
Modulation of Inflammatory Mediators by Polymeric Nanoparticles Loaded with Anti-Inflammatory Drugs

 , and
, and
Abstract
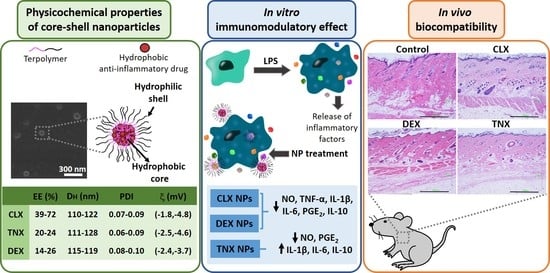
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
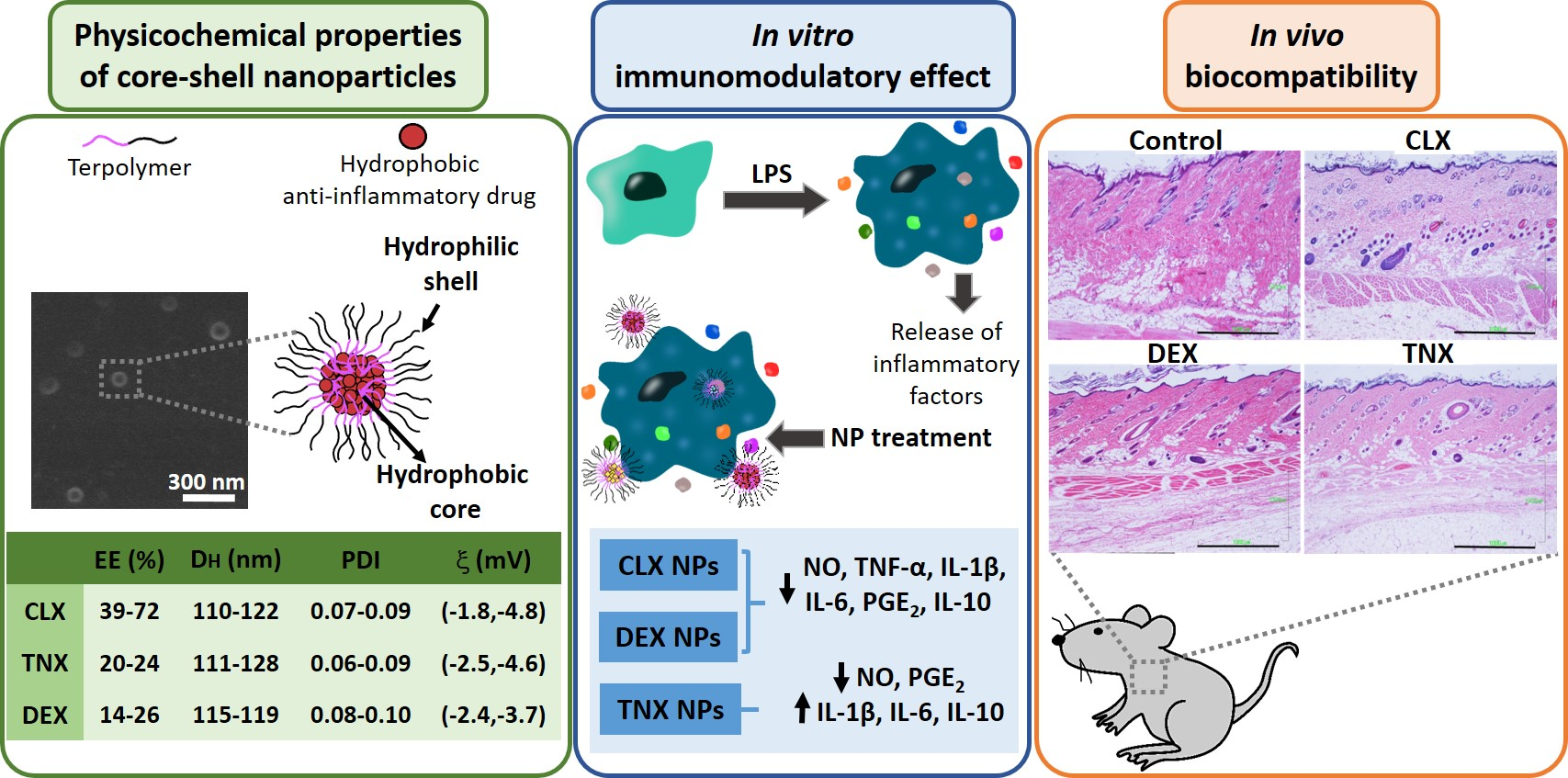
2.2. Preparation and Physicochemical Characterization of NPs
2.3. Cell Cultures and Biological Products
2.4. NP Cytotoxicity
2.5. Quantification of NO Cellular Release
2.6. Semi-Quantitative Inflammation Antibody Array
2.7. Quantification of TNF-α, IL-1β, IL-6, PGE2 and IL-10 Cellular Release
2.8. In Vivo Biocompatibility Evaluation
2.8.1. Animals
2.8.2. In Vivo Subcutaneous Injection of NPs
2.8.3. Histological Study
3. Results and Discussion
3.1. Preparation and Physicochemical Characterization of NPs
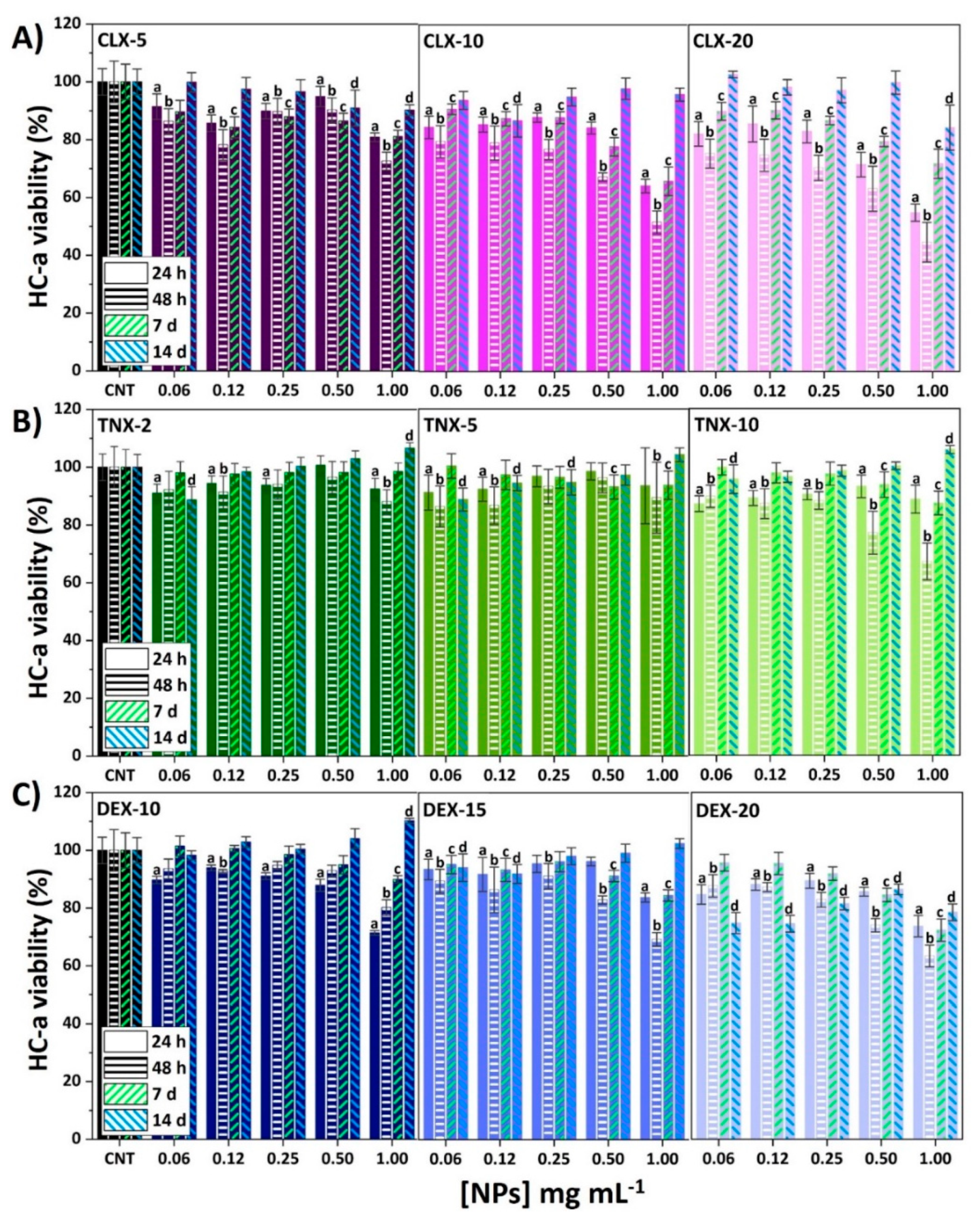
3.2. Effect on HC-a Viability
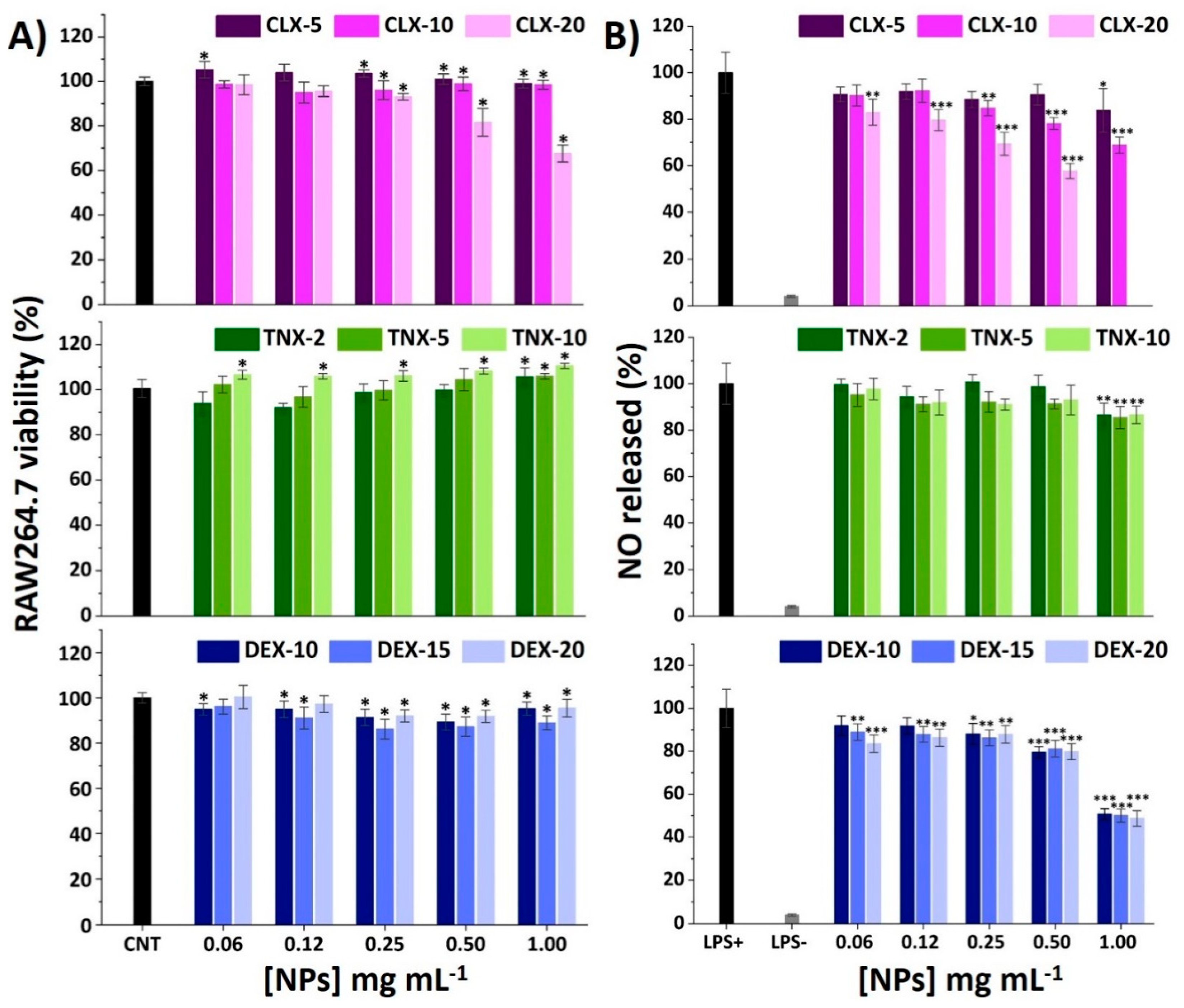
3.3. Effect on RAW264.7 Viability and NO Release
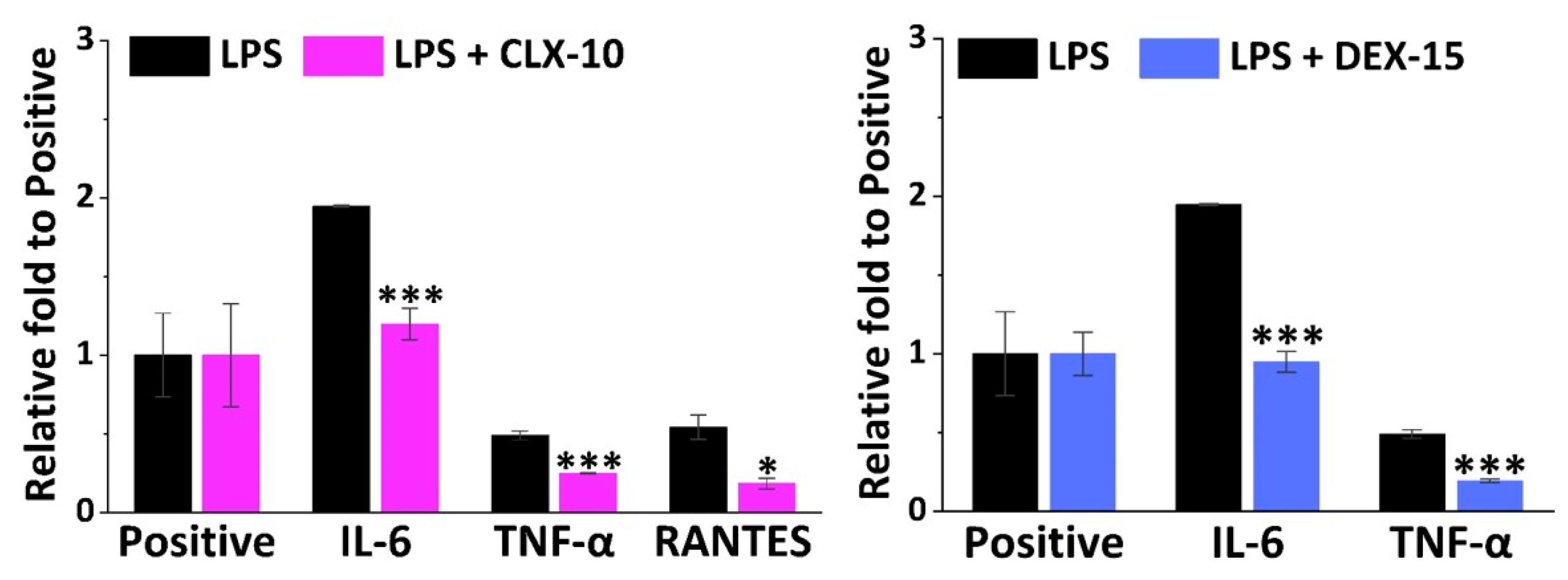
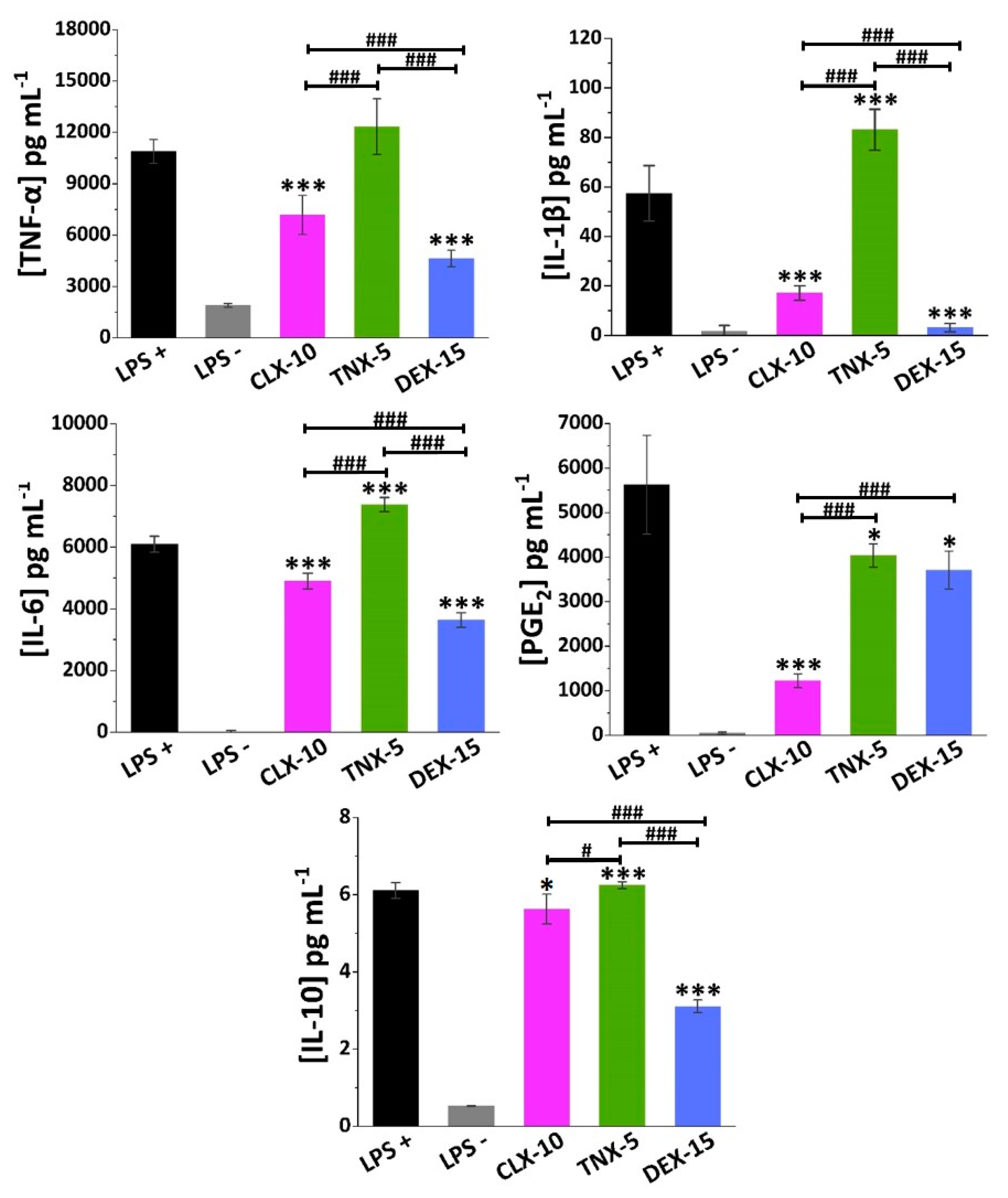
3.4. Effect on RAW264.7 Release of Inflammatory Factors
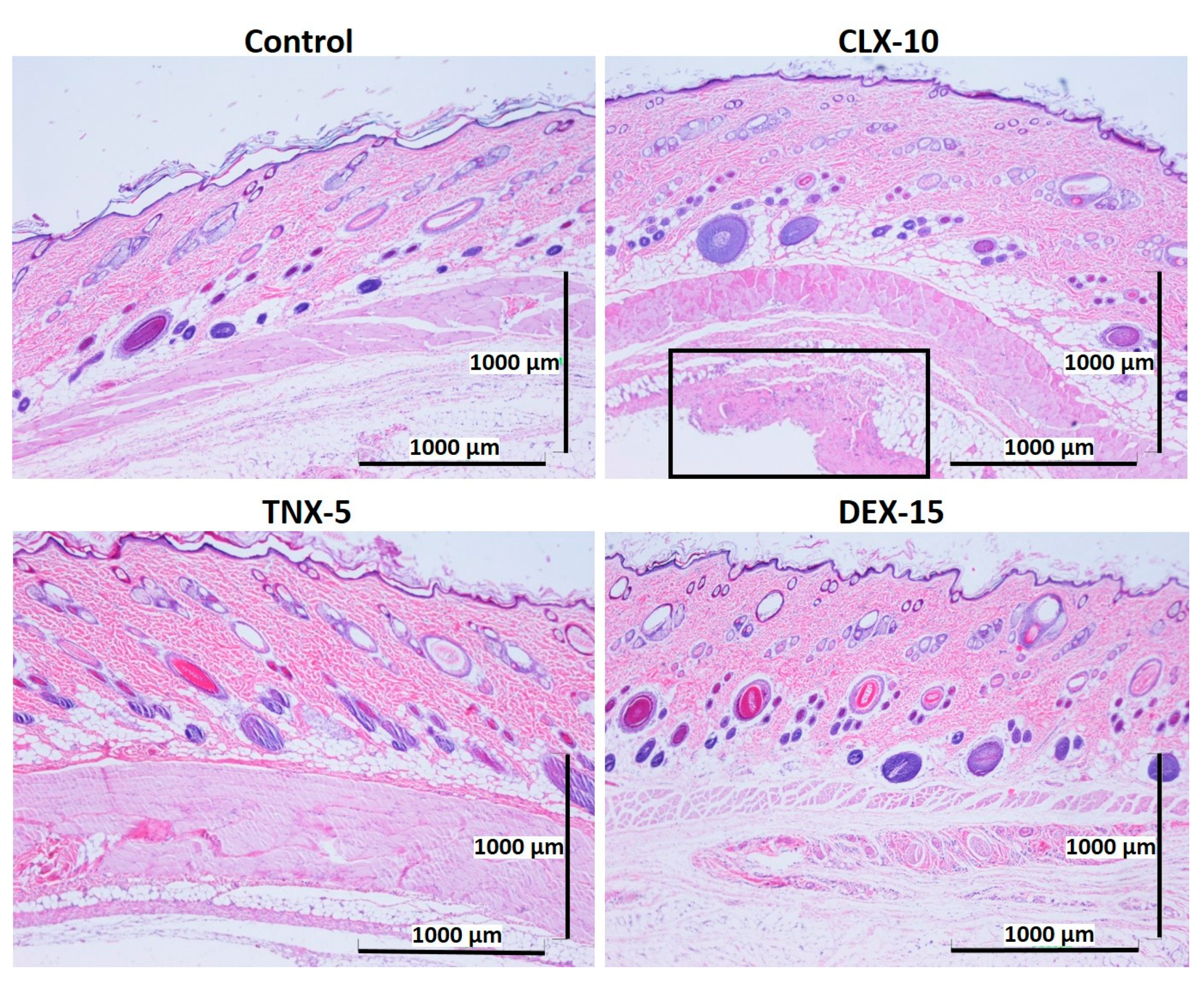
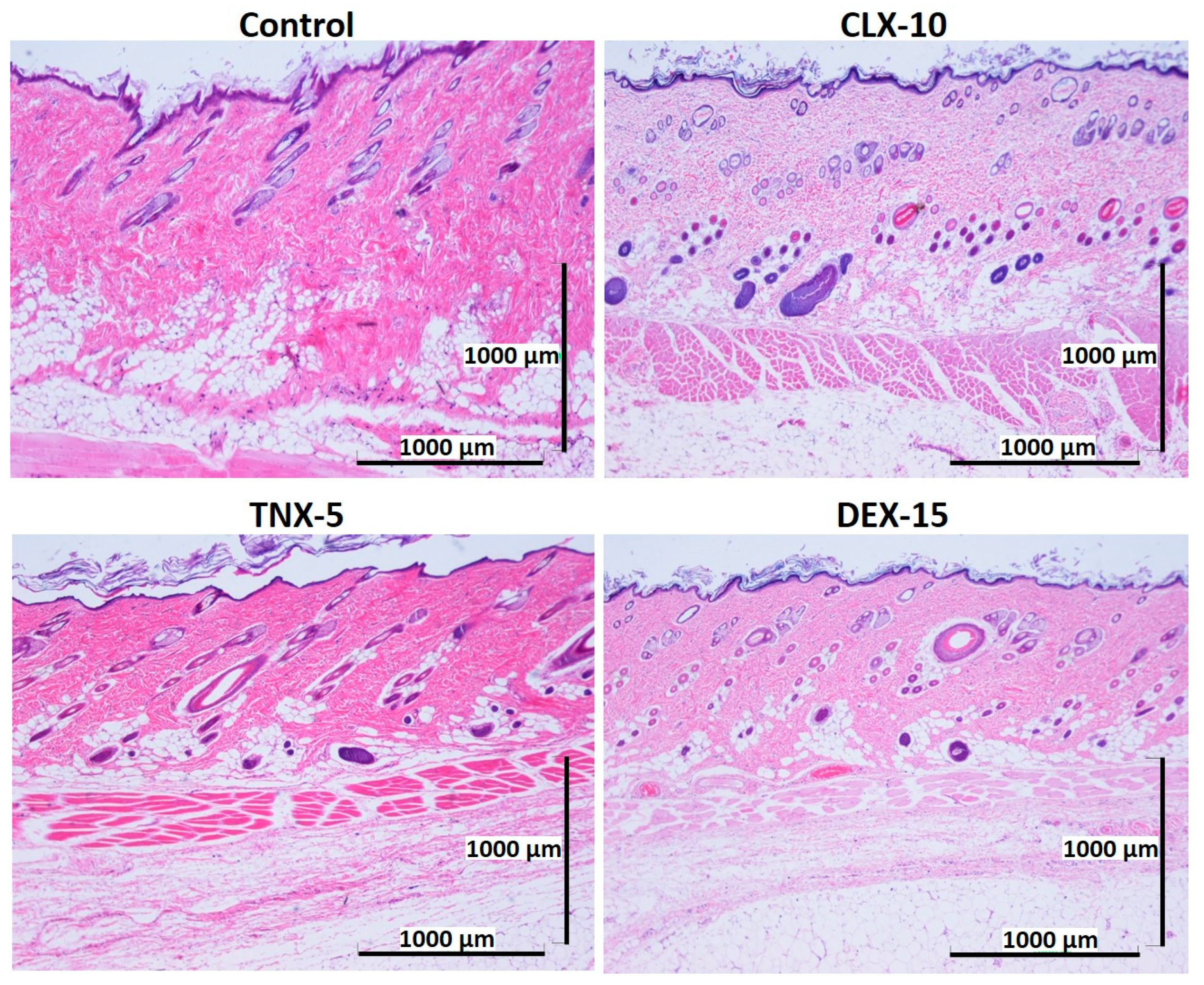
3.5. Histological Evaluation: In Vivo Biocompatibility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Berenbaum, F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthr. Cartil. 2013, 21, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef]
- Minguzzi, M.; Cetrullo, S.; D’Adamo, S.; Silvestri, Y.; Flamigni, F.; Borzi, R.M. Emerging Players at the Intersection of Chondrocyte Loss of Maturational Arrest, Oxidative Stress, Senescence and Low-Grade Inflammation in Osteoarthritis. Oxid. Med. Cell. Longev. 2018, 2018, 3075293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Cai, D.; Bai, X. Macrophages regulate the progression of osteoarthritis. Osteoarthr. Cartil. 2020, 28, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, Y.T.; Sillat, T.; Barreto, G.; Ainola, M.; Nordstrom, D.C. Osteoarthritis as an autoinflammatory disease caused by chondrocyte-mediated inflammatory responses. Arthritis Rheum. 2012, 64, 613–616. [Google Scholar] [CrossRef]
- Wu, C.L.; Harasymowicz, N.S.; Klimak, M.A.; Collins, K.H.; Guilak, F. The role of macrophages in osteoarthritis and cartilage repair. Osteoarthr. Cartil. 2020, 28, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chanalaris, A.; Troeberg, L. ADAMTS and ADAM metalloproteinases in osteoarthritis—Looking beyond the ‘usual suspects’. Osteoarthr. Cartil. 2017, 25, 1000–1009. [Google Scholar] [CrossRef] [Green Version]
- Mabey, T.; Honsawek, S. Cytokines as biochemical markers for knee osteoarthritis. World J. Orthop. 2015, 6, 95–105. [Google Scholar] [CrossRef]
- Pontes-Quero, G.M.; García-Fernández, L.; Aguilar, M.R.; Román, J.S.; Cano, J.P.; Vázquez-Lasa, B. Active viscosupplements for osteoarthritis treatment. Semin. Arthritis Rheum. 2019, 49, 171–183. [Google Scholar] [CrossRef]
- Tomić, M.; Micov, A.; Pecikoza, U.; Stepanović-Petrović, R. Clinical Uses of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) and Potential Benefits of NSAIDs Modified-Release Preparations. In Microsized and Nanosized Carriers for Nonsteroidal Anti-Inflammatory Drugs; Academic Press: New York, NY, USA, 2017; pp. 1–29. [Google Scholar]
- Niederberger, E.; Tegeder, I. NSAIDs, COX-Independent Actions. In Encyclopedia of Pain; Schmidt, R.F., Willis, W.D., Eds.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2007; pp. 1470–1473. [Google Scholar]
- Grzanka, A.; Misiołek, M.; Golusiński, W.; Jarząb, J. Molecular Mechanisms of Glucocorticoids Action: Implications for Treatment of Rhinosinusitis and Nasal Polyposis. Eur. Arch. Oto-Rhino-Laryngol. 2011, 268, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Deeks, J.J.; Smith, L.A.; Bradley, M.D. Efficacy, tolerability, and upper gastrointestinal safety of celecoxib for treatment of osteoarthritis and rheumatoid arthritis: Systematic review of randomised controlled trials. BMJ 2002, 325, 619. [Google Scholar] [CrossRef] [Green Version]
- Todd, P.A.; Clissold, S.P. Tenoxicam. An update of its pharmacology and therapeutic efficacy in rheumatic diseases. Drugs 1991, 41, 625–646. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Du, H.; Zhai, G. Progress in intra-articular drug delivery systems for osteoarthritis. Curr. Drug Targets 2014, 15, 888–900. [Google Scholar] [CrossRef] [PubMed]
- García-Couce, J.; Almirall, A.; Fuentes, G.; Kaijzel, E.; Chan, A.; Cruz, L.J. Targeting Polymeric Nanobiomaterials as a Platform for Cartilage Tissue Engineering. Curr. Pharm. Des. 2019, 25, 1915–1932. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Joseph, R.M.; Hunter, A.L.; Ray, D.W.; Dixon, W.G. Systemic glucocorticoid therapy and adrenal insufficiency in adults: A systematic review. Semin. Arthritis Rheum. 2016, 46, 133–141. [Google Scholar] [CrossRef]
- Ni, R.; Song, G.; Fu, X.; Song, R.; Li, L.; Pu, W.; Gao, J.; Hu, J.; Liu, Q.; He, F.; et al. Reactive oxygen species-responsive dexamethasone-loaded nanoparticles for targeted treatment of rheumatoid arthritis via suppressing the iRhom2/TNF-α/BAFF signaling pathway. Biomaterials 2020, 232, 119730. [Google Scholar] [CrossRef]
- Yu, C.; Li, X.; Hou, Y.; Meng, X.; Wang, D.; Liu, J.; Sun, F.; Li, Y. Hyaluronic Acid Coated Acid-Sensitive Nanoparticles for Targeted Therapy of Adjuvant-Induced Arthritis in Rats. Molecules 2019, 24, 146. [Google Scholar] [CrossRef] [Green Version]
- Lorscheider, M.; Tsapis, N.; Ur-Rehman, M.; Gaudin, F.; Stolfa, I.; Abreu, S.; Mura, S.; Chaminade, P.; Espeli, M.; Fattal, E. Dexamethasone palmitate nanoparticles: An efficient treatment for rheumatoid arthritis. J. Control. Release 2019, 296, 179–189. [Google Scholar] [CrossRef]
- Espinosa-Cano, E.; Aguilar, M.R.; Portilla, Y.; Barber, D.F.; Roman, J.S. Anti-Inflammatory Polymeric Nanoparticles Based on Ketoprofen and Dexamethasone. Pharmaceutics 2020, 12, 723. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, C.; Meng, X.; Hou, Y.; Cui, Y.; Zhu, T.; Li, Y.; Teng, L.; Sun, F.; Li, Y. Study of double-targeting nanoparticles loaded with MCL-1 siRNA and dexamethasone for adjuvant-induced arthritis therapy. Eur. J. Pharm. Biopharm. 2020, 154, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Bajpayee, A.G.; Quadir, M.A.; Hammond, P.T.; Grodzinsky, A.J. Charge based intra-cartilage delivery of single dose dexamethasone using Avidin nano-carriers suppresses cytokine-induced catabolism long term. Osteoarthr. Cartil. 2016, 24, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Ha, E.S.; Choo, G.H.; Baek, I.H.; Kim, M.S. Formulation, characterization, and in vivo evaluation of celecoxib-PVP solid dispersion nanoparticles using supercritical antisolvent process. Molecules 2014, 19, 20325–20339. [Google Scholar] [CrossRef] [Green Version]
- El-Gogary, R.I.; Khattab, M.A.; Abd-Allah, H. Intra-articular multifunctional celecoxib loaded hyaluronan nanocapsules for the suppression of inflammation in an osteoarthritic rat model. Int. J. Pharm. 2020, 583, 119378. [Google Scholar] [CrossRef]
- Crivelli, B.; Bari, E.; Perteghella, S.; Catenacci, L.; Sorrenti, M.; Mocchi, M.; Farago, S.; Tripodo, G.; Prina-Mello, A.; Torre, M.L. Silk fibroin nanoparticles for celecoxib and curcumin delivery: ROS-scavenging and anti-inflammatory activities in an in vitro model of osteoarthritis. Eur. J. Pharm. Biopharm. 2019, 137, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Khattab, A.; Abouhussein, D.M.N.; Mohammad, F.E. Development of injectable tenoxicam in situ forming microparticles based on sesame oil and poly-DL-lactide: Characterization, efficacy and acute toxicity. J. Drug Deliv. Sci. Technol. 2019, 51, 682–694. [Google Scholar] [CrossRef]
- Goindi, S.; Narula, M.; Kalra, A. Microemulsion-Based Topical Hydrogels of Tenoxicam for Treatment of Arthritis. AAPS PharmSciTech 2016, 17, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Ammar, H.O.; Ghorab, M.; El-Nahhas, S.A.; Higazy, I.M. Proniosomes as a carrier system for transdermal delivery of tenoxicam. Int. J. Pharm. 2011, 405, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Negi, L.M.; Chauhan, M.; Garg, A.K. Nano-appended transdermal gel of Tenoxicam via ultradeformable drug carrier system. J. Exp. Nanosci. 2013, 8, 657–669. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, C.; Palao-Suay, R.; Rodrigáñez, L.; Aguilar, M.R.; Martín-Saldaña, S.; Román, J.S.; Sanz-Fernández, R. α-Tocopheryl Succinate-Based Polymeric Nanoparticles for the Treatment of Head and Neck Squamous Cell Carcinoma. Biomolecules 2018, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Palao-Suay, R.; Aguilar, M.R.; Parra-Ruiz, F.J.; Maji, S.; Hoogenboom, R.; Rohner, N.A.; Thomas, S.N.; Román, J.S. α-TOS-based RAFT block copolymers and their NPs for the treatment of cancer. Polym. Chem. 2016, 7, 838–850. [Google Scholar] [CrossRef] [Green Version]
- Palao-Suay, R.; Martín-Saavedra, F.M.; Aguilar, M.R.; Escudero-Duch, C.; Martín-Saldaña, S.; Parra-Ruiz, F.J.; Rohner, N.A.; Thomas, S.N.; Vilaboa, N.; Román, J.S. Photothermal and photodynamic activity of polymeric nanoparticles based on α-tocopheryl succinate-RAFT block copolymers conjugated to IR-780. Acta Biomater. 2017, 57, 70–84. [Google Scholar] [CrossRef]
- Palao-Suay, R.; Aguilar, M.R.; Parra-Ruiz, F.J.; Martín-Saldaña, S.; Rohner, N.A.; Thomas, S.N.; Román, J.S. Multifunctional decoration of alpha-tocopheryl succinate-based NP for cancer treatment: Effect of TPP and LTVSPWY peptide. J. Mater. Sci. Mater. Med. 2017, 28, 152. [Google Scholar] [CrossRef]
- Martin-Saldana, S.; Palao-Suay, R.; Trinidad, A.; Aguilar, M.R.; Ramirez-Camacho, R.; Roman, J.S. Otoprotective properties of 6alpha-methylprednisolone-loaded nanoparticles against cisplatin: In vitro and in vivo correlation. Nanomedicine 2016, 12, 965–976. [Google Scholar] [CrossRef] [Green Version]
- Martin-Saldana, S.; Palao-Suay, R.; Aguilar, M.R.; Garcia-Fernandez, L.; Arevalo, H.; Trinidad, A.; Ramirez-Camacho, R.; Roman, J.S. pH-sensitive polymeric nanoparticles with antioxidant and anti-inflammatory properties against cisplatin-induced hearing loss. J. Control. Release 2018, 270, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Martín-Saldaña, S.; Palao-Suay, R.; Aguilar, M.R.; Ramírez-Camacho, R.; Román, J.S. Polymeric nanoparticles loaded with dexamethasone or α-tocopheryl succinate to prevent cisplatin-induced ototoxicity. Acta Biomater. 2017, 53, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Pontes-Quero, G.; Benito-Garzón, L.; Cano, J.P.; Aguilar, M.; Vázquez-Lasa, B. Amphiphilic polymeric nanoparticles encapsulating curcumin: Antioxidant, antiinflammatory and biocompatibility studies. Mater. Sci. Eng. C 2021, 121, 111793. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 2662, Celecoxib” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Celecoxib (accessed on 22 February 2021).
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 54677971, Tenoxicam” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Tenoxicam (accessed on 22 February 2021).
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 5743, Dexamethasone” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Dexamethasone (accessed on 22 February 2021).
- Liu, Y.; Yang, G.; Baby, T.; Tengjisi; Chen, D.; Weitz, D.A.; Zhao, C.X. Stable Polymer Nanoparticles with Exceptionally High Drug Loading by Sequential Nanoprecipitation. Angew. Chem.-Int. Ed. 2020, 59, 4720–4728. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, G.; Zou, D.; Hui, Y.; Nigam, K.; Middelberg, A.P.J.; Zhao, C.-X. Formulation of Nanoparticles Using Mixing-Induced Nanoprecipitation for Drug Delivery. Ind. Eng. Chem. Res. 2020, 59, 4134–4149. [Google Scholar] [CrossRef]
- Macedo, A.; Carvalho, E.O.; Cardoso, V.; Correia, D.M.; Tubio, C.; Fidalgo-Marijuan, A.; Botelho, G.; Lanceros-Méndez, S. Tailoring electroactive poly(vinylidene fluoride-co-trifluoroethylene) microspheres by a nanoprecipitation method. Mater. Lett. 2019, 261, 127018. [Google Scholar] [CrossRef]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Murugan, K.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. Parameters and characteristics governing cellular internalization and trans-barrier trafficking of nanostructures. Int. J. Nanomed. 2015, 10, 2191–2206. [Google Scholar]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kröger, M.; Liu, W.K. Shape effect in cellular uptake of PEGylated nanoparticles: Comparison between sphere, rod, cube and disk. Nanoscale 2015, 7, 16631–16646. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Liu, H.; He, H.; Ribbe, A.E.; Thayumanavan, S. Blended Assemblies of Amphiphilic Random and Block Copolymers for Tunable Encapsulation and Release of Hydrophobic Guest Molecules. Macromolecules 2020, 53, 2713–2723. [Google Scholar] [CrossRef]
- Imai, S.; Hirai, Y.; Nagao, C.; Sawamoto, M.; Terashima, T. Programmed Self-Assembly Systems of Amphiphilic Random Copolymers into Size-Controlled and Thermoresponsive Micelles in Water. Macromolecules 2018, 51, 398–409. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 969516, Curcumin” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Curcumin (accessed on 4 December 2020).
- Zhang, T.; Liu, H.; Li, Y.; Li, C.; Wan, G.; Chen, B.; Li, C.; Wang, Y. A pH-sensitive nanotherapeutic system based on a marine sulfated polysaccharide for the treatment of metastatic breast cancer through combining chemotherapy and COX-2 inhibition. Acta Biomater. 2019, 99, 412–425. [Google Scholar] [CrossRef]
- Albisa, A.; Piacentini, E.; Sebastian, V.; Arruebo, M.; Santamaria, J.; Giorno, L. Preparation of Drug-Loaded PLGA-PEG Nanoparticles by Membrane-Assisted Nanoprecipitation. Pharm. Res. 2017, 34, 1296–1308. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, E.; Pisani, S.; Colzani, B.; Dorati, R.; Conti, B.; Modena, T.; Braekmans, K.; Genta, I. Intra-Articular Formulation of GE11-PLGA Conjugate-Based NPs for Dexamethasone Selective Targeting-In Vitro Evaluation. Int. J. Mol. Sci. 2018, 19, 2304. [Google Scholar] [CrossRef] [Green Version]
- Ummarino, A.; Gambaro, F.M.; Kon, E.; Andon, F.T. Therapeutic Manipulation of Macrophages Using Nanotechnological Approaches for the Treatment of Osteoarthritis. Nanomaterials 2020, 10, 1562. [Google Scholar] [CrossRef]
- Pavillon, N.; Hobro, A.J.; Akira, S.; Smith, N.I. Noninvasive detection of macrophage activation with single-cell resolution through machine learning. Proc. Natl. Acad. Sci. USA 2018, 115, E2676–E2685. [Google Scholar] [CrossRef] [Green Version]
- Canton, J.; Khezri, R.; Glogauer, M.; Grinstein, S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol. Biol. Cell 2014, 25, 3330–3341. [Google Scholar] [CrossRef]
- Jones, E.; Adcock, I.M.; Ahmed, B.Y.; Punchard, N.A. Modulation of LPS stimulated NF-kappaB mediated Nitric Oxide production by PKCepsilon and JAK2 in RAW macrophages. J. Inflamm. 2007, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.J.; Kim, Y.W.; Park, Y.; Lee, H.J.; Kim, K.W. Anti-inflammatory effects of chlorogenic acid in lipopolysaccharide-stimulated RAW 264.7 cells. Inflamm. Res. 2014, 63, 81–90. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, R.; Lei, L.; Song, Q.; Li, X. High drug payload nanoparticles formed from dexamethasone-peptide conjugates for the treatment of endotoxin-induced uveitis in rabbit. Int. J. Nanomed. 2019, 14, 591–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Naeem, M.; Noh, J.-K.; Lee, E.H.; Yoo, J.-W. Dexamethasone phosphate-loaded folate-conjugated polymeric nanoparticles for selective delivery to activated macrophages and suppression of inflammatory responses. Macromol. Res. 2015, 23, 485–492. [Google Scholar] [CrossRef]
- Wojdasiewicz, P.; Poniatowski, Ł.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosset, M.; Berenbaum, F.; Levy, A.; Pigenet, A.; Thirion, S.; Cavadias, S.; Jacques, C. Mechanical stress and prostaglandin E2 synthesis in cartilage. Biorheology 2008, 45, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Bhardwaj, A.; Potdar, P.; Aggarwal, B.B. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-κB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene 2004, 23, 9247–9258. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiong, H.S.; Yong, Y.K.; Ahmad, Z.; Sulaiman, M.R.; Zakaria, Z.A.; Yuen, K.H.; Hakim, M.N. Cytoprotective and enhanced anti-inflammatory activities of liposomal piroxicam formulation in lipopolysaccharide-stimulated RAW 264.7 macrophages. Int. J. Nanomed. 2013, 8, 1245–1255. [Google Scholar]
- Nakata, K.; Hanai, T.; Take, Y.; Osada, T.; Tsuchiya, T.; Shima, D.; Fujimoto, Y. Disease-modifying effects of COX-2 selective inhibitors and non-selective NSAIDs in osteoarthritis: A systematic review. Osteoarthr. Cartil. 2018, 26, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Gallelli, L.; Galasso, O.; Falcone, D.; Southworth, S.; Greco, M.; Ventura, V.; Romualdi, P.; Corigliano, A.; Terracciano, R.; Savino, R.; et al. The effects of nonsteroidal anti-inflammatory drugs on clinical outcomes, synovial fluid cytokine concentration and signal transduction pathways in knee osteoarthritis. A randomized open label trial. Osteoarthr. Cartil. 2013, 21, 1400–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweers, M.C.; de Boer, T.N.; van Roon, J.; Bijlsma, J.W.J.; Lafeber, F.P.J.G.; Mastbergen, S.C. Celecoxib: Considerations regarding its potential disease-modifying properties in osteoarthritis. Arthritis Res. Ther. 2011, 13, 239. [Google Scholar] [CrossRef] [Green Version]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.M. Biological Responses to Materials. Annu. Rev. Mater. Res. 2001, 31, 81–110. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Celecoxib | Tenoxicam | Dexamethasone | |
|---|---|---|---|
| Type | Coxib, selective cyclooxygenase 2 (COX-2) Inhibitor | Oxicam, class of nonsteroidal anti-inflammatory drugs (NSAID) | Glucocorticoid, class of corticoid |
| Chemical structure |  |  |  |
| Main mechanism of action | Selective inhibition of COX-2 | Inhibition of both COX-1 and COX-2 | Binding to cellular glucocorticoid receptors, inducing or repressing the transcription of multiple genes |
| Solubility in water (mg L−1) | 4.3 [41] | 14.1 [42] | 89.0 [43] |
| LogP (partition coefficient) | 3.53 [41] | 1.9 [42] | 1.83 [43] |
| Most common administration route | Oral | Intra-articular injection, oral | Intra-articular injection, oral |
| Current used in OA | Inflammation and pain relief in mild or moderate OA | Post-operative pain relief | Inflammation and pain relief in mild or moderate OA |
| NP Code | [Drug] | EE(%) | [Encapsulated Drug] | ||
|---|---|---|---|---|---|
| % w/w | mg mL−1 | % w/w | mg mL−1 | ||
| CLX-5 | 5 | 0.10 | 72 ± 8 | 3.60 ± 0.29 | 0.072 ± 0.006 |
| CLX-10 | 10 | 0.20 | 50 ± 7 | 5.00 ± 0.35 | 0.100 ± 0.007 |
| CLX-20 | 20 | 0.40 | 39 ± 6 | 7.80 ± 0.47 | 0.156 ± 0.009 |
| TNX-2 | 2 | 0.04 | 22 ± 5 | 0.44 ± 0.03 | 0.009 ± 0.001 |
| TNX-5 | 5 | 0.10 | 20 ± 4 | 1.00 ± 0.07 | 0.020 ± 0.001 |
| TNX-10 | 10 | 0.20 | 24 ± 5 | 2.50 ± 0.18 | 0.050 ± 0.004 |
| DEX-10 | 10 | 0.20 | 26 ± 6 | 6.76 ± 0.34 | 0.052 ± 0.003 |
| DEX-15 | 15 | 0.30 | 16 ± 7 | 0.80 ± 0.10 | 0.048 ± 0.002 |
| DEX-20 | 20 | 0.40 | 14 ± 7 | 0.70 ± 0.10 | 0.056 ± 0.003 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pontes-Quero, G.M.; Benito-Garzón, L.; Pérez Cano, J.; Aguilar, M.R.; Vázquez-Lasa, B. Modulation of Inflammatory Mediators by Polymeric Nanoparticles Loaded with Anti-Inflammatory Drugs. Pharmaceutics 2021, 13, 290. https://doi.org/10.3390/pharmaceutics13020290
Pontes-Quero GM, Benito-Garzón L, Pérez Cano J, Aguilar MR, Vázquez-Lasa B. Modulation of Inflammatory Mediators by Polymeric Nanoparticles Loaded with Anti-Inflammatory Drugs. Pharmaceutics. 2021; 13(2):290. https://doi.org/10.3390/pharmaceutics13020290
Chicago/Turabian StylePontes-Quero, Gloria María, Lorena Benito-Garzón, Juan Pérez Cano, María Rosa Aguilar, and Blanca Vázquez-Lasa. 2021. "Modulation of Inflammatory Mediators by Polymeric Nanoparticles Loaded with Anti-Inflammatory Drugs" Pharmaceutics 13, no. 2: 290. https://doi.org/10.3390/pharmaceutics13020290