Selection: The Fremanezumab (AJOVY®) Journey to Select a Phase 3 Dose Using Pharmacokinetic Data from a Phase 1 Study. Pharmaceutics. 2021; 13(6):785. https://doi.org/10.3390/pharmaceutics13060785
Chicago/Turabian StyleJones, Aksana, Orit Cohen-Barak, Andrijana Radivojevic, and Jill Fiedler-Kelly. 2021. "Scaling Approaches for Pediatric Dose Selection: The Fremanezumab (AJOVY®) Journey to Select a Phase 3 Dose Using Pharmacokinetic Data from a Phase 1 Study" Pharmaceutics 13, no. 6: 785. https://doi.org/10.3390/pharmaceutics13060785