
Dose-Dependent Solubility–Permeability Interplay for Poorly Soluble Drugs under Non-Sink Conditions



Abstract
:1. Introduction
2. Theoretical Basis
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Flux Measurements
3.2.2. Sample Concentration Measurements by UHPLC
4. Results
4.1. Effect of SLS on Griseofulvin Solubility and Triamcinolone Solubility
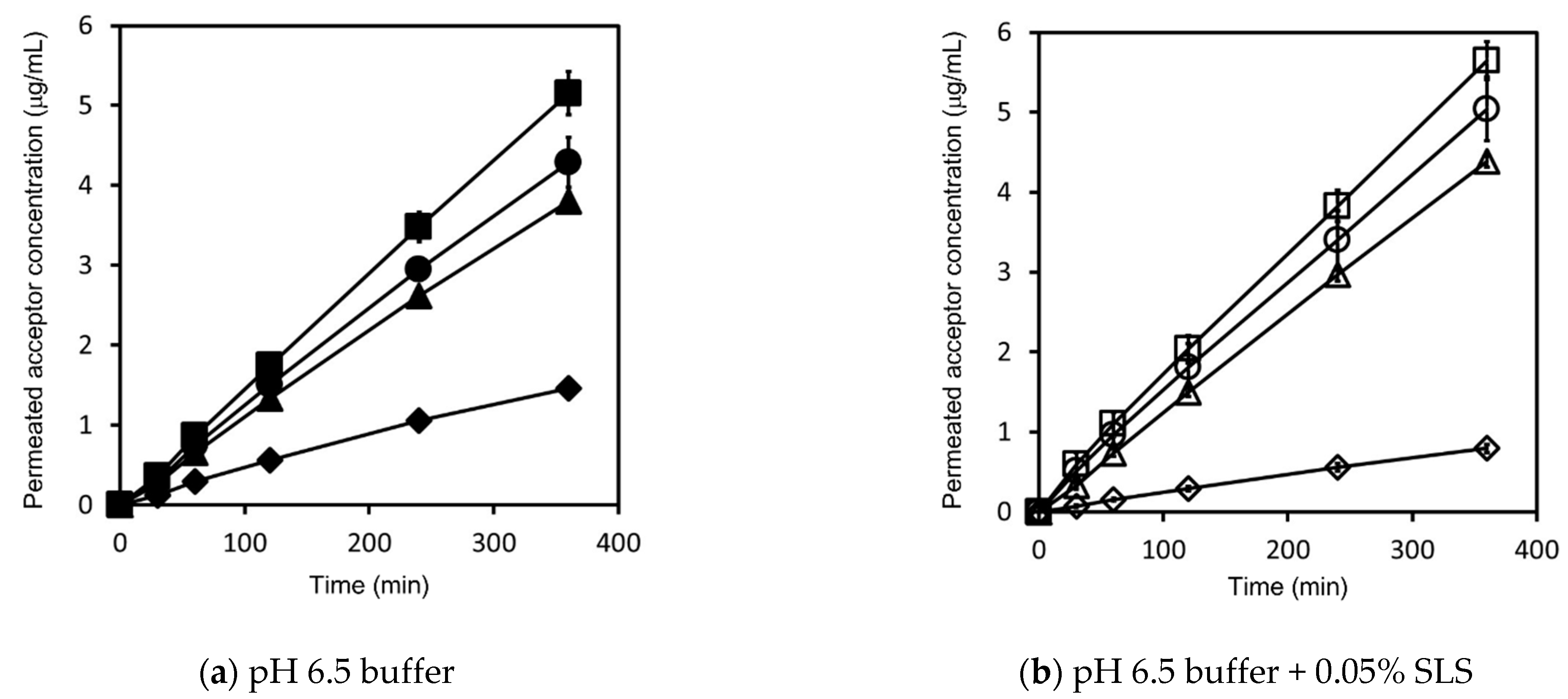
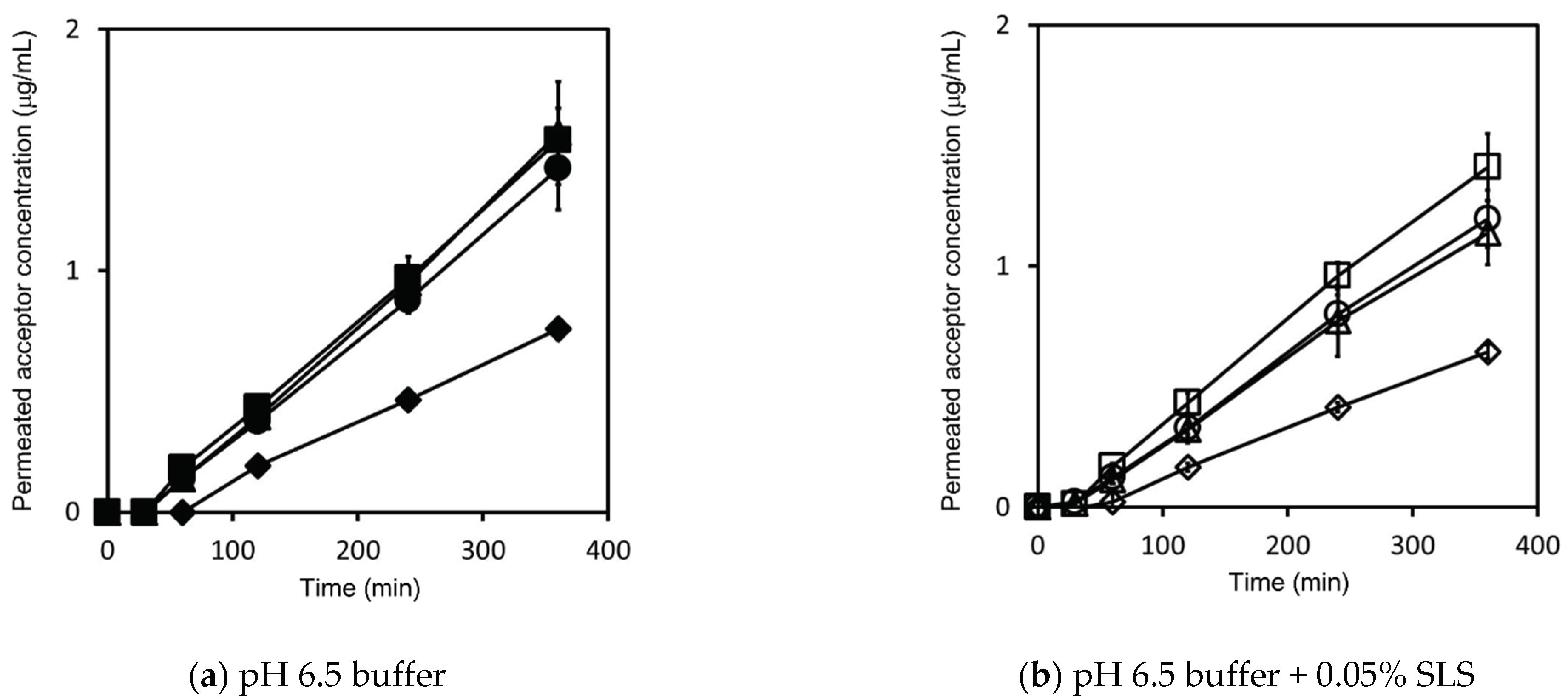
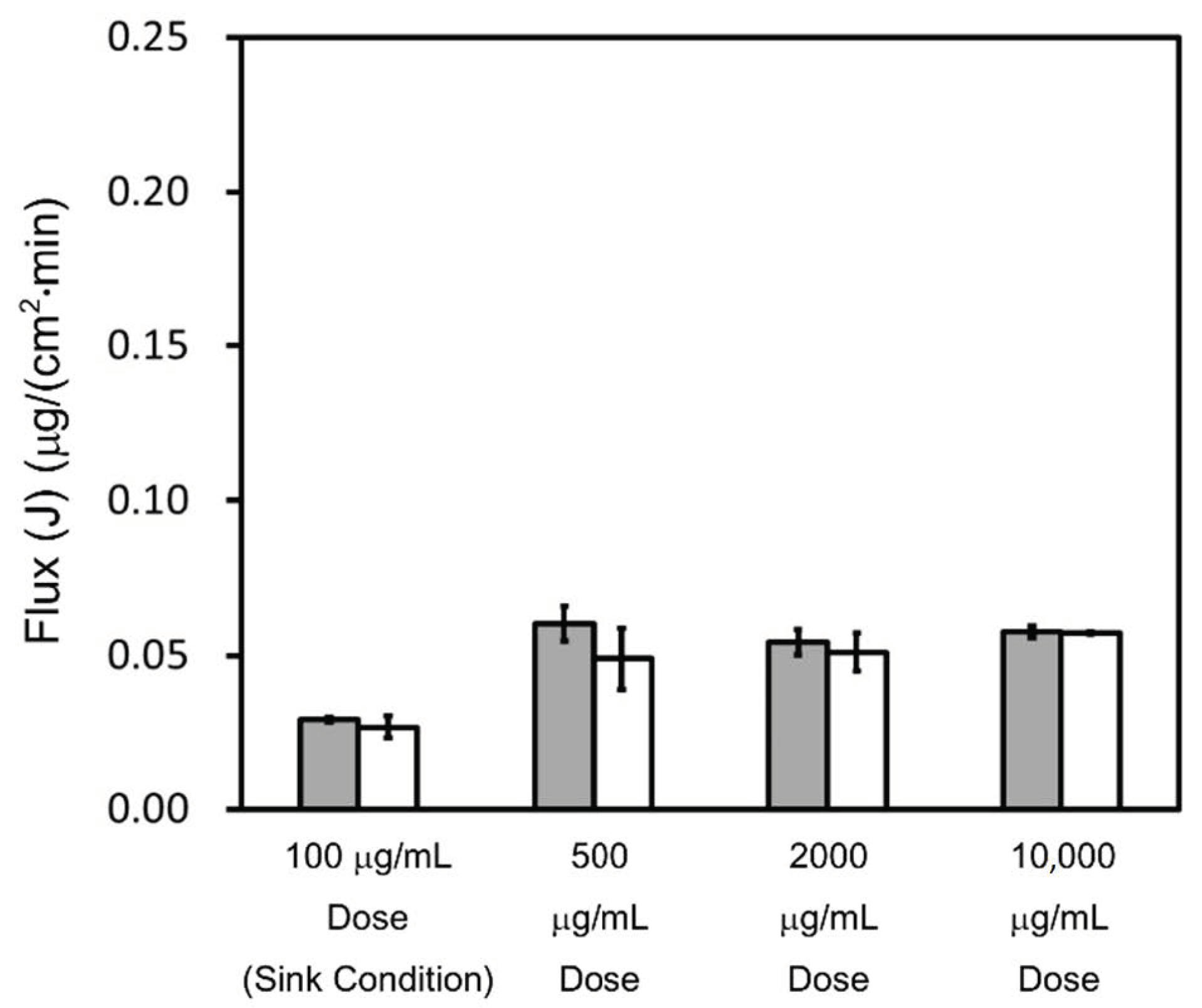
4.2. Effect of Dose Amount and SLS on Flux and Permeability
4.2.1. Griseofulvin
4.2.2. Triamcinolone
4.3. Theoretical Calculation about Permeability
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Boyd, B.J.; Bergström, C.A.; Vinarov, Z.; Kuentz, M.; Brouwers, J.; Augustijns, P.; Brandl, M.; Bernkop-Schnürch, A.; Shrestha, N.; Préat, V.; et al. Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur. J. Pharm. Sci. 2019, 137, 104967. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Dressman, J.B. The Developability Classification System: Application of Biopharmaceutics Concepts to Formulation Development. J. Pharm. Sci. 2010, 99, 4940–4954. [Google Scholar] [CrossRef]
- Rosenberger, J.; Butler, J.; Dressman, J. A Refined Developability Classification System. J. Pharm. Sci. 2018, 107, 2020–2032. [Google Scholar] [CrossRef]
- Rosenberger, J.; Butler, J.; Muenster, U.; Dressman, J. Application of a Refined Developability Classification System. J. Pharm. Sci. 2019, 108, 1090–1100. [Google Scholar] [CrossRef]
- Lennernäs, H.; Aarons, L.; Augustijns, P.; Beato, S.; Bolger, M.; Box, K.; Brewster, M.; Butler, J.; Dressman, J.; Holm, R.; et al. Oral biopharmaceutics tools—Time for a new initiative—An introduction to the IMI project OrBiTo. Eur. J. Pharm. Sci. 2014, 57, 292–299. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Abrahamsson, B.; Brewster, M.; Brouwers, J.; Butler, J.; Carlert, S.; Dickinson, P.A.; Dressman, J.; Holm, R.; Klein, S.; et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 342–366. [Google Scholar] [CrossRef]
- Lennernäs, H.; Lindahl, A.; Van Peer, A.; Ollier, C.; Flanagan, T.; Lionberger, R.; Nordmark, A.; Yamashita, S.; Yu, L.; Amidon, G.L.; et al. In Vivo Predictive Dissolution (IPD) and Biopharmaceutical Modeling and Simulation: Future Use of Modern Approaches and Methodologies in a Regulatory Context. Mol. Pharm. 2017, 14, 1307–1314. [Google Scholar] [CrossRef]
- Tsume, Y.; Mudie, D.M.; Langguth, P.; Amidon, G.E.; Amidon, G.L. The Biopharmaceutics Classification System: Subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur. J. Pharm. Sci. 2014, 57, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Food and Drug Administration. Guidance for Industry. Immediate Release Solid Oral Dosage Forms, Scale-Up and Post-Approval Changes: Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation; FDA: Rockville, MD, USA, 1995.
- Ministry of Health, Labour and Welfare (MHLW). Guideline for Bioequivalence Studies of Generic Products (Revision); MHLW: Tokyo, Japan, 2012. [Google Scholar]
- MHLW. Guideline for Bioequivalence Studies of Generic Products for Different Strengths of Oral Solid Dosage Forms (Revision); MHLW: Tokyo, Japan, 2012.
- European Medicines Agency. Guideline on the Investigation of Bioequivalence; EMA: London, UK, 2010.
- Kuribayashi, R.; Takishita, T.; Mikami, K. Regulatory Considerations of Bioequivalence Studies for Oral Solid Dosage Forms in Japan. J. Pharm. Sci. 2016, 105, 2270–2277. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances; European Medicines Agency: London, UK, 2000. [Google Scholar]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Van Der Merwe, J.; Steenekamp, J.; Steyn, D.; Hamman, J. The Role of Functional Excipients in Solid Oral Dosage Forms to Overcome Poor Drug Dissolution and Bioavailability. Pharmaceutics 2020, 12, 393. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef]
- Sun, D.D.; Wen, H.; Taylor, L.S. Non-Sink Dissolution Conditions for Predicting Product Quality and In Vivo Performance of Supersaturating Drug Delivery Systems. J. Pharm. Sci. 2016, 105, 2477–2488. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.D.; Lee, P.I. Haste Makes Waste: The Interplay Between Dissolution and Precipitation of Supersaturating Formulations. AAPS J. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Phillips, D.J.; Pygall, S.R.; Cooper, V.B.; Mann, J.C. Overcoming sink limitations in dissolution testing: A review of traditional methods and the potential utility of biphasic systems. J. Pharm. Pharmacol. 2012, 64, 1549–1559. [Google Scholar] [CrossRef]
- Miyaji, Y.; Fujii, Y.; Takeyama, S.; Kawai, Y.; Kataoka, M.; Takahashi, M.; Yamashita, S. Advantage of the Dissolution/Permeation System for Estimating Oral Absorption of Drug Candidates in the Drug Discovery Stage. Mol. Pharm. 2016, 13, 1564–1574. [Google Scholar] [CrossRef]
- Sun, D.; Hu, M.; Browning, M.; Friedman, R.L.; Jiang, W.; Zhao, L.; Wen, H. Dissolution Failure of Solid Oral Drug Products in Field Alert Reports. J. Pharm. Sci. 2017, 106, 1302–1309. [Google Scholar] [CrossRef]
- Dahan, A.; Miller, J.M. The Solubility–Permeability Interplay and Its Implications in Formulation Design and Development for Poorly Soluble Drugs. AAPS J. 2012, 14, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Dahan, A.; Beig, A.; Lindley, D.; Miller, J.M. The solubility–permeability interplay and oral drug formulation design: Two heads are better than one. Adv. Drug Deliv. Rev. 2016, 101, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Porat, D.; Dahan, A. Active intestinal drug absorption and the solubility-permeability interplay. Int. J. Pharm. 2018, 537, 84–93. [Google Scholar] [CrossRef]
- Nainwal, N.; Singh, R.; Jawla, S.; Saharan, V.A. The Solubility-Permeability Interplay for Solubility-Enabling Oral Formulations. Curr. Drug Targets 2019, 20, 1434–1446. [Google Scholar] [CrossRef]
- Dahlgren, D.; Lennernäs, H. Intestinal Permeability and Drug Absorption: Predictive Experimental, Computational and In Vivo Approaches. Pharmaceutics 2019, 11, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Lu, L.; Wang, S.; Wu, J.; Shi, J.; Yan, T.; Xie, C.; Li, Q.; Hu, M.; Liu, Z. Chapter 11. Oral Absorption Basics: Pathways and Physicochemical and Biological Factors Affecting Absorption. In Developing Solid Oral Dosage Forms, 2nd ed.; Qiu, Y., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 297–329. [Google Scholar]
- Raina, S.A.; Zhang, G.G.Z.; Alonzo, D.E.; Wu, J.; Zhu, D.; Catron, N.D.; Gao, Y.; Taylor, L.S. Impact of Solubilizing Additives on Supersaturation and Membrane Transport of Drugs. Pharm. Res. 2015, 32, 3350–3364. [Google Scholar] [CrossRef] [PubMed]
- Shore, P.A.; Brodie, B.B.; Hogben, C.A. The gastric secretion of drugs: A phpartition hypothesis. J. Pharmacol. Exp. Ther. 1957, 119, 361–369. [Google Scholar] [PubMed]
- Beig, A.; Agbaria, R.; Dahan, A. Oral Delivery of Lipophilic Drugs: The Tradeoff between Solubility Increase and Permeability Decrease When Using Cyclodextrin-Based Formulations. PLoS ONE 2013, 8, e68237. [Google Scholar] [CrossRef]
- Beig, A.; Miller, J.M.; Dahan, A. The interaction of nifedipine with selected cyclodextrins and the subsequent solubility–permeability trade-off. Eur. J. Pharm. Biopharm. 2013, 85, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Beig, A.; Agbaria, R.; Dahan, A. The use of captisol (SBE7-β-CD) in oral solubility-enabling formulations: Comparison to HPβCD and the solubility–permeability interplay. Eur. J. Pharm. Sci. 2015, 77, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Beig, A.; Krieg, B.J.; Carr, R.A.; Borchardt, T.B.; Amidon, G.E.; Amidon, G.L.; Dahan, A. The Solubility–Permeability Interplay: Mechanistic Modeling and Predictive Application of the Impact of Micellar Solubilization on Intestinal Permeation. Mol. Pharm. 2011, 8, 1848–1856. [Google Scholar] [CrossRef]
- Hens, B.; Brouwers, J.; Corsetti, M.; Augustijns, P. Gastrointestinal behavior of nano- and microsized fenofibrate: In vivo evaluation in man and in vitro simulation by assessment of the permeation potential. Eur. J. Pharm. Sci. 2015, 77, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.E.; Higuchi, W.I.; Ho, N.F.H. Theoretical and Experimental Studies of Transport of Micelle-Solubilized Solutes. J. Pharm. Sci. 1982, 71, 77–84. [Google Scholar] [CrossRef]
- Sugano, K. Possible reduction of effective thickness of intestinal unstirred water layer by particle drifting effect. Int. J. Pharm. 2010, 387, 103–109. [Google Scholar] [CrossRef]
- Imono, M.; Uchiyama, H.; Yoshida, S.; Miyazaki, S.; Tamura, N.; Tsutsumimoto, H.; Kadota, K.; Tozuka, Y. The elucidation of key factors for oral absorption enhancement of nanocrystal formulations: In vitro–in vivo correlation of nanocrystals. Eur. J. Pharm. Biopharm. 2020, 146, 84–92. [Google Scholar] [CrossRef]
- Arce, F.A.; Setiawan, N.; Campbell, H.R.; Lu, X.; Nethercott, M.J.; Bummer, P.; Su, Y.; Marsac, P.J. Toward Developing Discriminating Dissolution Methods for Formulations Containing Nanoparticulates in Solution: The Impact of Particle Drift and Drug Activity in Solution. Mol. Pharm. 2020, 17, 4125–4140. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Elzinga, P.A.; Hageman, M.; Herron, J.N. Rapid Throughput Solubility Screening Method for BCS Class II Drugs in Animal GI Fluids and Simulated Human GI Fluids Using a 96-well Format. J. Pharm. Sci. 2008, 97, 1427–1442. [Google Scholar] [CrossRef] [PubMed]
- Mithani, S.D.; Bakatselou, V.; TenHoor, C.N.; Dressman, J.B. Estimation of the Increase in Solubility of Drugs as a Function of Bile Salt Concentration. Pharm. Res. 1996, 13, 163–167. [Google Scholar] [CrossRef]
- Bakatselou, V.; Oppenheim, R.C.; Dressman, J.B. Solubilization and Wetting Effects of Bile Salts on the Dissolution of Steroids. Pharm. Res. 1991, 8, 1461–1469. [Google Scholar] [CrossRef]
- Tsinman, K.; Tsinman, O.; Lingamaneni, R.; Zhu, S.; Riebesehl, B.; Grandeury, A.; Juhnke, M.; Van Eerdenbrugh, B. Ranking Itraconazole Formulations Based on the Flux through Artificial Lipophilic Membrane. Pharm. Res. 2018, 35, 161. [Google Scholar] [CrossRef]
- Avdeef, A. Leakiness and Size Exclusion of Paracellular Channels in Cultured Epithelial Cell Monolayers–Interlaboratory Comparison. Pharm. Res. 2010, 27, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Miller, J.M.; Hoffman, A.; Amidon, G.E.; Amidon, G.L. The Solubility–Permeability Interplay in Using Cyclodextrins as Pharmaceutical Solubilizers: Mechanistic Modeling and Application to Progesterone. J. Pharm. Sci. 2010, 99, 2739–2749. [Google Scholar] [CrossRef] [Green Version]
- Sugano, K. Aqueous Boundary Layers Related to Oral Absorption of a Drug: From Dissolution of a Drug to Carrier Mediated Transport and Intestinal Wall Metabolism. Mol. Pharm. 2010, 7, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Korjamo, T.; Heikkinen, A.T.; Waltari, P.; Mönkkönen, J. The Asymmetry of the Unstirred Water Layer in Permeability Experiments. Pharm. Res. 2008, 25, 1714–1722. [Google Scholar] [CrossRef] [PubMed]
- Safety Data in IPEC Japan. Available online: http://www.jpec.gr.jp/detail=normal&date=safetydata/ra/dara3.html (accessed on 30 November 2020).
- Schiller, C.; Frohlich, C.-P.; Giessmann, T.; Siegmund, W.; Monnikes, H.; Hosten, N.; Weitschies, W. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment. Pharmacol. Ther. 2005, 22, 971–979. [Google Scholar] [CrossRef]
- Mudie, D.M.; Murray, K.; Hoad, C.L.; Pritchard, S.E.; Garnett, M.C.; Amidon, G.L.; Gowland, P.A.; Spiller, R.C.; Amidon, G.E.; Marciani, L. Quantification of Gastrointestinal Liquid Volumes and Distribution Following a 240 mL Dose of Water in the Fasted State. Mol. Pharm. 2014, 11, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A. Chapter 7. Permeability—PAMPA. In Absorption and Drug Development: Solubility, Permeability, and Charge State, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 319–498. [Google Scholar]
- Turro, N.J.; Yekta, A. Luminescent probes for detergent solutions. A simple procedure for determination of the mean aggregation number of micelles. J. Am. Chem. Soc. 1978, 100, 5951–5952. [Google Scholar] [CrossRef]
- Doyle-McCullough, M.; Smyth, S.; Moyes, S.; Carr, K. Factors influencing intestinal microparticle uptake in vivo. Int. J. Pharm. 2007, 335, 79–89. [Google Scholar] [CrossRef]
- Limpanussorn, J.; Simon, L.; Dayan, A.D. Transepithelial Transport of Large Particles in Rat: A New Model for the Quantitative Study of Particle Uptake. J. Pharm. Pharmacol. 1998, 50, 753–760. [Google Scholar] [CrossRef]
- Norris, D.A.; Puri, N.; Sinko, P.J. The effect of physical barriers and properties on the oral absorption of particulates. Adv. Drug Deliv. Rev. 1998, 34, 135–154. [Google Scholar] [CrossRef]
- Smyth, S.; Feldhaus, S.; Schumacher, U.; Carr, K. Uptake of inert microparticles in normal and immune deficient mice. Int. J. Pharm. 2008, 346, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, J.; Flanagan, D.R. Chapter 9. Fundamentals of Diffusion and Dissolution. In Developing Solid Oral Dosage Forms, 2nd ed.; Qiu, Y., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 253–270. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Properties | Griseofulvin | Triamcinolone |
|---|---|---|
| Molecular weight (MW) | 352.77 | 394.43 |
| Ionization properties | Neutral | Neutral |
| Log P 1 | 2.18 [40,41] | 1.03 [40,42] |
| Aqueous solubility (μg/mL) 2 | 29.9 [40,41] | 158 [40,42] |
| Compound | Test Media | Sample Dose Amount (μg/mL) |
|---|---|---|
| Griseofulvin | pH 6.5 phosphate buffer (pH 6.5 buffer) | 5, 50, 200, and 1000 |
| pH 6.5 phosphate buffer with 0.05% (w/w) sodium lauryl sulfate (SLS) (pH 6.5 buffer + 0.05% SLS) | 5, 50, 200, and 1000 | |
| Triamcinolone | pH 6.5 buffer | 100, 500, 2000, and 10,000 |
| pH 6.5 buffer + 0.05% SLS | 100, 500, 2000, and 10,000 |
| Compound | Test Media | Solubility (μg/mL) 1 | UFD Amount (μg/mL) | Fraction of UFD (FU) |
|---|---|---|---|---|
| Griseofulvin | pH 6.5 buffer | 10.75 ± 0.38 | 10.75 | 1.00 |
| pH 6.5 buffer + 0.05% SLS | 27.40 ± 0.07 | 10.75 | 0.39 | |
| Triamcinolone | pH 6.5 buffer | 205.04 ± 10.34 | 205.04 | 1.00 |
| pH 6.5 buffer + 0.05% SLS | 210.07 ± 6.54 | 205.04 | 0.98 |
| Compound | Griseofulvin | Triamcinolone |
|---|---|---|
| MW | 352.77 | 394.43 |
| Measured Papp (U) (cm/min) 1 | 0.0148 ± 0.0007 | 0.000304 ± 0.000010 |
| Calculated Daq (U) (cm2/s) | 5.18 × 10−6 | 4.93 × 10−6 |
| Calculated PUWL (U) (cm/min) | 0.0311 | 0.0296 |
| Calculated Pm (U) (cm/min) | 0.0284 | 0.000307 |
| Calculated Papp (U) (cm/min) | 0.0148 | 0.000304 |
| Compound | Griseofulvin | Triamcinolone |
|---|---|---|
| Measured Papp (cm/min) 1 | 0.00648 ± 0.00026 | 0.000263 ± 0.000028 |
| Calculated Pm (app) (cm/min) | 0.0112 | 0.000300 |
| Calculated PUWL (app) (cm/min) | 0.0153 | 0.0212 |
| Calculated Papp (cm/min) | 0.00648 | 0.000263 |
| Calculated Daq (app) (cm2/s) | 2.56 × 10−6 | 3.54 × 10−7 |
| Calculated Daq (B) (cm2/s) | 8.37 × 10−7 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugita, K.; Takata, N.; Yonemochi, E. Dose-Dependent Solubility–Permeability Interplay for Poorly Soluble Drugs under Non-Sink Conditions. Pharmaceutics 2021, 13, 323. https://doi.org/10.3390/pharmaceutics13030323
Sugita K, Takata N, Yonemochi E. Dose-Dependent Solubility–Permeability Interplay for Poorly Soluble Drugs under Non-Sink Conditions. Pharmaceutics. 2021; 13(3):323. https://doi.org/10.3390/pharmaceutics13030323
Chicago/Turabian StyleSugita, Kazuya, Noriyuki Takata, and Etsuo Yonemochi. 2021. "Dose-Dependent Solubility–Permeability Interplay for Poorly Soluble Drugs under Non-Sink Conditions" Pharmaceutics 13, no. 3: 323. https://doi.org/10.3390/pharmaceutics13030323