
Repurposing of Sitagliptin- Melittin Optimized Nanoformula against SARS-CoV-2; Antiviral Screening and Molecular Docking Studies

 , , ,
, , ,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Factorial Design for Development and Optimization of SIT-MEL Nano-Sized Systems
2.3. Preparation of SIT-MEL Formulations
2.4. Particle Size and Zeta Potential Determination
2.5. Optimization of SIT-MEL NPs
2.6. Fourier-Transform Infrared Spectroscopy of the Optimized SIT-MEL NPs
2.7. IC50 Calculation Using Crystal Violet Assay
2.8. In Vtro Mpro 3CL-protease Inhibition Test
2.9. Molecular Docking Study
2.10. Statistical Analysis
3. Results
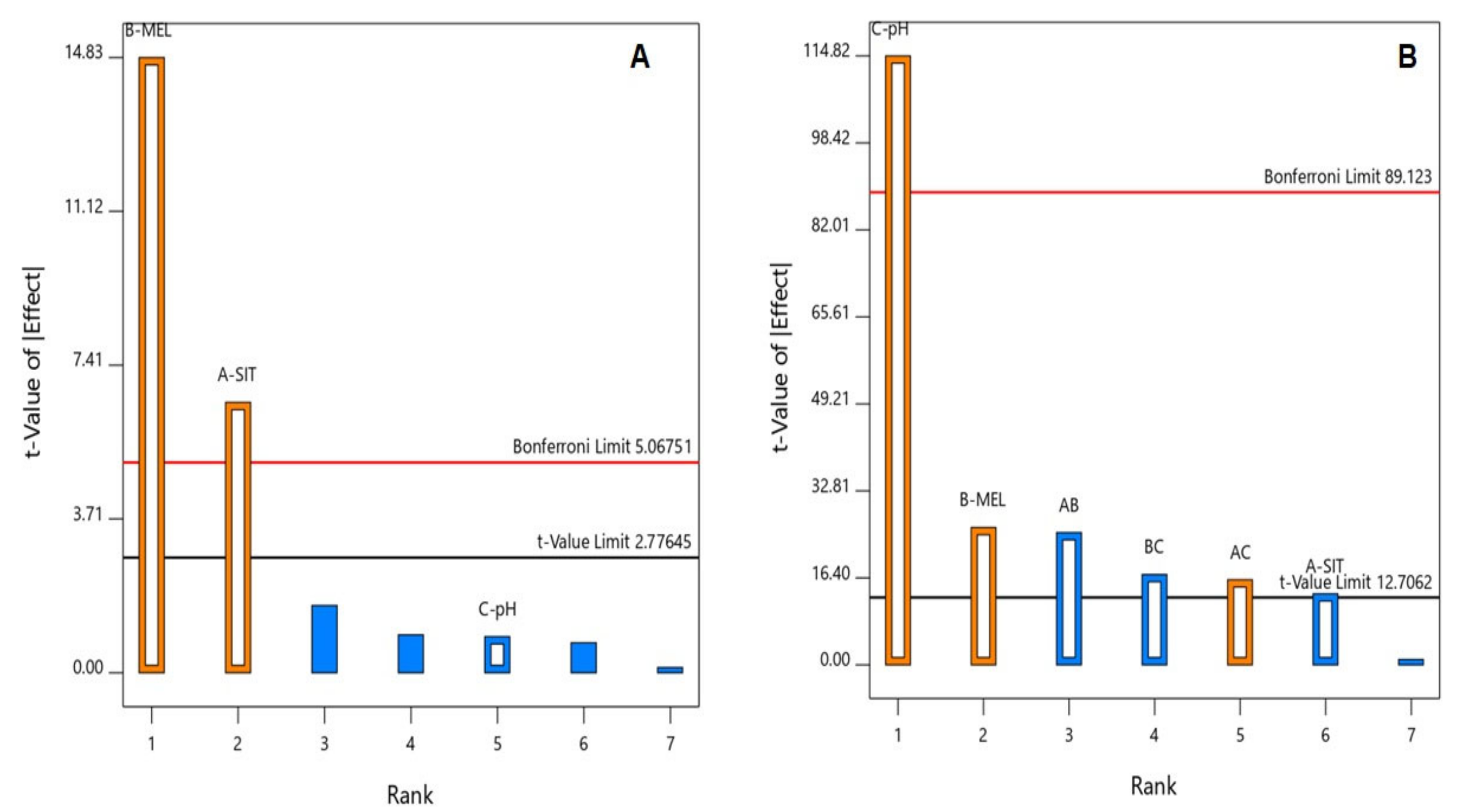
3.1. Statistical Analysis of the Factorial Design
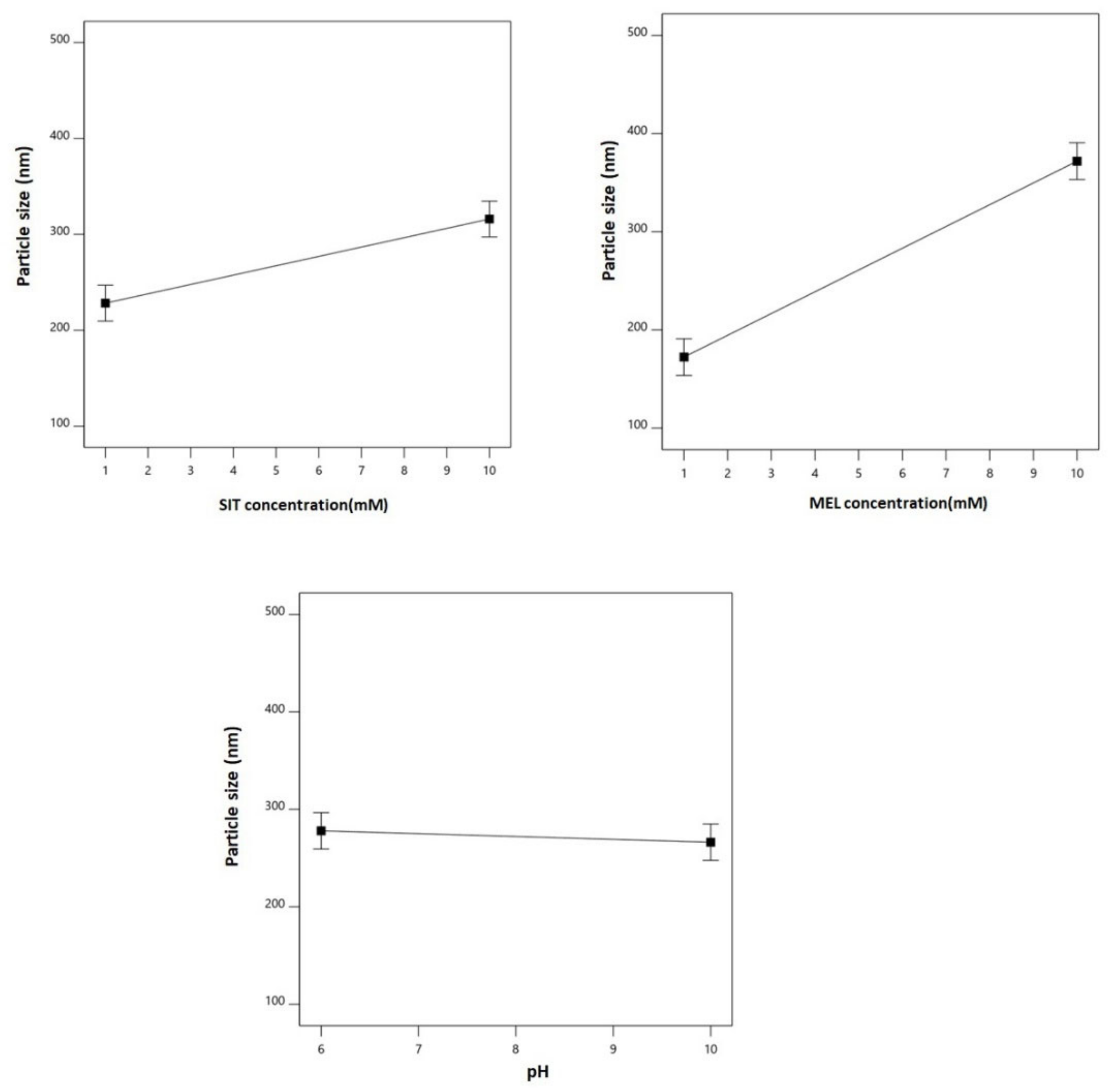
3.2. Effect of Variables on Particle Size (Y1)
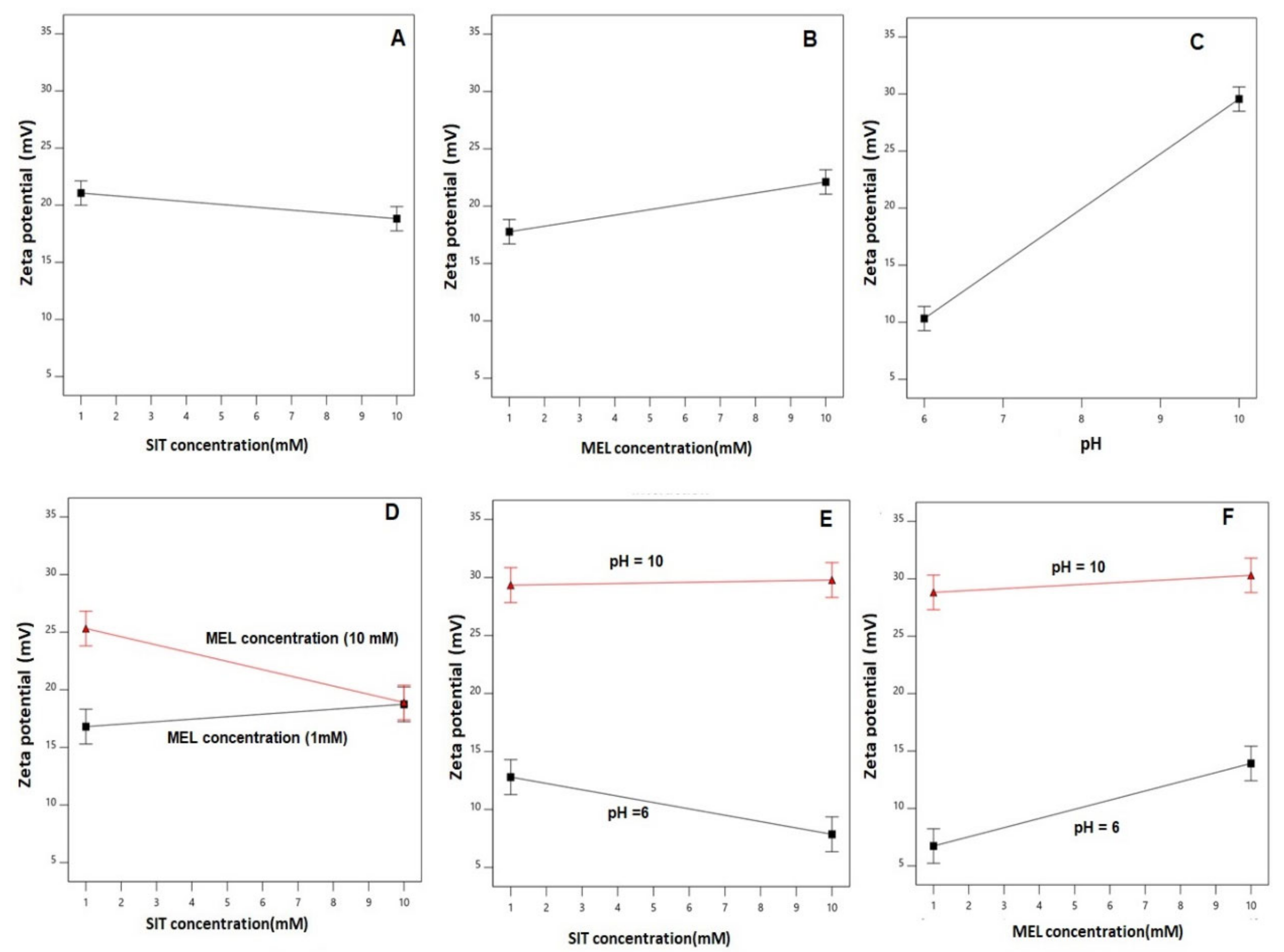
3.3. Effect of Variables on Zeta Potential (Y2)
3.4. Selection of the Optimized SIT-MEL Nano-Sized Systems
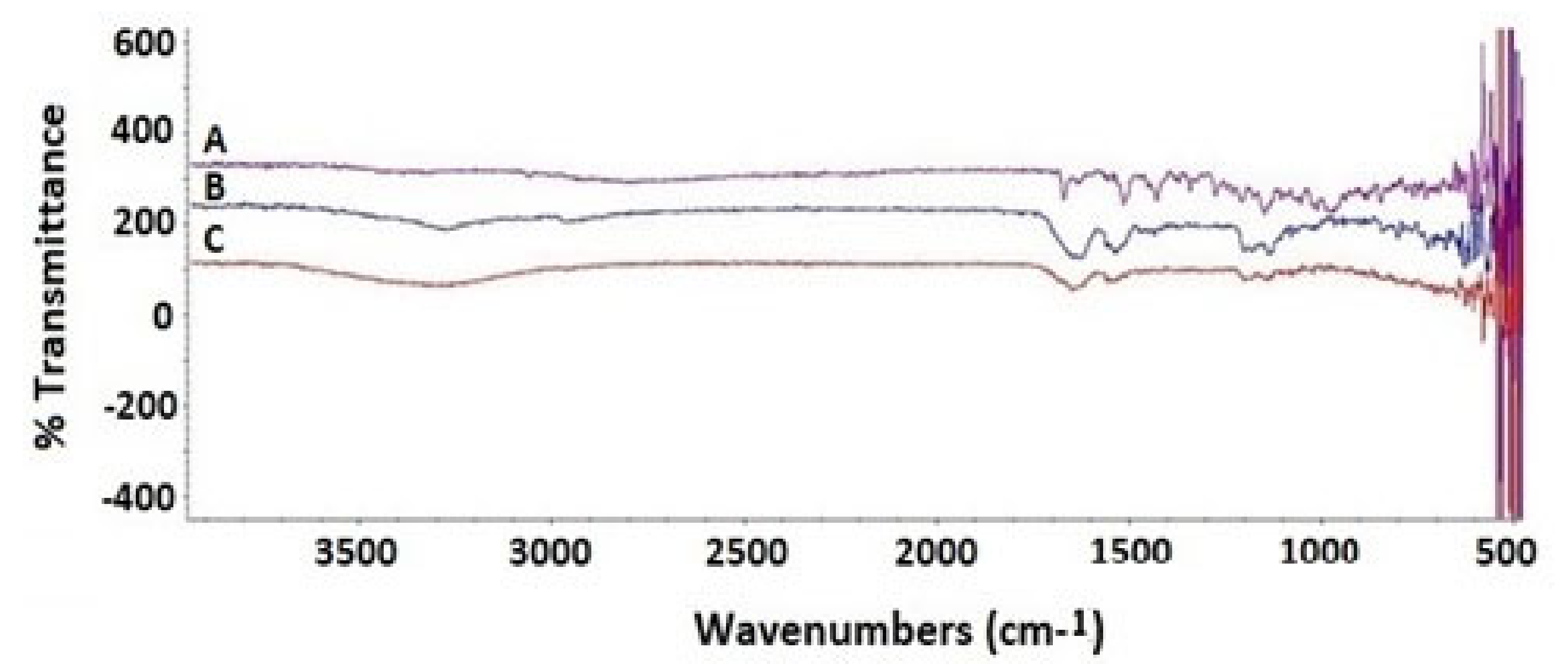
3.5. Fourier-Transform Infrared Spectroscopy Investigation of the Optimized SIT-MEL NPs
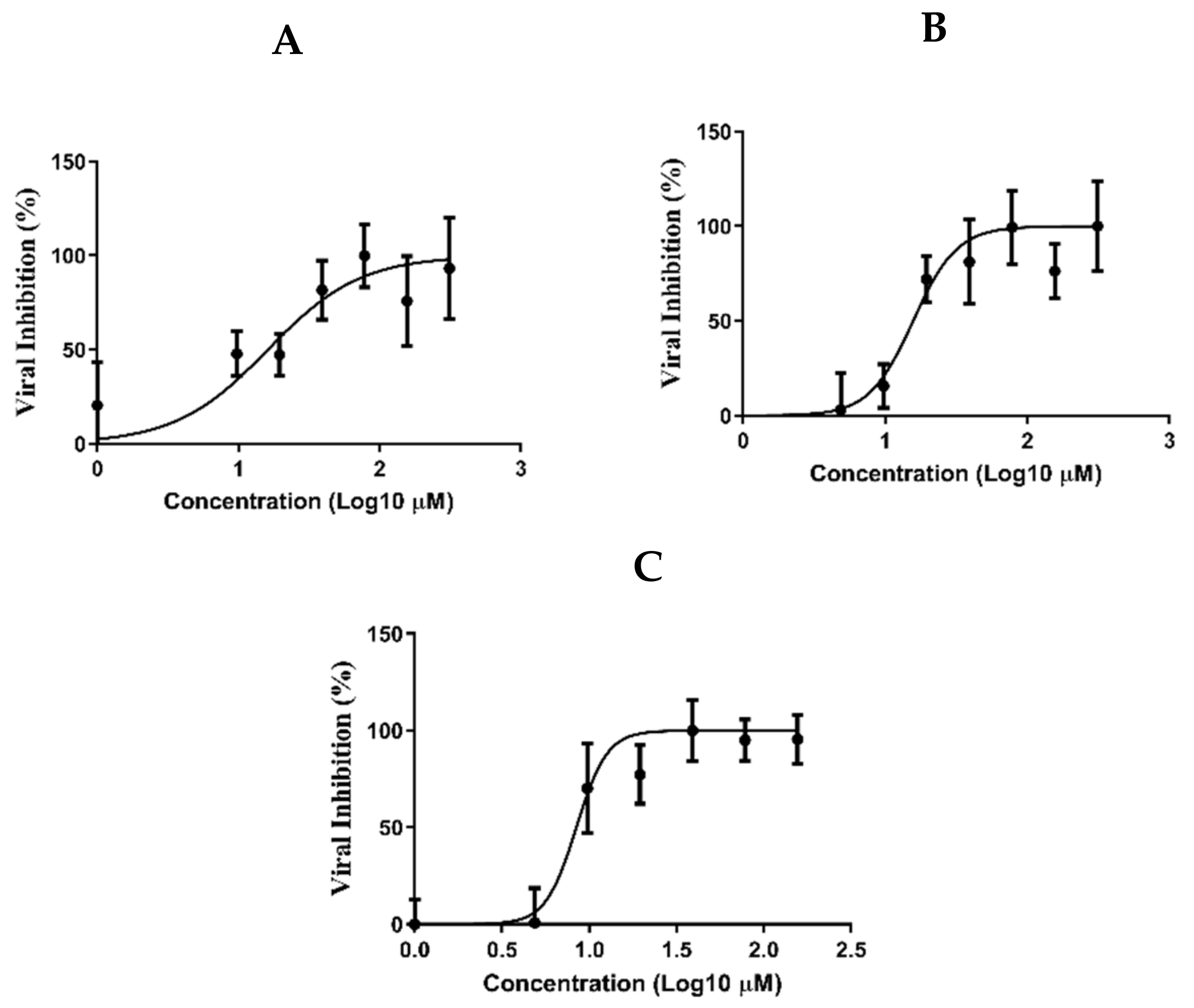
3.6. Determination of the Antiviral Activity
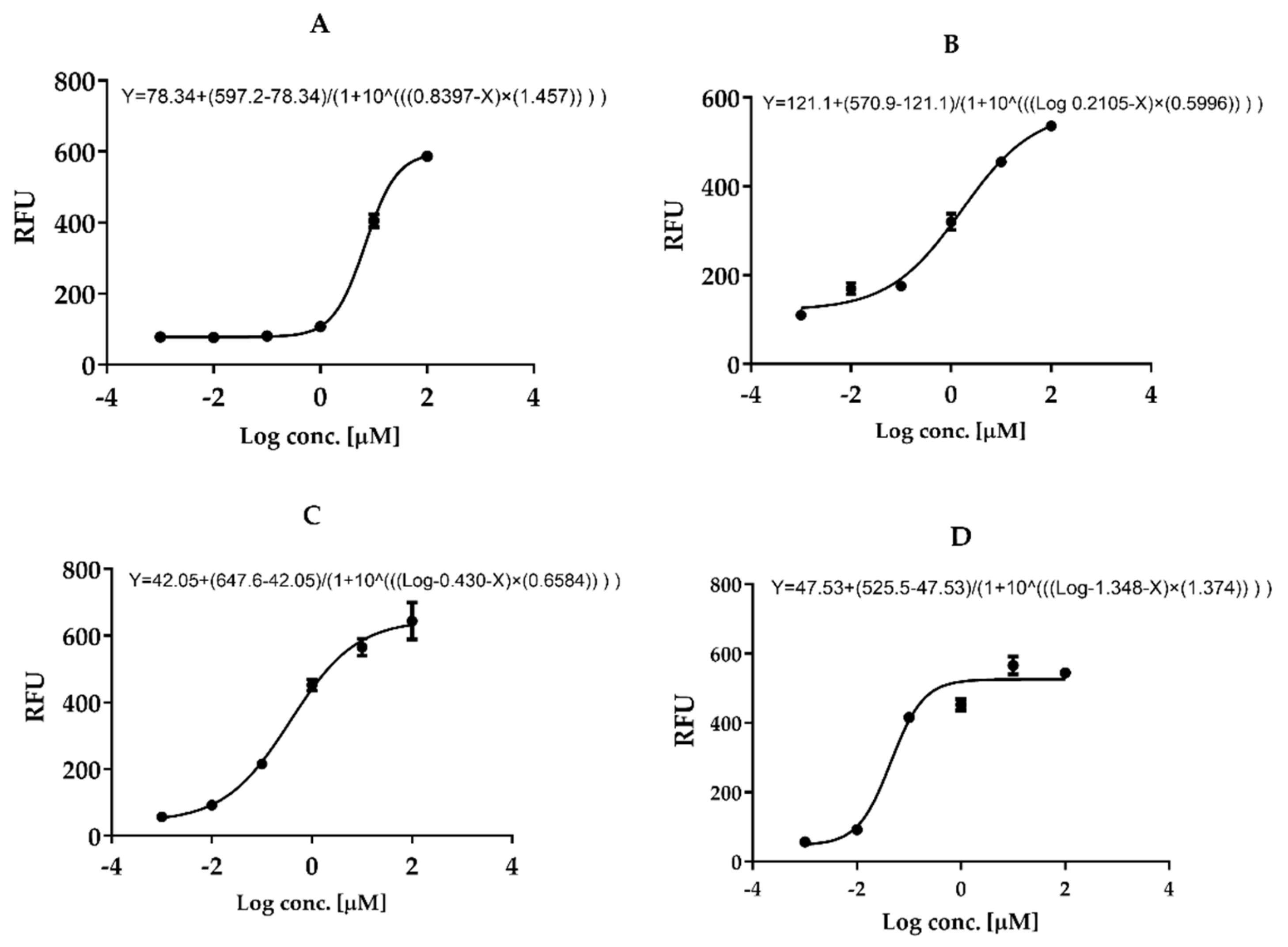
3.7. In Vitro Mpro 3CL-Protease Inhibition
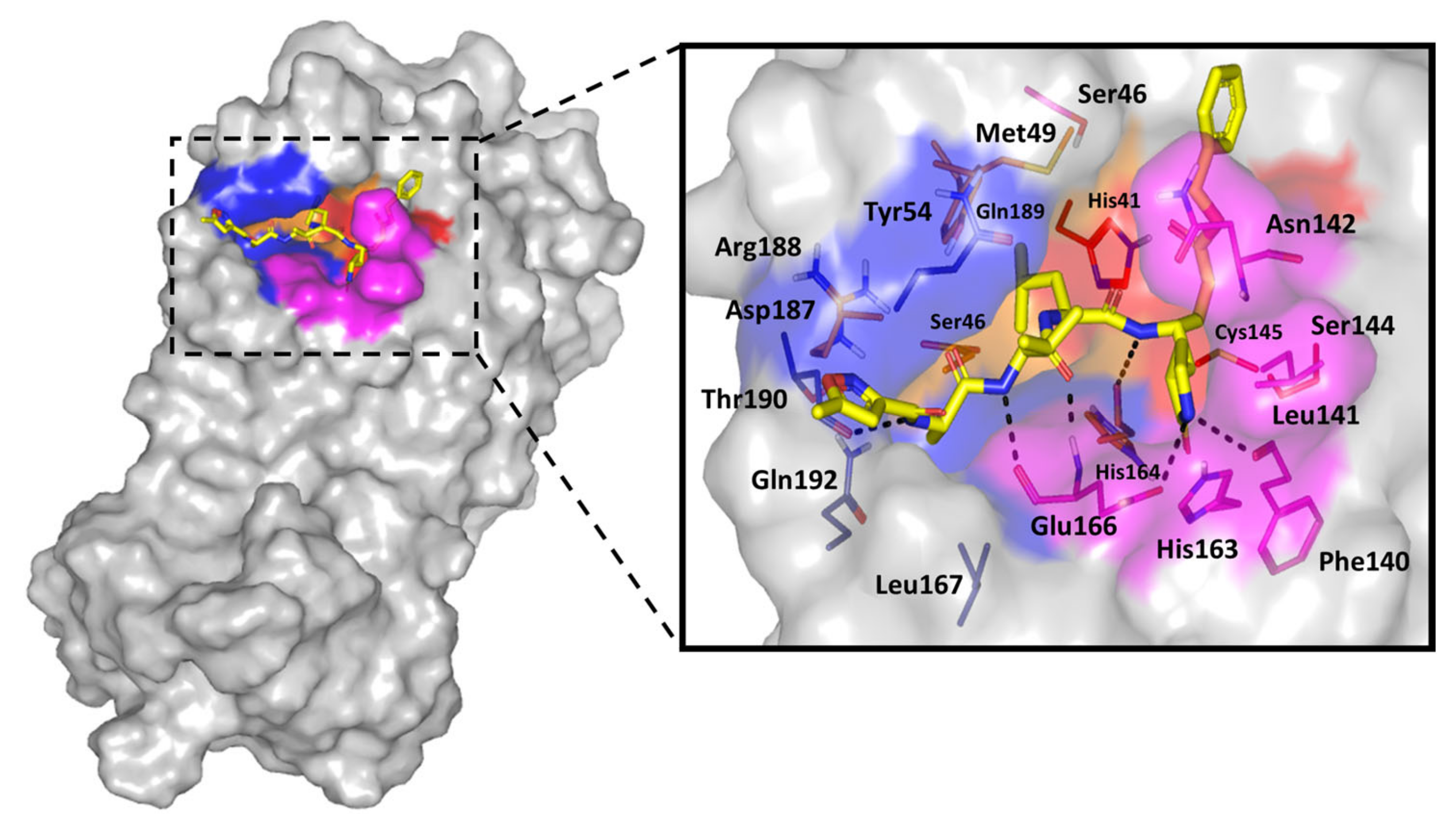
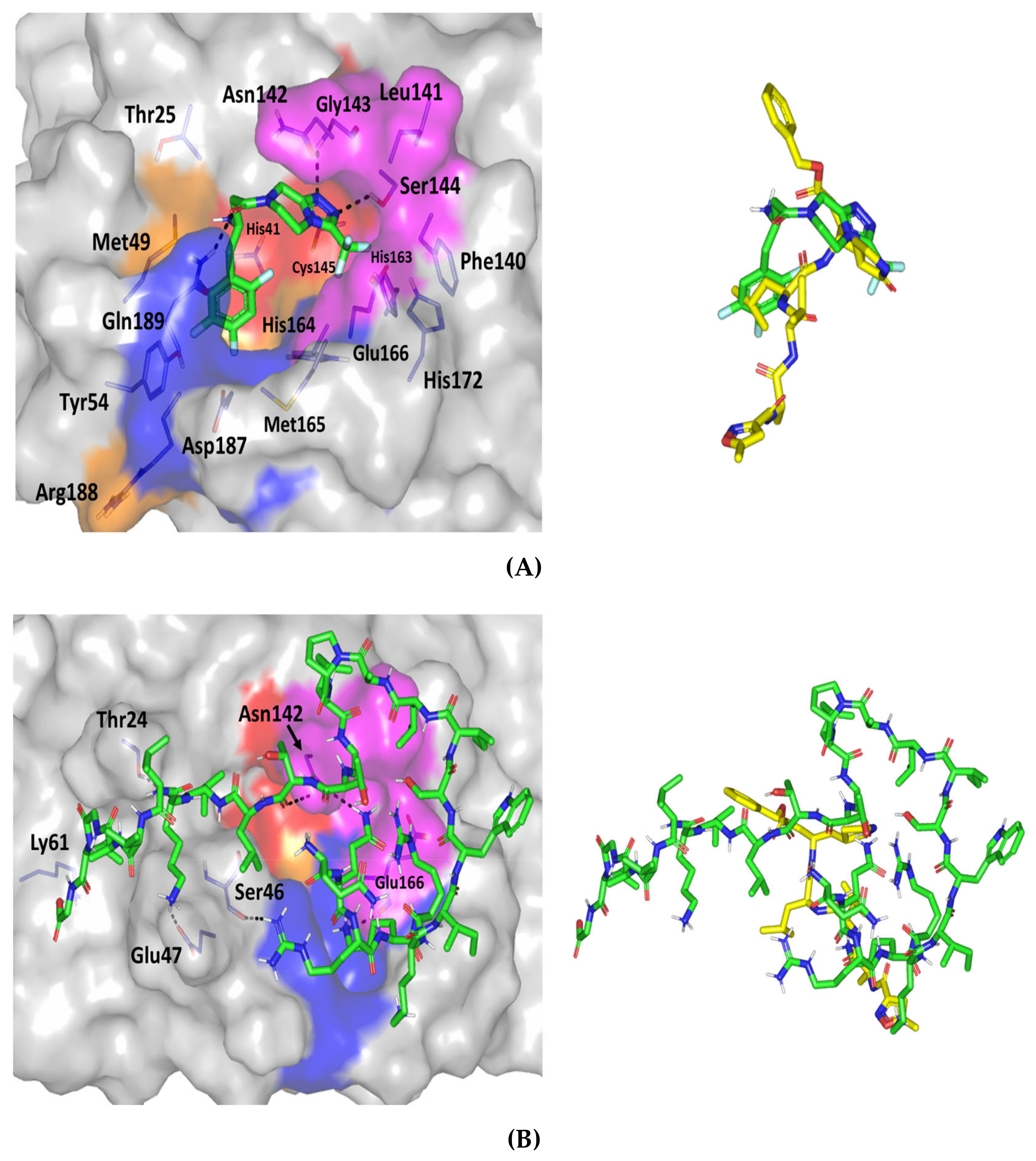
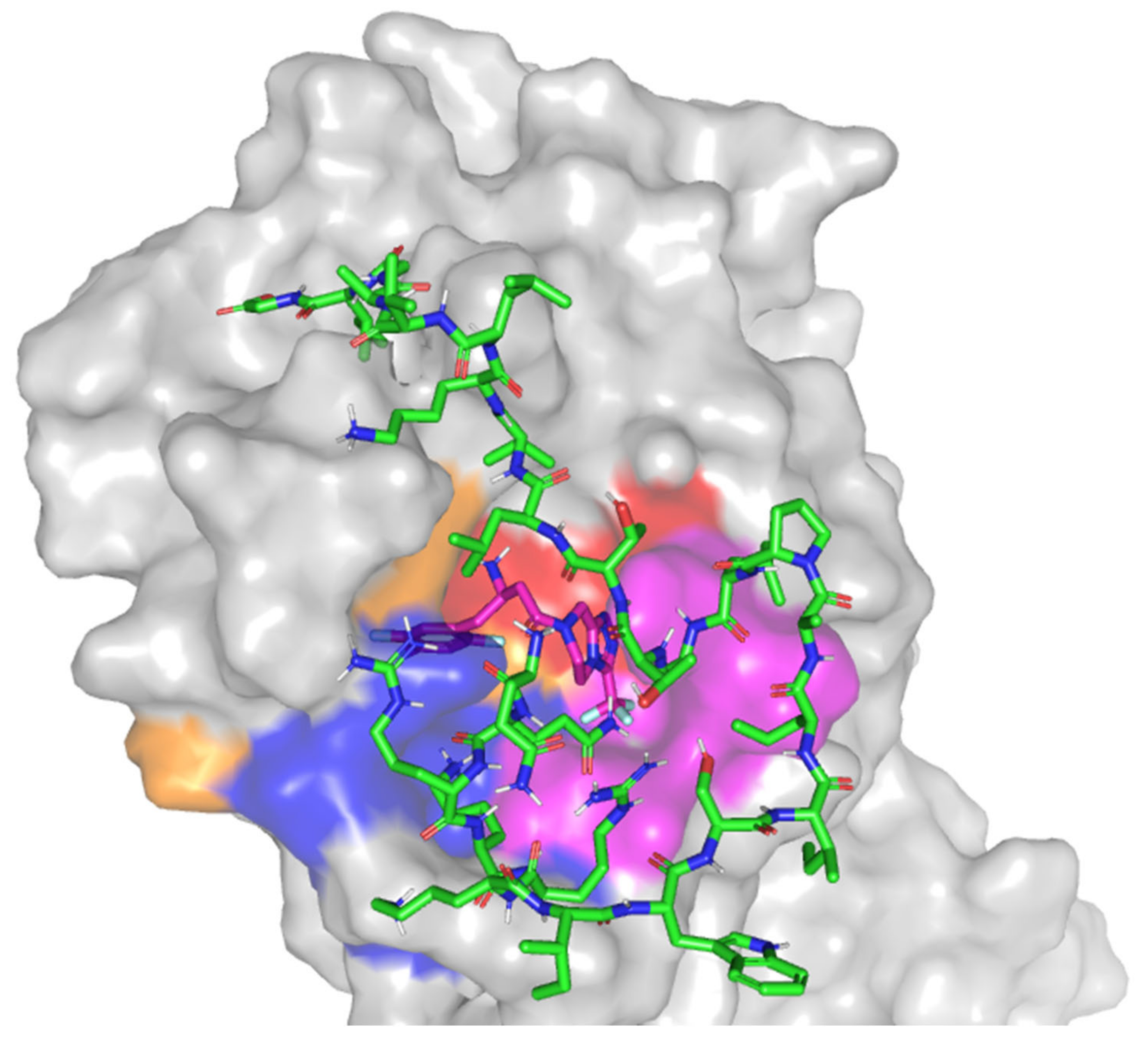
3.8. Molecular Docking Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chakraborty, I.; Maity, P. COVID-19 outbreak: Migration, effects on society, global environment and prevention. Sci. Total Environ. 2020, 728, 138882. [Google Scholar] [CrossRef]
- Schindler, S.; Jepson, N.; Cui, W. Covid-19, China and the future of global development. Res. Glob. 2020, 2, 100020. [Google Scholar] [CrossRef]
- WHO. Naming the Coronavirus Disease (COVID-19) and the Virus That Causes It. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it (accessed on 3 February 2021).
- Kontoangelos, K.; Economou, M.; Papageorgiou, C. Mental health effects of COVID-19 pandemia: A review of clinical and psychological traits. Psychiatry Investig. 2020, 17, 491–505. [Google Scholar] [CrossRef]
- Möhn, N.; Pul, R.; Kleinschnitz, C.; Prüss, H.; Witte, T.; Stangel, M.; Skripuletz, T. Implications of COVID-19 Outbreak on Immune Therapies in Multiple Sclerosis Patients-Lessons Learned from SARS and MERS. Front. Immunol. 2020, 1, 1059. [Google Scholar] [CrossRef]
- Florindo, H.F.; Kleiner, R.; Vaskovich-Koubi, D.; Acúrcio, R.C.; Carreira, B.; Yeini, E.; Tiram, G.; Liubomirski, Y.; Satchi-Fainaro, R. Immune-mediated approaches against COVID-19. Nat. Nanotechnol. 2020, 15, 630–645. [Google Scholar] [CrossRef]
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 2020, 109, 531–538. [Google Scholar] [CrossRef]
- Rodilla, E.; Saura, A.; Jiménez, I.; Mendizábal, A.; Pineda-Cantero, A.; Lorenzo-Hernández, E.; Fidalgo-Montero, M.P.; López-Cuervo, J.F.; Gil-Sánchez, R.; Rabadán-Pejenaute, E.; et al. Association of Hypertension with All-Cause Mortality among Hospitalized Patients with COVID-19. J. Clin. Med. 2020, 9, 3136. [Google Scholar] [CrossRef] [PubMed]
- Pantea Stoian, A.; Pricop-Jeckstadt, M.; Pana, A.; Ileanu, B.V.; Schitea, R.; Geanta, M.; Catrinoiu, D.; Suceveanu, A.I.; Serafinceanu, C.; Pituru, S.; et al. Death by SARS-CoV 2: A Romanian COVID-19 multi-centre comorbidity study. Sci. Rep. 2020, 10, 21613. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.; Mustafa, F.; Rizvi, T.A.; Loney, T.; Al Suwaidi, H.; Al-Marzouqi, A.H.H.; Eldin, A.K.; Alsabeeha, N.; Adrian, T.E.; Stefanini, C.; et al. SARS-CoV-2/COVID-19: Viral genomics, epidemiology, vaccines, and therapeutic interventions. Viruses 2020, 12, 526. [Google Scholar] [CrossRef]
- Berger, J.R.; Brandstadter, R.; Bar-Or, A. COVID-19 and MS disease-modifying therapies. Neurol Neuroimmunol. Neuroinflamm. 2020, 7, 761. [Google Scholar] [CrossRef] [PubMed]
- Valencia, I.; Peiró, C.; Lorenzo, Ó.; Sánchez-Ferrer, C.F.; Eckel, J.; Romacho, T. DPP4 and ACE2 in Diabetes and COVID-19: Therapeutic Targets for Cardiovascular Complications? Front. Pharmacol. 2020, 11, 1161. [Google Scholar] [CrossRef]
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B. Global pandemics interconnected—obesity, impaired metabolic health and COVID-19. Nat. Rev. Endocrinol. 2021, 17, 135–149. [Google Scholar] [CrossRef]
- Popkin, B.M.; Du, S.; Green, W.D.; Beck, M.A.; Algaith, T.; Herbst, C.H.; Alsukait, R.F.; Alluhidan, M.; Alazemi, N.; Shekar, M. Individuals with obesity and COVID-19: A global perspective on the epidemiology and biological relationships. Obes. Rev. 2020, 21, e13128. [Google Scholar] [CrossRef] [PubMed]
- Mozafari, N.; Azadi, S.; Mehdi-Alamdarlou, S.; Ashrafi, H.; Azadi, A. Inflammation: A bridge between diabetes and COVID-19, and possible management with sitagliptin. Med. Hypotheses 2020, 143, 110111. [Google Scholar] [CrossRef]
- Du, F.; Liu, B.; Zhang, S. COVID-19: The role of excessive cytokine release and potential ACE2 down-regulation in promoting hypercoagulable state associated with severe illness. J. Thromb. Thrombolysis 2021, 51, 313–329. [Google Scholar] [CrossRef]
- Bardaweel, S.K.; Hajjo, R.; Sabbah, D.A. Sitagliptin: A potential drug for the treatment of COVID-19? Acta Pharm. 2021, 71, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Apicella, M.; Campopiano, M.C.; Mantuano, M.; Mazoni, L.; Coppelli, A.; Del Prato, S. COVID-19 in people with diabetes: Understanding the reasons for worse outcomes. Lancet Diabetes Endocrinol. 2020, 8, 782–792. [Google Scholar] [CrossRef]
- Hussain, A.; Bhowmik, B.; do Vale Moreira, N.C. COVID-19 and diabetes: Knowledge in progress. Diabetes Res. Clin. Pract. 2020, 162, 108142. [Google Scholar] [CrossRef]
- Begum, J.; Mir, N.A.; Dev, K.; Buyamayum, B.; Wani, M.Y.; Raza, M. Challenges and prospects of COVID-19 vaccine development based on the progress made in SARS and MERS vaccine development. Transbound. Emerg. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Garner-Spitzer, E.; Jasinska, J.; Kollaritsch, H.; Stiasny, K.; Kundi, M.; Wiedermann, U. Age-related differences in humoral and cellular immune responses after primary immunisation: Indications for stratified vaccination schedules. Sci. Rep. 2018, 8, 9825. [Google Scholar] [CrossRef] [Green Version]
- Fulton, R.B.; Varga, S.M. Effects of aging on the adaptive immune response to respiratory virus infections. Aging Health 2009, 5, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindarchuk, J. Repurposing Drugs is Key to Fighting the Coronavirus Pandemic, This Virologist Explains. Available online: https://www.forbes.com/sites/coronavirusfrontlines/2020/05/08/repurposing-drugs-is-key-to-fighting-the-coronavirus-pandemic-this-virologist-explains/?sh=5c6db0ae17ce (accessed on 3 February 2021).
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef]
- Center for Drug Evaluation; Research Clinical Pharmacology and Biopharmaceutics Review (S). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202343Orig1s000ClinPharmR.pdf (accessed on 4 February 2021).
- Kim, S.J.; Park, J.H.; Kim, K.H.; Lee, W.R.; Kim, K.S.; Park, K.K. Melittin inhibits atherosclerosis in LPS/high-fat treated mice through atheroprotective actions. J. Atheroscler. Thromb. 2011, 18, 1117–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Aarag, B.; Magdy, M.; AlAjmi, M.F.; Khalifa, S.A.M.; El-Seedi, H.R. Melittin exerts beneficial effects on paraquat-induced lung injuries in mice by modifying oxidative stress and apoptosis. Molecules 2019, 24, 1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Yu, M.; He, Y.; Xiao, L.; Wang, F.; Song, C.; Sun, S.; Ling, C.; Xu, Z. Melittin prevents liver cancer cell metastasis through inhibition of the Rac1-dependent pathway. Hepatology 2008, 47, 1964–1973. [Google Scholar] [CrossRef]
- Wehbe, R.; Frangieh, J.; Rima, M.; El Obeid, D.; Sabatier, J.-M.; Fajloun, Z. Bee venom: Overview of main compounds and bioactivities for therapeutic interests. Molecules 2019, 24, 2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, N.W.; Medzhitov, R. Role of the inflammasome in defense against venoms. Proc. Natl. Acad. Sci. USA 2013, 110, 1809–1814. [Google Scholar] [CrossRef] [Green Version]
- Memariani, H.; Memariani, M.; Shahidi-Dadras, M.; Nasiri, S.; Akhavan, M.M.; Moravvej, H. Melittin: From honeybees to superbugs. Appl. Microbiol. Biotechnol. 2019, 103, 3265–3276. [Google Scholar] [CrossRef]
- Gajski, G.; Garaj-Vrhovac, V. Melittin: A lytic peptide with anticancer properties. Environ. Toxicol. Pharmacol. 2013, 36, 697–705. [Google Scholar] [CrossRef]
- Uddin, M.B.; Lee, B.H.; Nikapitiya, C.; Kim, J.H.; Kim, T.H.; Lee, H.C.; Kim, C.G.; Lee, J.S.; Kim, C.J. Inhibitory effects of bee venom and its components against viruses in vitro and in vivo. J. Microbiol. 2016, 54, 853–866. [Google Scholar] [CrossRef]
- Caramalho, I.; Melo, A.; Pedro, E.; Barbosa, M.M.P.; Victorino, R.M.M.; Pereira Santos, M.C.; Sousa, A.E. Bee venom enhances the differentiation of human regulatory T cells. Allergy Eur. J. Allergy Clin. Immunol. 2015, 70, 1340–1345. [Google Scholar] [CrossRef]
- Kasozi, K.I.; Niedbała, G.; Alqarni, M.; Zirintunda, G.; Ssempijja, F.; Musinguzi, S.P.; Usman, I.M.; Matama, K.; Hetta, H.F.; Mbiydzenyuy, N.E.; et al. Bee Venom—A Potential Complementary Medicine Candidate for SARS-CoV-2 Infections. Front. Public Health 2020, 8, 594458. [Google Scholar] [CrossRef]
- Rady, I.; Siddiqui, I.A.; Rady, M.; Mukhtar, H. Melittin, a major peptide component of bee venom, and its conjugates in cancer therapy. Cancer Lett. 2017, 402, 16–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Lu, X.; Deng, Z.; Xiao, S.; Yuan, B.; Yang, K. How melittin inserts into cell membrane: Conformational changes, inter-peptide cooperation, and disturbance on the membrane. Molecules 2019, 24, 1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allolio, C.; Magarkar, A.; Jurkiewicz, P.; Baxová, K.; Javanainen, M.; Mason, P.E.; Šachl, R.; Cebecauer, M.; Hof, M.; Horinek, D.; et al. Arginine-rich cell-penetrating peptides induce membrane multilamellarity and subsequently enter via formation of a fusion pore. Proc. Natl. Acad. Sci. USA 2018, 115, 11923–11928. [Google Scholar] [CrossRef] [Green Version]
- Farkhani, S.M.; Valizadeh, A.; Karami, H.; Mohammadi, S.; Sohrabi, N.; Badrzadeh, F. Cell penetrating peptides: Efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides 2014, 57, 78–94. [Google Scholar] [CrossRef]
- Gräslund, A.; Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S. Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar]
- Jobin, M.L.; Alves, I.D. On the importance of electrostatic interactions between cell penetrating peptides and membranes: A pathway toward tumor cell selectivity? Biochimie 2014, 107, 154–159. [Google Scholar] [CrossRef]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef]
- Takeuchi, T.; Futaki, S. Current understanding of direct translocation of arginine-rich cell-penetrating peptides and its internalization mechanisms. Chem. Pharm. Bull. 2016, 64, 1431–1437. [Google Scholar] [CrossRef] [Green Version]
- Takechi-Haraya, Y.; Saito, H. Current Understanding of Physicochemical Mechanisms for Cell Membrane Penetration of Arginine-rich Cell Penetrating Peptides: Role of Glycosaminoglycan Interactions. Curr. Protein Pept. Sci. 2018, 19, 623–630. [Google Scholar] [CrossRef]
- Dupont, E.; Prochiantz, A.; Joliot, A. Penetratin Story: An Overview. In Methods in Molecular Biology; Clifton, N.J., Ed.; Humana Press: Totowa, NJ, USA, 2011; Volume 683, pp. 21–29. [Google Scholar]
- Walrant, A.; Cardon, S.; Burlina, F.; Sagan, S. Membrane Crossing and Membranotropic Activity of Cell-Penetrating Peptides: Dangerous Liaisons? Acc. Chem. Res. 2017, 50, 2968–2975. [Google Scholar] [CrossRef]
- Mostafa, A.; Kandeil, A.; Elshaier, Y.A.M.M.; Kutkat, O.; Moatasim, Y.; Rashad, A.A.; Shehata, M.; Gomaa, M.R.; Mahrous, N.; Mahmoud, S.H.; et al. FDA-Approved Drugs with Potent In Vitro Antiviral Activity against Severe Acute Respiratory Syndrome Coronavirus 2. Pharmaceuticals 2020, 13, 443. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malebari, A.M.; Ibrahim, T.S.; Salem, I.M.; Salama, I.; Khayyat, A.N.; Mostafa, S.M.; El-Sabbagh, O.I.; Darwish, K.M. The anticancer activity for the bumetanide-based analogs via targeting the tumor-associated membrane-bound human carbonic anhydrase-IX enzyme. Pharmaceuticals 2020, 13, 252. [Google Scholar] [CrossRef] [PubMed]
- Inc, M. Comprehensive System for Life Science Education. Available online: https://www.molsis.co.jp/wp-content/themes/molsis/pdf/moe_eng.pdf (accessed on 18 February 2021).
- Attique, S.A.; Hassan, M.; Usman, M.; Atif, R.M.; Mahboob, S.; Al-Ghanim, K.A.; Bilal, M.; Nawaz, M.Z. A molecular docking approach to evaluate the pharmacological properties of natural and synthetic treatment candidates for use against hypertension. Int. J. Environ. Res. Public Health 2019, 16, 923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadie, M.A.; Kishk, S.M.; Darwish, K.M.; Mostafa, S.M.; Elgawish, M.S. Simultaneous Determination of Losartan and Rosuvastatin in Rat Plasma Using Liquid Chromatography–Tandem Mass Spectrometric Technique for Application into Pharmacokinetic and Drug–Drug Interaction Studies. Chromatographia 2020, 83, 1477–1494. [Google Scholar] [CrossRef]
- Chemical Computing Group (CCG) Research. Available online: https://www.chemcomp.com/Research-Citing_MOE.htm (accessed on 15 December 2020).
- Edelsbrunner, H.; Facello, M.; Fu, P.; Liang, J. Measuring proteins and voids in proteins. In Proceedings of the 28th Annual Hawaii International Conference on System Sciences, Wailea, HI, USA, 3–6 January 1995; IEEE Computer Society: Piscataway, NJ, USA, 1995; Volume 5, pp. 256–264. [Google Scholar]
- Liang, J.; Edelsbrunner, H.; Woodward, C. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. 1998, 7, 1884–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soga, S.; Shirai, H.; Koborv, M.; Hirayama, N. Use of amino acid composition to predict ligand-binding sites. J. Chem. Inf. Model. 2007, 47, 400–406. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Román, J.; Castillo, A.; Mahn, A. Molecular Docking of Potential Inhibitors of Broccoli Myrosinase. Molecules 2018, 23, 1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; He, F.; Liu, D.; Fang, M.; Wu, Z.; Xu, D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. bioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, T.A.; Badr-Eldin, S.M.; Ahmed, O.A.A.; Aldawsari, H. Intranasal optimized solid lipid nanoparticles loaded in situ gel for enhancing trans-mucosal delivery of simvastatin. J. Drug Deliv. Sci. Technol. 2018, 48, 499–508. [Google Scholar] [CrossRef]
- Aldawsari, H.M.; Fahmy, U.A.; Abd-Allah, F.; Ahmed, O.A.A. Formulation and Optimization of Avanafil Biodegradable Polymeric Nanoparticles: A Single-Dose Clinical Pharmacokinetic Evaluation. Pharmaceutics 2020, 12, 596. [Google Scholar] [CrossRef]
- Aldawsari, H.M.; Alhakamy, N.A.; Padder, R.; Husain, M.; Md, S. Preparation and characterization of chitosan coated plga nanoparticles of resveratrol: Improved stability, antioxidant and apoptotic activities in H1299 lung cancer cells. Coatings 2020, 10, 439. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal violet assay for determining viability of cultured cells. Cold Spring Harb. Protoc. 2016, 2016, 343–346. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Hung, H.-C.; Ke, Y.-Y.; Huang, S.Y.; Huang, P.-N.; Kung, Y.-A.; Chang, T.-Y.; Yen, K.-J.; Peng, T.-T.; Chang, S.-E.; Huang, C.-T.; et al. Discovery of M protease inhibitors encoded by SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, e00872–e00920. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Lamas, M.S.; Maggini, J. Functional and druggability analysis of the SARS-CoV-2 proteome. Eur. J. Pharmacol. 2021, 890, 173705. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Yiu, C.P.B.; Wong, K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Owis, A.I.; El-Hawary, M.S.; El Amir, D.; Aly, O.M.; Abdelmohsen, U.R.; Kamel, M.S. Molecular docking reveals the potential ofSalvadora persicaflavonoids to inhibit COVID-19 virus main protease. RSC Adv. 2020, 10, 19570–19575. [Google Scholar] [CrossRef]
- Ahmed, O.A.A.; Badr-Eldin, S.M. Development of an optimized avanafil-loaded invasomal transdermal film: Ex vivo skin permeation and in vivo evaluation. Int. J. Pharm. 2019, 570, 118657. [Google Scholar] [CrossRef]
- Badr-Eldin, S.M.; Ahmed, O.A.A. Optimized nano-transfersomal films for enhanced sildenafil citrate transdermal delivery: Ex vivo and in vivo evaluation. Drug Des. Dev. Ther. 2016, 10, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Algandaby, M.M.; Breikaa, R.M.; Eid, B.G.; Neamatallah, T.A.; Abdel-Naim, A.B.; Ashour, O.M. Icariin protects against thioacetamide-induced liver fibrosis in rats: Implication of anti-angiogenic and anti-autophagic properties. Pharmacol. Rep. 2017, 69, 616–624. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Al-Saleem, M.S.M.; Ahmed, O.A.A.; Fahmy, U.A.; Alhakamy, N.A.; Eid, B.G.; Abdel-Naim, A.B.; Abdel-Mageed, W.M.; Alrasheed, M.M.; Shazly, G.A. Optimized conjugation of fluvastatin to hiv-1 tat displays enhanced pro-apoptotic activity in hepg2 cells. Int. J. Mol. Sci. 2020, 21, 4138. [Google Scholar] [CrossRef]
- Hosny, K.M.; Ahmed, O.A.A.; Fahmy, U.A.; Alkhalidi, H.M. Nanovesicular systems loaded with a recently approved second generation type-5 phospodiesterase inhibitor (avanafil): I. Plackett-Burman screening and characterization. J. Drug Deliv. Sci. Technol. 2018, 43, 154–159. [Google Scholar] [CrossRef]
- Fahmy, U.A.; Ahmed, O.A.A.; Alhakamy, N.A. Augmentation of Alendronate Cytotoxicity against Breast Cancer Cells by Complexation with Trans-activating Regulatory Protein. Int. J. Pharmacol. 2019, 15, 731–737. [Google Scholar] [CrossRef]
- Alhakamy, N.A.; Ahmed, O.A.A.; Aldawsari, H.M.; Alfaifi, M.Y.; Eid, B.G.; Abdel-Naim, A.B.; Fahmy, U.A. Encapsulation of Lovastatin in Zein Nanoparticles Exhibits Enhanced Apoptotic Activity in HepG2 Cells. Int. J. Mol. Sci. 2019, 20, 5788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memariani, H.; Memariani, M.; Moravvej, H.; Shahidi-Dadras, M. Melittin: A venom-derived peptide with promising anti-viral properties. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 5–17. [Google Scholar] [CrossRef]
- Akaji, K.; Konno, H.; Mitsui, H.; Teruya, K.; Shimamoto, Y.; Hattori, Y.; Ozaki, T.; Kusunoki, M.; Sanjoh, A. Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J. Med. Chem. 2011, 54, 7962–7973. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Levels in Coded Units | |
| (−1) | (+1) | |
| X1: SIT concentration (mM) | 1 | 10 |
| X2: MEL concentration (mM) | 1 | 10 |
| X3: pH | 6 | 10 |
| Responses | Desirability constraints | |
| Y1: particle size (nm) | Minimize | |
| Y2: zeta potential (mV) | Maximize | |
| Experimental Run | Independent Variables | PS ± SD | ZP ± SD | ||
|---|---|---|---|---|---|
| SIT Concentration (mM) | MEL Concentration (mM) | pH | |||
| F-1 | 10 | 10 | 6 | 432.11± 5.12 | 9.11 ± 0.32 |
| F-2 | 10 | 1 | 10 | 231.43 ± 3.21 | 31.21 ± 1.67 |
| F-3 | 10 | 10 | 10 | 387.19 ± 4.91 | 28.35 ± 1.44 |
| F-4 | 10 | 1 | 6 | 213.25 ± 2.98 | 6.27 ± 0.33 |
| F-5 | 1 | 1 | 6 | 121.31 ± 2.11 | 7.19 ± 0.25 |
| F-6 | 1 | 10 | 10 | 323.16 ± 4.99 | 32.25 ± 1.15 |
| F-7 | 1 | 10 | 6 | 345.29 ± 4.31 | 18.39 ± 0.77 |
| F-8 | 1 | 1 | 10 | 123.41 ± 2.11 | 26.42 ± 0.98 |
| Responses | Process Order | p-Value | R2 | Adjusted R2 | Predicted R2 | Adequate Precision | Significant Factors and Interactions |
|---|---|---|---|---|---|---|---|
| Y1: particle size (nm) | Main effects | 0.0004 | 0.9851 | 0.9738 | 0.9405 | 22.21 | X1, X2 |
| Y2: zeta potential (mV) | 2FI | 0.0152 | 0.9999 | 0.9995 | 0.9958 | 116.49 | X1, X2, X3, X1X2, X1X3, X2X3 |
| Ligand | S a (Kcal·moL-1) | Rescoring (Kcal·moL-1) | RMSD b (Å) | Ligand-Target Interaction Descriptive Data (Type; Length Å; Angle °; Binding Residues; Ligand’s Partner) |
|---|---|---|---|---|
| Sitagliptin | −5.699 | −5.819 | 1.456 | Hydrogen bond; 2.39 Å; 139.1°; Gly143 (NHCO) main chain with N1 triazole ring. Hydrogen bond; 2.37 Å; 124.6°; Ser144 (OH) side chain with N2 triazole ring Hydrogen bond; 2.31 Å; 158.1°; Gln189 (CONHH) side chain amide linker (-C=O)π-π stacking; 3.59 Å; His41 with trifluorophenyl ring |
| Melittin | −8.618 | −8.821 | 3.139 | Hydrogen bond; 2.3 Å; 121.8°; Thr24 (OH) side chain with C-terminal Melittin-Val8 (NHCO) main chain Hydrogen bond; 2.4 Å; 113.0°; Ser46 (NHCO) main chain with N-terminal Melittin-Arg3 (=NHH) side chain Hydrogen bond; 1.8 Å; 151.5°; Glu47 (COO) sidechain with C-terminal Melittin-Lys7 (NHH) side chain. Hydrogen bond; 2.2 Å; 127.8°; Lys61 (NH3+) side chain with C-terminal Melittin-Gly1 (COO) main chain Hydrogen bond; 2.2 Å; 177.2°; Asn142 (NHCO) side chain with N-terminal Melittin-Gln2 (NHH) side chain Hydrogen bond; 2.9 Å; 102.7°; Asn142 (NHCO) side chain with C-terminal Melittin-Thr10 (NHCO) main chain Hydrogen bond; 1.9 Å; 125.8°; Glu166 (NHCO) main chain with N-terminal Melittin-Lys4 (NHH) side chain |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Rabia, M.W.; Alhakamy, N.A.; Ahmed, O.A.A.; Eljaaly, K.; Alaofi, A.L.; Mostafa, A.; Asfour, H.Z.; Aldarmahi, A.A.; Darwish, K.M.; Ibrahim, T.S.; et al. Repurposing of Sitagliptin- Melittin Optimized Nanoformula against SARS-CoV-2; Antiviral Screening and Molecular Docking Studies. Pharmaceutics 2021, 13, 307. https://doi.org/10.3390/pharmaceutics13030307
Al-Rabia MW, Alhakamy NA, Ahmed OAA, Eljaaly K, Alaofi AL, Mostafa A, Asfour HZ, Aldarmahi AA, Darwish KM, Ibrahim TS, et al. Repurposing of Sitagliptin- Melittin Optimized Nanoformula against SARS-CoV-2; Antiviral Screening and Molecular Docking Studies. Pharmaceutics. 2021; 13(3):307. https://doi.org/10.3390/pharmaceutics13030307
Chicago/Turabian StyleAl-Rabia, Mohammed W., Nabil A. Alhakamy, Osama A. A. Ahmed, Khalid Eljaaly, Ahmed L. Alaofi, Ahmed Mostafa, Hani Z. Asfour, Ahmed A. Aldarmahi, Khaled M. Darwish, Tarek S. Ibrahim, and et al. 2021. "Repurposing of Sitagliptin- Melittin Optimized Nanoformula against SARS-CoV-2; Antiviral Screening and Molecular Docking Studies" Pharmaceutics 13, no. 3: 307. https://doi.org/10.3390/pharmaceutics13030307