
Nanotherapeutics for Nose-to-Brain Drug Delivery: An Approach to Bypass the Blood Brain Barrier



Abstract
:1. Introduction
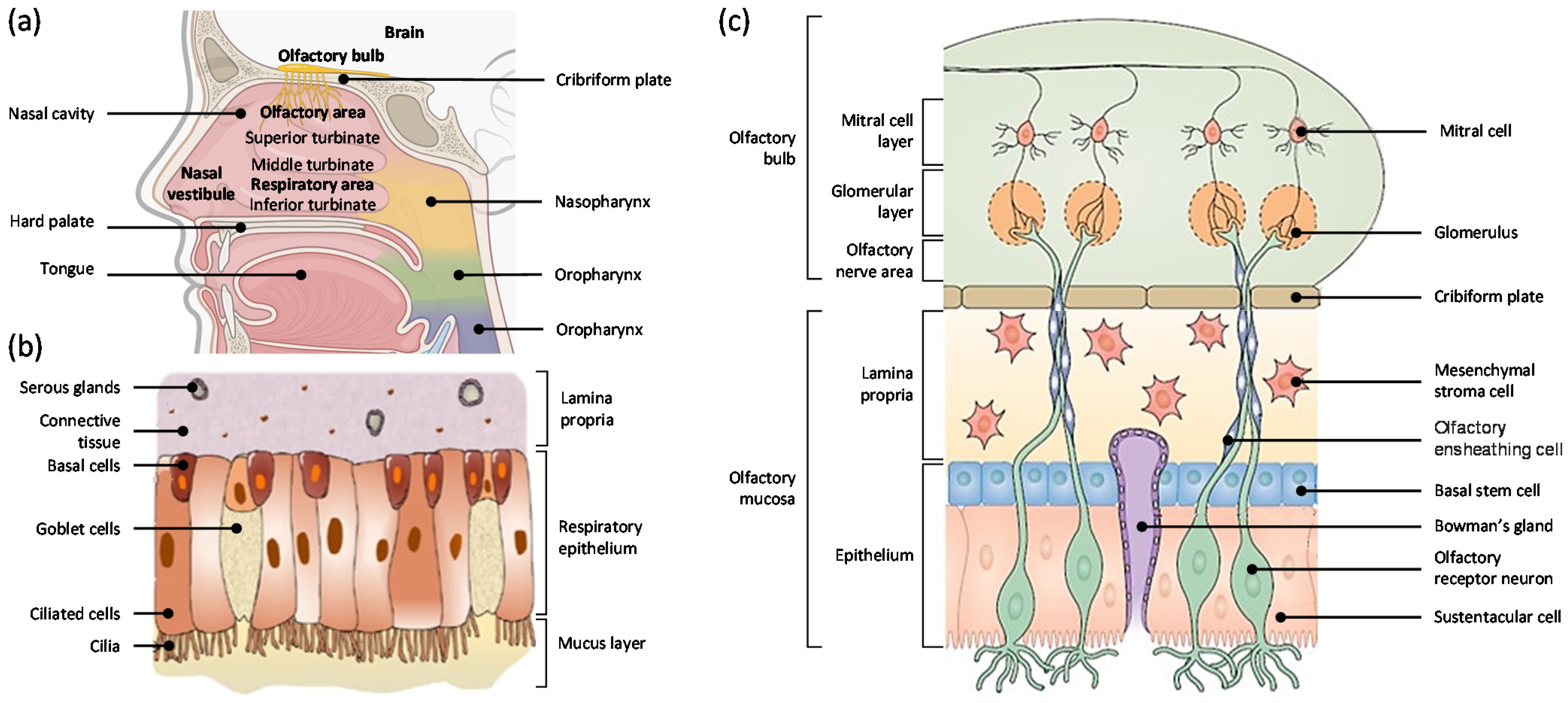
2. Nasal Cavity
2.1. Anatomy of the Nasal Cavity
2.2. Respiratory Mucosa
2.3. Olfactory Mucosa
3. Pathways for Nose-To-Brain Delivery
3.1. Olfactory Pathway
3.2. Trigeminal Pathway
3.3. Systemic Pathway
4. Potential Challenges for Nose-To-Brain Delivery
4.1. Cilia and Nasal Mucus Transport
4.2. Mucociliary Clearance
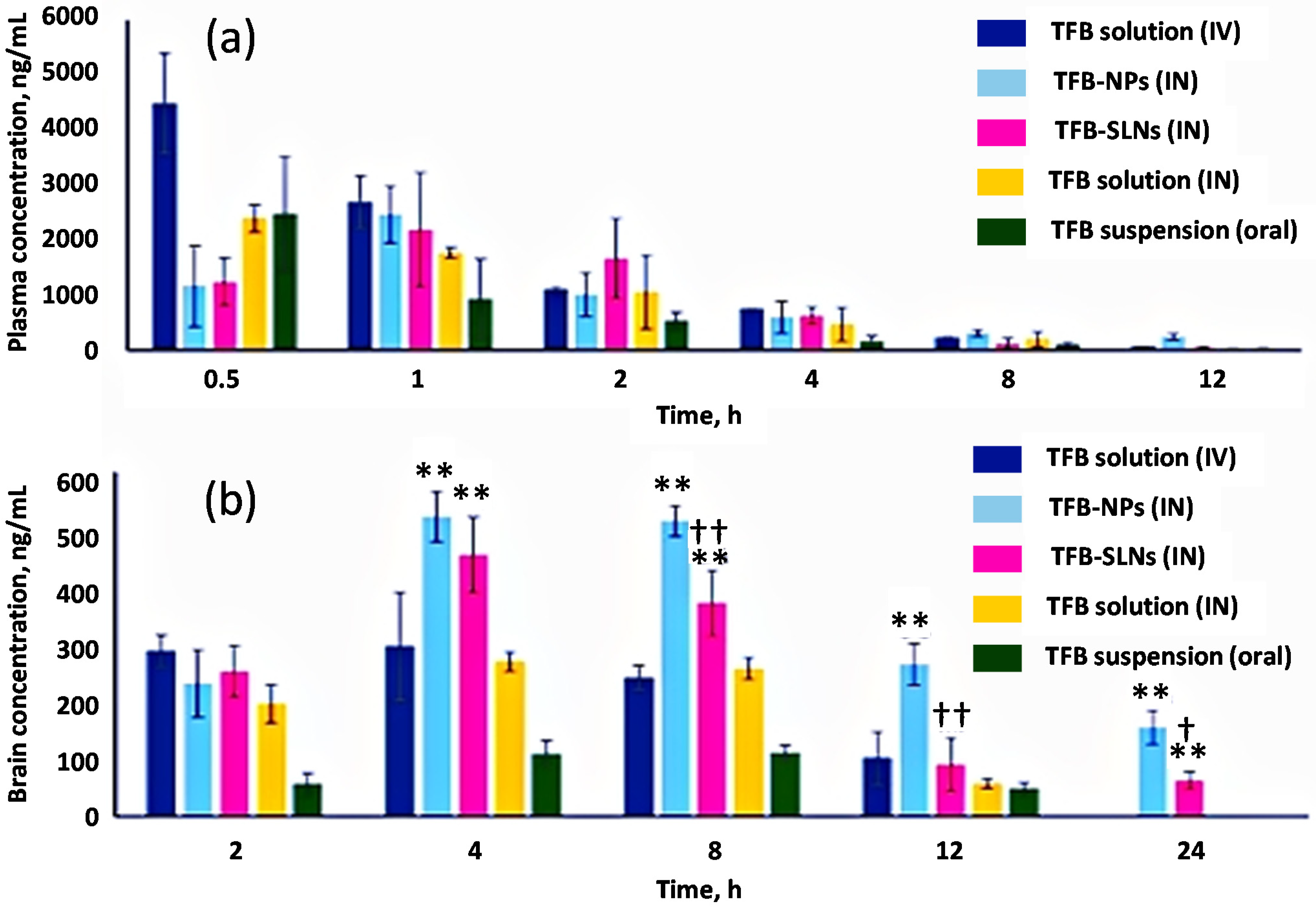
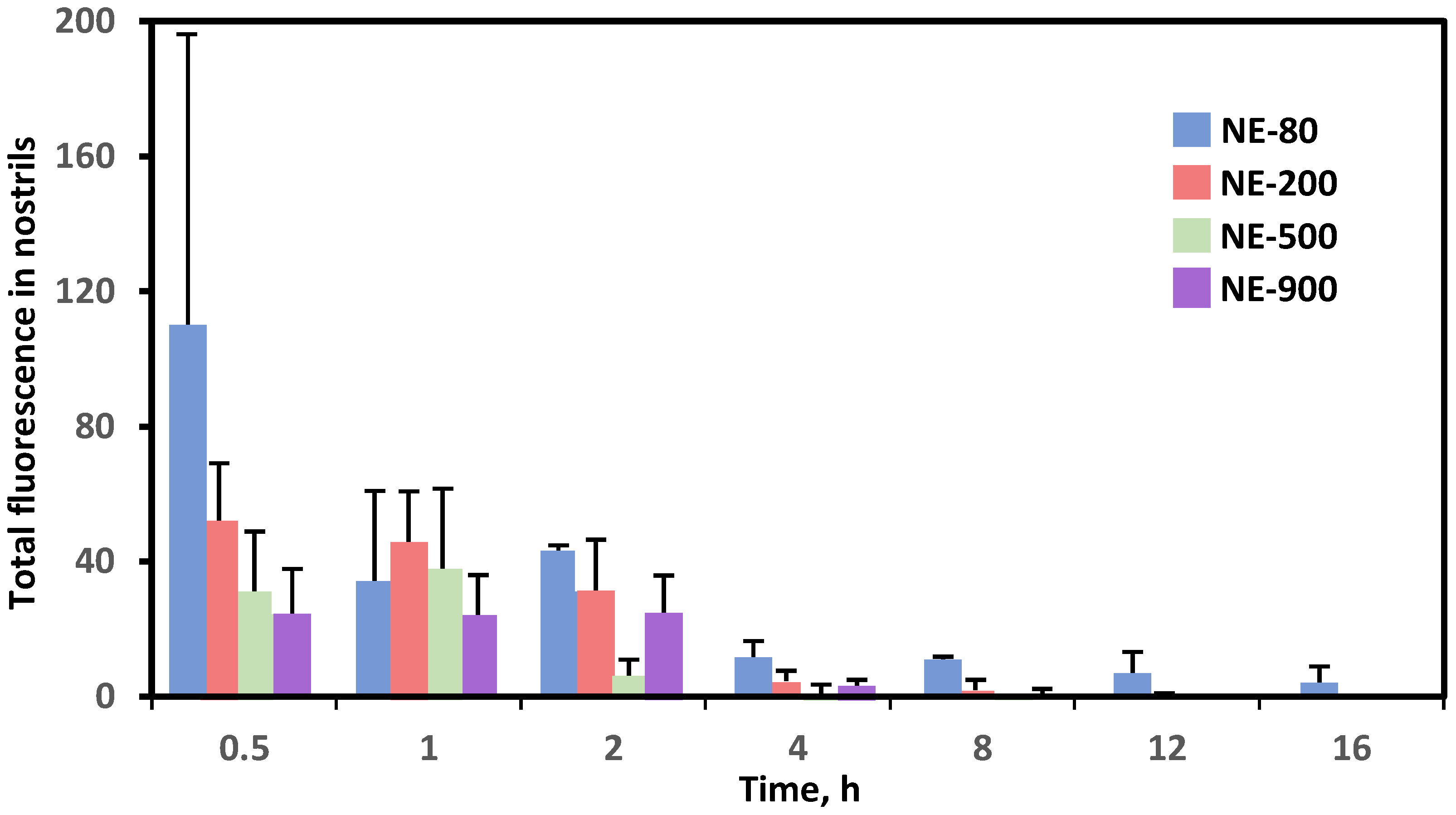
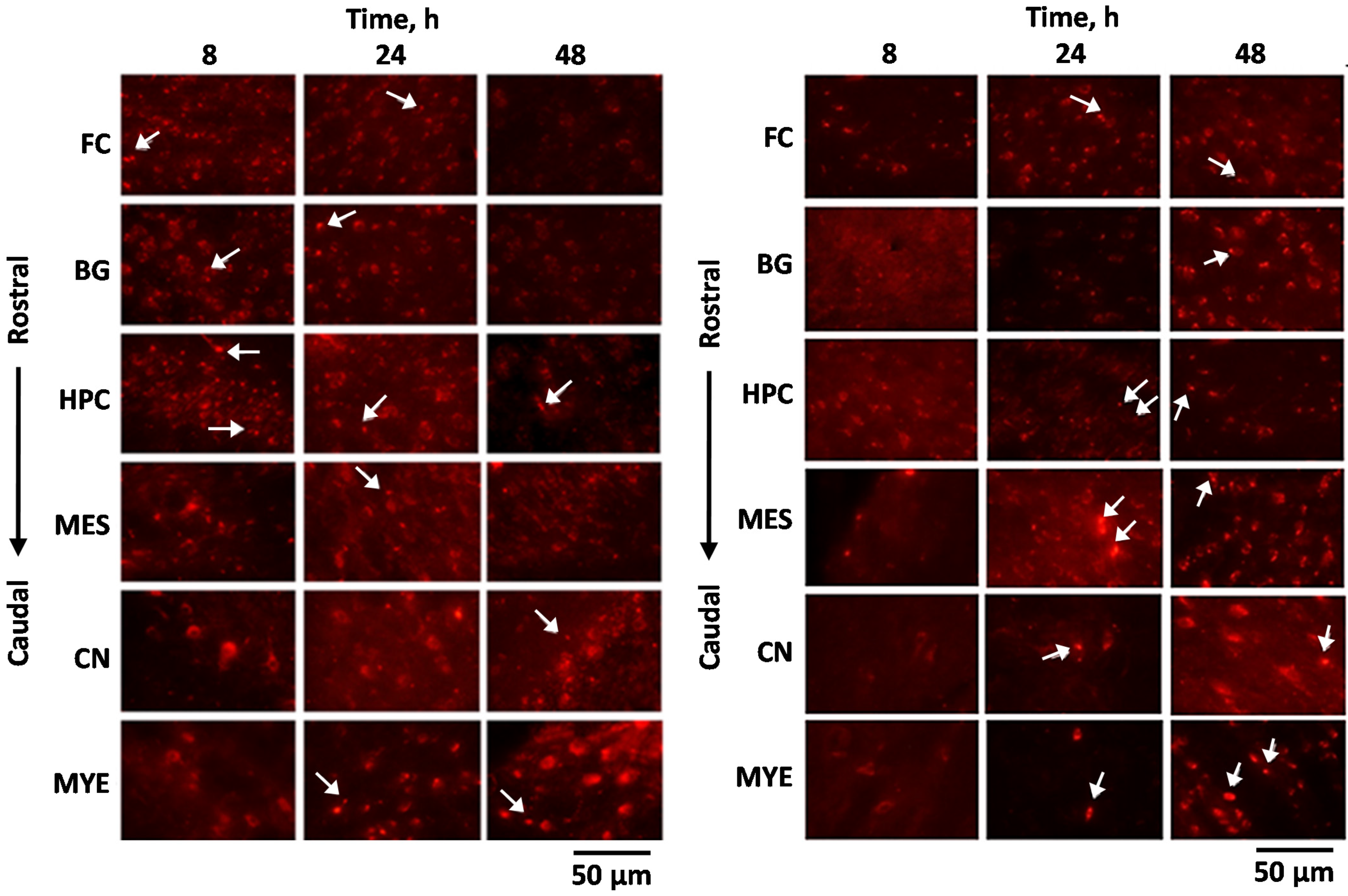
5. Pharmacokinetics of Nose-To-Brain Delivery
6. The Potential Role of Nanotechnology for Nose-To-Brain Delivery
6.1. Lipid-Based Nanoparticles
6.1.1. Liposomes
6.1.2. Solid Lipid Nanoparticles
6.1.3. Nanostructured Lipid Carriers
6.1.4. Nanoemulsions
6.2. Polymer-Based Nanoparticles
6.2.1. Natural Polymer-Based Nanoparticles
6.2.2. Synthetic Polymer-Based Nanoparticles
7. Physicochemical Properties That Can Affect Nose-To-Brain Delivery
7.1. Particle Size
7.2. Surface Charge
7.3. Lipophilicity
8. Therapeutic Applications of Nose-To-Brain Delivery
8.1. Epilepsy
8.2. Alzheimer’s Disease
8.3. Parkinson’s Disease
8.4. Stroke
8.5. Schizophrenia
8.6. Depression
8.7. Other CNS/Neurological Disorders
9. Challenges and Potential Future Directions
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexander, A.; Agrawal, M.; Uddin, A.; Siddique, S.; Shehata, A.M.; Shaker, M.A.; Ata Ur Rahman, S.; Abdul, M.I.M.; Shaker, M.A. Recent expansions of novel strategies towards the drug targeting into the brain. Int. J. Nanomed. 2019, 14, 5895–5909. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B.; Sorokin, L. The blood-brain and the blood-cerebrospinal fluid barriers: Function and dysfunction. Semin. Immunopathol. 2009, 31, 497–511. [Google Scholar] [CrossRef] [Green Version]
- Koutsari, C.; Dilworth, T.J.; Holt, J.; Elshaboury, R.; Rotschafer, J.C. Central Nervous System Infections. In Pharmacotherapy: A Pathopysiologic Approach, 11e; DiPiro, J.T., Yee, G.C., Posey, L., Haines, S.T., Nolin, T.D., Ellingrod, V., Eds.; McGraw-Hill Education: New York, NY, USA, 2020. [Google Scholar]
- GBD 2019 Collaborators. Global mortality from dementia: Application of a new method and results from the Global Burden of Disease Study 2019. Alzheimers Dement. 2021, 7, e12200. [Google Scholar] [CrossRef]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Rickards, H. Depression in neurological disorders: Parkinson’s disease, multiple sclerosis, and stroke. J. Neurol. Neurosurg. Psychiatry 2005, 76, i48–i52. [Google Scholar] [CrossRef] [Green Version]
- Simonato, M.; Bennett, J.; Boulis, N.M.; Castro, M.G.; Fink, D.J.; Goins, W.F.; Gray, S.J.; Lowenstein, P.R.; Vandenberghe, L.H.; Wilson, T.J.; et al. Progress in gene therapy for neurological disorders. Nat. Rev. Neurol. 2013, 9, 277–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasi, P.; Giovagnoli, S.; Schoubben, A.; Ricci, M.; Rossi, C. Solid lipid nanoparticles for targeted brain drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 454–477. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.S. Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol. Sci. 2010, 31, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Potschka, H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2005, 2, 86–98. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Terasaki, T. Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm. Res. 2007, 24, 1745–1758. [Google Scholar] [CrossRef] [PubMed]
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barar, J.; Rafi, M.A.; Pourseif, M.M.; Omidi, Y. Blood-brain barrier transport machineries and targeted therapy of brain diseases. Bioimpacts 2016, 6, 225–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, R.A.; De Felice, F.G. The crosstalk between brain and periphery: Implications for brain health and disease. Neuropharmacology 2021, 197, 108728. [Google Scholar] [CrossRef]
- Cook, A.M.; Mieure, K.D.; Owen, R.D.; Pesaturo, A.B.; Hatton, J. Intracerebroventricular administration of drugs. Pharmacotherapy 2009, 29, 832–845. [Google Scholar] [CrossRef]
- Groiss, S.J.; Wojtecki, L.; Südmeyer, M.; Schnitzler, A. Deep brain stimulation in Parkinson’s disease. Ther. Adv. Neurol. Disord. 2009, 2, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, R.; Iwata, H.; Sakata, Y.; Muramoto, K.; Matsuoka, T. Safety of Gliadel Implant for Malignant Glioma: Report of Postmarketing Surveillance in Japan. Neurol. Med. Chir. 2021, 61, 536–548. [Google Scholar] [CrossRef]
- Shibahara, I.; Miyasaka, K.; Sekiguchi, A.; Ishiyama, H.; Inukai, M.; Yasui, Y.; Watanabe, T.; Sato, S.; Hide, T.; Kumabe, T. Long-term follow-up after BCNU wafer implantation in patients with newly diagnosed glioblastoma. J. Clin. Neurosci. 2021, 86, 202–210. [Google Scholar] [CrossRef]
- Pires, A.; Fortuna, A.; Alves, G.; Falcão, A. Intranasal drug delivery: How, why and what for? J. Pharm. Pharm. Sci. 2009, 12, 288–311. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Shang, Y.; Chen, R.; Bai, R.; Chen, C.; Inthavong, K.; Tu, J. Correlation of regional deposition dosage for inhaled nanoparticles in human and rat olfactory. Part. Fibre Toxicol. 2019, 16, 6. [Google Scholar] [CrossRef]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H., II. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Karakosta, P.; Alexopoulos, A.H.; Kiparissides, C. Computational model of particle deposition in the nasal cavity under steady and dynamic flow. Comput Methods Biomech Biomed. Engin 2015, 18, 514–526. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Cloyd, J.C.; Siegel, R.A. A review of intranasal formulations for the treatment of seizure emergencies. J. Control. Release 2016, 237, 147–159. [Google Scholar] [CrossRef]
- Bourganis, V.; Kammona, O.; Alexopoulos, A.; Kiparissides, C. Recent advances in carrier mediated nose-to-brain delivery of pharmaceutics. Eur. J. Pharm. Biopharm. 2018, 128, 337–362. [Google Scholar] [CrossRef] [PubMed]
- Gänger, S.; Schindowski, K. Tailoring Formulations for Intranasal Nose-to-Brain Delivery: A Review on Architecture, Physico-Chemical Characteristics and Mucociliary Clearance of the Nasal Olfactory Mucosa. Pharmaceutics 2018, 10, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Byun, S.W.; Lee, S.S. Various tumors in the nasal vestibule. Int. J. Clin. Exp. Pathol. 2013, 6, 2713–2718. [Google Scholar]
- Illum, L. Transport of drugs from the nasal cavity to the central nervous system. Eur. J. Pharm. Sci. 2000, 11, 1–18. [Google Scholar] [CrossRef]
- Chatterjee, B.; Gorain, B.; Mohananaidu, K.; Sengupta, P.; Mandal, U.K.; Choudhury, H. Targeted drug delivery to the brain via intranasal nanoemulsion: Available proof of concept and existing challenges. Int. J. Pharm. 2019, 565, 258–268. [Google Scholar] [CrossRef]
- Lindsay, S.L.; McCanney, G.A.; Willison, A.G.; Barnett, S.C. Multi-target approaches to CNS repair: Olfactory mucosa-derived cells and heparan sulfates. Nat. Rev. Neurol. 2020, 16, 229–240. [Google Scholar] [CrossRef]
- Rock, J.R.; Randell, S.H.; Hogan, B.L. Airway basal stem cells: A perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech. 2010, 3, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Lillehoj, E.P.; Kato, K.; Lu, W.; Kim, K.C. Cellular and molecular biology of airway mucins. Int. Rev. Cell Mol. Biol. 2013, 303, 139–202. [Google Scholar] [CrossRef] [Green Version]
- Lillehoj, E.R.; Kim, K.C. Airway mucus: Its components and function. Arch. Pharm. Res. 2002, 25, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef] [Green Version]
- Md, S.; Mustafa, G.; Baboota, S.; Ali, J. Nanoneurotherapeutics approach intended for direct nose to brain delivery. Drug Dev. Ind. Pharm. 2015, 41, 1922–1934. [Google Scholar] [CrossRef] [PubMed]
- Stoeckelhuber, M.; Olzowy, B.; Ihler, F.; Matthias, C.; Scherer, E.Q.; Babaryka, G.; Loeffelbein, D.J.; Rohleder, N.H.; Nieberler, M.; Kesting, M.R. Immunolocalization of antimicrobial and cytoskeletal components in the serous glands of human sinonasal mucosa. Histol. Histopathol. 2014, 29, 1315–1324. [Google Scholar] [CrossRef]
- Walker, H.K. Cranial Nerve I: The Olfactory Nerve. In Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed.; Walker, H.K., Hall, W.D., Hurst, J.W., Eds.; Butterworths: Boston, MA, USA, 1990. [Google Scholar]
- Harkema, J.R.; Carey, S.A.; Wagner, J.G. The nose revisited: A brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol. Pathol. 2006, 34, 252–269. [Google Scholar] [CrossRef] [PubMed]
- Trimmer, C.; Snyder, L.L.; Mainland, J.D. High-throughput analysis of mammalian olfactory receptors: Measurement of receptor activation via luciferase activity. J. Vis. Exp. 2014, 88, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Landis, M.S.; Boyden, T.; Pegg, S. Nasal-to-CNS drug delivery: Where are we now and where are we heading? An industrial perspective. Ther. Deliv. 2012, 3, 195–208. [Google Scholar] [CrossRef]
- Gizurarson, S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr. Drug Deliv. 2012, 9, 566–582. [Google Scholar] [CrossRef] [Green Version]
- Cowan, C.M.; Roskams, A.J. Apoptosis in the mature and developing olfactory neuroepithelium. Microsc. Res. Tech. 2002, 58, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Caggiano, M.; Kauer, J.S.; Hunter, D.D. Globose basal cells are neuronal progenitors in the olfactory epithelium: A lineage analysis using a replication-incompetent retrovirus. Neuron 1994, 13, 339–352. [Google Scholar] [CrossRef]
- Mackay-Sim, A. Stem cells and their niche in the adult olfactory mucosa. Arch. Ital. Biol. 2010, 148, 47–58. [Google Scholar]
- Iwai, N.; Zhou, Z.; Roop, D.R.; Behringer, R.R. Horizontal basal cells are multipotent progenitors in normal and injured adult olfactory epithelium. Stem. Cells 2008, 26, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Liang, F. Olfactory receptor neuronal dendrites become mostly intra-sustentacularly enwrapped upon maturity. J. Anat. 2018, 232, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R.; Kachramanoglou, C.; Li, D.; Andrews, P.; Choi, D. Anatomy and cellular constituents of the human olfactory mucosa: A review. J. Neurol. Surg. B Skull Base 2014, 75, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Maurya, D.K.; Henriques, T.; Marini, M.; Pedemonte, N.; Galietta, L.J.; Rock, J.R.; Harfe, B.D.; Menini, A. Development of the Olfactory Epithelium and Nasal Glands in TMEM16A−/− and TMEM16A+/+ Mice. PLoS ONE 2015, 10, e0129171. [Google Scholar] [CrossRef] [Green Version]
- Solbu, T.T.; Holen, T. Aquaporin pathways and mucin secretion of Bowman’s glands might protect the olfactory mucosa. Chem. Senses 2012, 37, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Tan, Z.; Huang, X.; Cheng, X.; Yuan, Y.; Qin, S.; Wang, D.; Hu, X.; Gu, Y.; Qian, W.-J.; et al. Direct neuronal reprogramming of olfactory ensheathing cells for CNS repair. Cell Death Dis. 2019, 10, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, D.; Ali, A.; Md, S.; Baboota, S.; Sahni, J.K.; Ali, J. Insights into direct nose to brain delivery: Current status and future perspective. Drug Deliv. 2014, 21, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Bannister, L.H.; Dodson, H.C. Endocytic pathways in the olfactory and vomeronasal epithelia of the mouse: Ultrastructure and uptake of tracers. Microsc. Res. Tech. 1992, 23, 128–141. [Google Scholar] [CrossRef]
- Morrison, E.E.; Costanzo, R.M. Morphology of olfactory epithelium in humans and other vertebrates. Microsc. Res. Tech. 1992, 23, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Buchner, K.; Seitz-Tutter, D.; Schönitzer, K.; Weiss, D.G. A quantitative study of anterograde and retrograde axonal transport of exogenous proteins in olfactory nerve C-fibers. Neuroscience 1987, 22, 697–707. [Google Scholar] [CrossRef]
- Selvaraj, K.; Gowthamarajan, K.; Karri, V. Nose to brain transport pathways an overview: Potential of nanostructured lipid carriers in nose to brain targeting. Artif. Cells Nanomed. Biotechnol. 2018, 46, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Marianecci, C.; Rinaldi, F.; Hanieh, P.N.; Paolino, D.; Marzio, L.D.; Carafa, M. Nose to Brain Delivery: New Trends in Amphiphile-Based “Soft” Nanocarriers. Curr. Pharm. Des. 2015, 21, 5225–5232. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, C.V.; Belgamwar, V.S. Direct nose to brain drug delivery via integrated nerve pathways bypassing the blood-brain barrier: An excellent platform for brain targeting. Expert Opin. Drug Deliv. 2013, 10, 957–972. [Google Scholar] [CrossRef]
- Bathla, G.; Hegde, A.N. The trigeminal nerve: An illustrated review of its imaging anatomy and pathology. Clin. Radiol. 2013, 68, 203–213. [Google Scholar] [CrossRef]
- Schaefer, M.L.; Böttger, B.; Silver, W.L.; Finger, T.E. Trigeminal collaterals in the nasal epithelium and olfactory bulb: A potential route for direct modulation of olfactory information by trigeminal stimuli. J. Comp. Neurol. 2002, 444, 221–226. [Google Scholar] [CrossRef]
- Ishikawa, T. Axoneme Structure from Motile Cilia. Cold Spring Harb. Perspect. Biol. 2017, 9, a028076. [Google Scholar] [CrossRef] [Green Version]
- Romanelli, M.C.; Gelardi, M.; Fiorella, M.L.; Tattoli, L.; Di Vella, G.; Solarino, B. Nasal ciliary motility: A new tool in estimating the time of death. Int. J. Legal Med. 2012, 126, 427–433. [Google Scholar] [CrossRef]
- Kaliner, M.; Marom, Z.; Patow, C.; Shelhamer, J. Human respiratory mucus. J. Allergy Clin. Immunol. 1984, 73, 318–323. [Google Scholar] [CrossRef]
- Cone, R.A. Barrier properties of mucus. Adv. Drug Deliv. Rev. 2009, 61, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Lieleg, O.; Ribbeck, K. Biological hydrogels as selective diffusion barriers. Trends Cell Biol. 2011, 21, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugwoke, M.I.; Agu, R.U.; Verbeke, N.; Kinget, R. Nasal mucoadhesive drug delivery: Background, applications, trends and future perspectives. Adv. Drug Deliv. Rev. 2005, 57, 1640–1665. [Google Scholar] [CrossRef]
- Williams, O.W.; Sharafkhaneh, A.; Kim, V.; Dickey, B.F.; Evans, C.M. Airway mucus: From production to secretion. Am. J. Respir. Cell Mol. Biol. 2006, 34, 527–536. [Google Scholar] [CrossRef]
- Merkus, F.W.; Verhoef, J.C.; Schipper, N.G.; Marttin, E. Nasal mucociliary clearance as a factor in nasal drug delivery. Adv. Drug Deliv. Rev. 1998, 29, 13–38. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and Mucociliary Clearance. Cold Spring Harb. Perspect. Biol. 2017, 9, a028241. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.C.; Santos, A.O. Nanosystems in nose-to-brain drug delivery: A review of non-clinical brain targeting studies. J. Control. Release 2018, 270, 89–100. [Google Scholar] [CrossRef]
- Kozlovskaya, L.; Abou-Kaoud, M.; Stepensky, D. Quantitative analysis of drug delivery to the brain via nasal route. J. Control. Release 2014, 189, 133–140. [Google Scholar] [CrossRef]
- Kuzmov, A.; Minko, T. Nanotechnology approaches for inhalation treatment of lung diseases. J. Control. Release 2015, 219, 500–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, A.M.; Minko, T. Pharmacokinetics of inhaled nanotherapeutics for pulmonary delivery. J. Control. Release 2020, 326, 222–244. [Google Scholar] [CrossRef] [PubMed]
- Sonvico, F.; Clementino, A.; Buttini, F.; Colombo, G.; Pescina, S.; Stanisçuaski Guterres, S.; Raffin Pohlmann, A.; Nicoli, S. Surface-Modified Nanocarriers for Nose-to-Brain Delivery: From Bioadhesion to Targeting. Pharmaceutics 2018, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Shannahan, J. The biocorona: A challenge for the biomedical application of nanoparticles. Nanotechnol. Rev. 2017, 6, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitman, M.; Larsen, J. The characterization of self-assembled nanostructures in whole blood. Anal. Methods 2020, 12, 2068–2081. [Google Scholar] [CrossRef]
- Lima, T.; Bernfur, K.; Vilanova, M.; Cedervall, T. Understanding the Lipid and Protein Corona Formation on Different Sized Polymeric Nanoparticles. Sci. Rep. 2020, 10, 1129. [Google Scholar] [CrossRef]
- Müller, L.K.; Simon, J.; Rosenauer, C.; Mailänder, V.; Morsbach, S.; Landfester, K. The Transferability from Animal Models to Humans: Challenges Regarding Aggregation and Protein Corona Formation of Nanoparticles. Biomacromolecules 2018, 19, 374–385. [Google Scholar] [CrossRef]
- Chinen, A.B.; Guan, C.M.; Ko, C.H.; Mirkin, C.A. The Impact of Protein Corona Formation on the Macrophage Cellular Uptake and Biodistribution of Spherical Nucleic Acids. Small 2017, 13, 1603847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranowska-Wójcik, E.; Szwajgier, D.; Oleszczuk, P.; Winiarska-Mieczan, A. Effects of Titanium Dioxide Nanoparticles Exposure on Human Health-a Review. Biol. Trace Elem. Res. 2020, 193, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Sahu, S.C.; Hayes, A.W. Toxicity of nanomaterials found in human environment: A literature review. Toxicol. Res. Appl. 2017, 1, 2397847317726352. [Google Scholar] [CrossRef]
- Feng, Y.; He, H.; Li, F.; Lu, Y.; Qi, J.; Wu, W. An update on the role of nanovehicles in nose-to-brain drug delivery. Drug Discov. Today 2018, 23, 1079–1088. [Google Scholar] [CrossRef]
- Ray, S.; Cheng, C.A.; Chen, W.; Li, Z.; Zink, J.I.; Lin, Y.Y. Magnetic Heating Stimulated Cargo Release with Dose Control using Multifunctional MR and Thermosensitive Liposome. Nanotheranostics 2019, 3, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Luo, Y. Biological fate of ingested lipid-based nanoparticles: Current understanding and future directions. Nanoscale 2019, 11, 11048–11063. [Google Scholar] [CrossRef] [PubMed]
- Taratula, O.; Kuzmov, A.; Shah, M.; Garbuzenko, O.B.; Minko, T. Nanostructured lipid carriers as multifunctional nanomedicine platform for pulmonary co-delivery of anticancer drugs and siRNA. J. Control. Release 2013, 171, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Liu, C.; Ji, X.; Joseph, J.; Tang, Z.; Ouyang, J.; Xiao, Y.; Kong, N.; Joshi, N.; Farokhzad, O.C.; et al. Stanene-Based Nanosheets for β-Elemene Delivery and Ultrasound-Mediated Combination Cancer Therapy. Angew. Chem. Int. Ed. 2021, 60, 7155–7164. [Google Scholar] [CrossRef]
- Bodratti, A.M.; Alexandridis, P. Formulation of Poloxamers for Drug Delivery. J. Funct. Biomater. 2018, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, E.; Paolino, D.; Fresta, M.; Cosco, D. Mucosal Applications of Poloxamer 407-Based Hydrogels: An Overview. Pharmaceutics 2018, 10, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.Z.; Li, X.; Lu, C.T.; Lin, M.; Chen, L.J.; Xiang, Q.; Zhang, M.; Jin, R.R.; Jiang, X.; Shen, X.T.; et al. Gelatin nanostructured lipid carriers-mediated intranasal delivery of basic fibroblast growth factor enhances functional recovery in hemiparkinsonian rats. Nanomedicine 2014, 10, 755–764. [Google Scholar] [CrossRef]
- Bondì, M.L.; Di Gesù, R.; Craparo, E.F. Lipid nanoparticles for drug targeting to the brain. Methods Enzymol. 2012, 508, 229–251. [Google Scholar] [CrossRef]
- He, Q.; Liu, J.; Liang, J.; Liu, X.; Li, W.; Liu, Z.; Ding, Z.; Tuo, D. Towards Improvements for Penetrating the Blood–Brain Barrier—Recent Progress from a Material and Pharmaceutical Perspective. Cells 2018, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Dow, S. Liposome-nucleic acid immunotherapeutics. Expert Opin. Drug Deliv. 2008, 5, 11–24. [Google Scholar] [CrossRef]
- Garbuzenko, O.B.; Mainelis, G.; Taratula, O.; Minko, T. Inhalation treatment of lung cancer: The influence of composition, size and shape of nanocarriers on their lung accumulation and retention. Cancer Biol. Med. 2014, 11, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Taratula, O.; Garbuzenko, O.B.; Patil, M.L.; Savla, R.; Zhang, M.; Minko, T. Genotoxicity of different nanocarriers: Possible modifications for the delivery of nucleic acids. Curr. Drug Discov. Technol. 2013, 10, 8–15. [Google Scholar] [PubMed]
- Sanchez-Purra, M.; Ramos, V.; Petrenko, V.A.; Torchilin, V.P.; Borros, S. Double-targeted polymersomes and liposomes for multiple barrier crossing. Int. J. Pharm. 2016, 511, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, H.K.; Fan, Y.; Kim, J.; Amiji, M.M. Intranasal Delivery and Transfection of mRNA Therapeutics in the Brain Using Cationic Liposomes. Mol. Pharm. 2020, 17, 1996–2005. [Google Scholar] [CrossRef]
- Al Asmari, A.K.; Ullah, Z.; Tariq, M.; Fatani, A. Preparation, characterization, and in vivo evaluation of intranasally administered liposomal formulation of donepezil. Drug Des. Devel. Ther. 2016, 10, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutlu, N.B.; Değim, Z.; Yilmaz, Ş.; Eşsiz, D.; Nacar, A. New perspective for the treatment of Alzheimer diseases: Liposomal rivastigmine formulations. Drug Dev. Ind. Pharm. 2011, 37, 775–789. [Google Scholar] [CrossRef]
- Hoekman, J.D.; Srivastava, P.; Ho, R.J. Aerosol-stable peptide-coated liposome nanoparticles: A proof-of-concept study with opioid fentanyl in enhancing analgesic effects and reducing plasma drug exposure. J. Pharm. Sci. 2014, 103, 2231–2239. [Google Scholar] [CrossRef] [Green Version]
- Migliore, M.M.; Vyas, T.K.; Campbell, R.B.; Amiji, M.M.; Waszczak, B.L. Brain delivery of proteins by the intranasal route of administration: A comparison of cationic liposomes versus aqueous solution formulations. J. Pharm. Sci. 2010, 99, 1745–1761. [Google Scholar] [CrossRef]
- Zheng, X.; Shao, X.; Zhang, C.; Tan, Y.; Liu, Q.; Wan, X.; Zhang, Q.; Xu, S.; Jiang, X. Intranasal H102 Peptide-Loaded Liposomes for Brain Delivery to Treat Alzheimer’s Disease. Pharm. Res. 2015, 32, 3837–3849. [Google Scholar] [CrossRef] [PubMed]
- Pashirova, T.N.; Zueva, I.V.; Petrov, K.A.; Lukashenko, S.S.; Nizameev, I.R.; Kulik, N.V.; Voloshina, A.D.; Almasy, L.; Kadirov, M.K.; Masson, P.; et al. Mixed cationic liposomes for brain delivery of drugs by the intranasal route: The acetylcholinesterase reactivator 2-PAM as encapsulated drug model. Colloids Surf. B Biointerfaces 2018, 171, 358–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makowski, L.; Olson-Sidford, W.; W-Weisel, J. Biological and Clinical Consequences of Integrin Binding via a Rogue RGD Motif in the SARS CoV-2 Spike Protein. Viruses 2021, 13, 146. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ray, S.; Thakur, R.S. Solid lipid nanoparticles: A modern formulation approach in drug delivery system. Indian J. Pharm. Sci. 2009, 71, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Puri, A.; Loomis, K.; Smith, B.; Lee, J.H.; Yavlovich, A.; Heldman, E.; Blumenthal, R. Lipid-based nanoparticles as pharmaceutical drug carriers: From concepts to clinic. Crit. Rev. Ther. Drug Carrier Syst. 2009, 26, 523–580. [Google Scholar] [CrossRef] [Green Version]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288–303. [Google Scholar] [CrossRef]
- Badilli, U.; Gumustas, M.; Uslu, B.; Ozkan, S.A. Chapter 9—Lipid-based nanoparticles for dermal drug delivery. In Organic Materials as Smart Nanocarriers for Drug Delivery; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 369–413. [Google Scholar]
- Sajid, M.; Cameotra, S.S.; Ahmad Khan, M.S.; Ahmad, I. Chapter 23—Nanoparticle-Based Delivery of Phytomedicines: Challenges and Opportunities. In New Look to Phytomedicine; Ahmad Khan, M.S., Ahmad, I., Chattopadhyay, D., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 597–623. [Google Scholar]
- Patel, S.; Chavhan, S.; Soni, H.; Babbar, A.K.; Mathur, R.; Mishra, A.K.; Sawant, K. Brain targeting of risperidone-loaded solid lipid nanoparticles by intranasal route. J. Drug Target 2011, 19, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.; Sarmento, B.; Ferreira, D.C.; Souto, E.B. Lipid-based colloidal carriers for peptide and protein delivery--liposomes versus lipid nanoparticles. Int. J. Nanomedicine 2007, 2, 595–607. [Google Scholar]
- Agrawal, M.; Saraf, S.; Saraf, S.; Dubey, S.K.; Puri, A.; Patel, R.J.; Ajazuddin; Ravichandiran, V.; Murty, U.S.; Alexander, A. Recent strategies and advances in the fabrication of nano lipid carriers and their application towards brain targeting. J. Control. Release 2020, 321, 372–415. [Google Scholar] [CrossRef]
- Tapeinos, C.; Battaglini, M.; Ciofani, G. Advances in the design of solid lipid nanoparticles and nanostructured lipid carriers for targeting brain diseases. J. Control. Release 2017, 264, 306–332. [Google Scholar] [CrossRef] [PubMed]
- Madane, R.G.; Mahajan, H.S. Curcumin-loaded nanostructured lipid carriers (NLCs) for nasal administration: Design, characterization, and in vivo study. Drug Deliv. 2016, 23, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Eral, H.B.; Hatton, T.A.; Doyle, P.S. Nanoemulsions: Formation, properties and applications. Soft Matter 2016, 12, 2826–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, D.G.; Ye, R.; Dunlap, R.N.; Anunciado, D.B.; Pingali, S.V.; O’Neill, H.M.; Urban, V.S. Bicontinuous microemulsions as a biomembrane mimetic system for melittin. Biochim. Biophys. Acta Biomembr. 2018, 1860, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; Dudhe, R.; Sharma, P.K. Nanoemulsion: An advanced mode of drug delivery system. 3 Biotech 2015, 5, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Moreira, J.N.; Amaral, M.H.; Sousa Lobo, J.M.; Silva, A.C. Nose-to-brain delivery of lipid-based nanosystems for epileptic seizures and anxiety crisis. J. Control. Release 2019, 295, 187–200. [Google Scholar] [CrossRef]
- Iqbal, R.; Ahmed, S.; Jain, G.K.; Vohora, D. Design and development of letrozole nanoemulsion: A comparative evaluation of brain targeted nanoemulsion with free letrozole against status epilepticus and neurodegeneration in mice. Int. J. Pharm. 2019, 565, 20–32. [Google Scholar] [CrossRef]
- Harden, C.; MacLusky, N.J. Aromatase inhibitors as add-on treatment for men with epilepsy. Expert Rev. Neurother. 2005, 5, 123–127. [Google Scholar] [CrossRef]
- Iqbal, R.; Jain, G.K.; Siraj, F.; Vohora, D. Aromatase inhibition by letrozole attenuates kainic acid-induced seizures but not neurotoxicity in mice. Epilepsy Res. 2018, 143, 60–69. [Google Scholar] [CrossRef]
- Elieh-Ali-Komi, D.; Hamblin, M.R. Chitin and Chitosan: Production and Application of Versatile Biomedical Nanomaterials. Int. J. Adv. Res. 2016, 4, 411–427. [Google Scholar]
- Nurunnabi, M.; Revuri, V.; Huh, K.M.; Lee, Y.-k. Chapter 14—Polysaccharide based nano/microformulation: An effective and versatile oral drug delivery system. In Nanostructures for Oral Medicine; Andronescu, E., Grumezescu, A.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 409–433. [Google Scholar]
- England, R.J.; Homer, J.J.; Knight, L.C.; Ell, S.R. Nasal pH measurement: A reliable and repeatable parameter. Clin. Otolaryngol. Allied Sci. 1999, 24, 67–68. [Google Scholar] [CrossRef] [Green Version]
- Deli, M.A. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. Biochim. Biophys. Acta 2009, 1788, 892–910. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.M.; Dornish, M.; Wood, E.J. Involvement of protein kinase C in chitosan glutamate-mediated tight junction disruption. Biomaterials 2005, 26, 3269–3276. [Google Scholar] [CrossRef]
- Gonçalves, C.; Ferreira, N.; Lourenço, L. Production of Low Molecular Weight Chitosan and Chitooligosaccharides (COS): A Review. Polymers 2021, 13, 2466. [Google Scholar] [CrossRef]
- Cardia, M.C.; Carta, A.R.; Caboni, P.; Maccioni, A.M.; Erbì, S.; Boi, L.; Meloni, M.C.; Lai, F.; Sinico, C. Trimethyl Chitosan Hydrogel Nanoparticles for Progesterone Delivery in Neurodegenerative Disorders. Pharmaceutics 2019, 11, 657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Pandey, R.S.; Patra, K.C.; Jain, S.K.; Soni, M.L.; Dangi, J.S.; Madan, J. Evaluation of neuropeptide loaded trimethyl chitosan nanoparticles for nose to brain delivery. Int. J. Biol. Macromol. 2013, 61, 189–195. [Google Scholar] [CrossRef]
- Singh, D.; Rashid, M.; Hallan, S.S.; Mehra, N.K.; Prakash, A.; Mishra, N. Pharmacological evaluation of nasal delivery of selegiline hydrochloride-loaded thiolated chitosan nanoparticles for the treatment of depression. Artif. Cells Nanomed. Biotechnol. 2016, 44, 865–877. [Google Scholar] [CrossRef]
- Patel, D.; Naik, S.; Chuttani, K.; Mathur, R.; Mishra, A.K.; Misra, A. Intranasal delivery of cyclobenzaprine hydrochloride-loaded thiolated chitosan nanoparticles for pain relief. J. Drug Target 2013, 21, 759–769. [Google Scholar] [CrossRef]
- Tønnesen, H.H.; Karlsen, J. Alginate in drug delivery systems. Drug Dev. Ind. Pharm. 2002, 28, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Md, S.; Sahni, J.K.; Ali, J.; Baboota, S. Development and evaluation of brain targeted intranasal alginate nanoparticles for treatment of depression. J. Psychiatr. Res. 2014, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xiong, G.; Tsang, W.C.; Schätzlein, A.G.; Uchegbu, I.F. Nose-to-Brain Delivery. J. Pharmacol. Exp. Ther. 2019, 370, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, W.Y.; Shalini, S.M.; Costantino, L. Nose-to-brain drug delivery by nanoparticles in the treatment of neurological disorders. Curr. Med. Chem. 2014, 21, 4247–4256. [Google Scholar] [CrossRef] [PubMed]
- McCall, R.L.; Sirianni, R.W. PLGA nanoparticles formed by single- or double-emulsion with vitamin E-TPGS. J. Vis. Exp. 2013, 82, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muntimadugu, E.; Dhommati, R.; Jain, A.; Challa, V.G.; Shaheen, M.; Khan, W. Intranasal delivery of nanoparticle encapsulated tarenflurbil: A potential brain targeting strategy for Alzheimer’s disease. Eur. J. Pharm. Sci. 2016, 92, 224–234. [Google Scholar] [CrossRef]
- Mistry, A.; Glud, S.Z.; Kjems, J.; Randel, J.; Howard, K.A.; Stolnik, S.; Illum, L. Effect of physicochemical properties on intranasal nanoparticle transit into murine olfactory epithelium. J. Drug Target 2009, 17, 543–552. [Google Scholar] [CrossRef]
- Ahmad, E.; Feng, Y.; Qi, J.; Fan, W.; Ma, Y.; He, H.; Xia, F.; Dong, X.; Zhao, W.; Lu, Y.; et al. Evidence of nose-to-brain delivery of nanoemulsions: Cargoes but not vehicles. Nanoscale 2017, 9, 1174–1183. [Google Scholar] [CrossRef]
- Law, S.L.; Huang, K.J.; Chou, H.Y. Preparation of desmopressin-containing liposomes for intranasal delivery. J. Control. Release 2001, 70, 375–382. [Google Scholar] [CrossRef]
- Gabal, Y.M.; Kamel, A.O.; Sammour, O.A.; Elshafeey, A.H. Effect of surface charge on the brain delivery of nanostructured lipid carriers in situ gels via the nasal route. Int. J. Pharm. 2014, 473, 442–457. [Google Scholar] [CrossRef]
- Bonaccorso, A.; Musumeci, T.; Serapide, M.F.; Pellitteri, R.; Uchegbu, I.F.; Puglisi, G. Nose to brain delivery in rats: Effect of surface charge of rhodamine B labeled nanocarriers on brain subregion localization. Colloids Surf. B Biointerfaces 2017, 154, 297–306. [Google Scholar] [CrossRef]
- Sosnik, A.; das Neves, J.; Sarmento, B. Mucoadhesive polymers in the design of nano-drug delivery systems for administration by non-parenteral routes: A review. Prog. Polym. Sci. 2014, 39, 2030–2075. [Google Scholar] [CrossRef]
- Kanazawa, T.; Kaneko, M.; Niide, T.; Akiyama, F.; Kakizaki, S.; Ibaraki, H.; Shiraishi, S.; Takashima, Y.; Suzuki, T.; Seta, Y. Enhancement of nose-to-brain delivery of hydrophilic macromolecules with stearate- or polyethylene glycol-modified arginine-rich peptide. Int. J. Pharm. 2017, 530, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Epilepsy. In Pharmacotherapy Quick Guide; Wells, B.G.; DiPiro, J.T.; Schwinghammer, T.L.; DiPiro, C.V. (Eds.) McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- Milligan, T.A. Epilepsy: A Clinical Overview. Am. J. Med. 2021, 134, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, K.A.; Lopez Ramos, C.; Buch, V.P.; Mekary, R.A.; Amundson, J.R.; Shah, M.; Rattani, A.; Dewan, M.C.; Park, K.B. An estimation of global volume of surgically treatable epilepsy based on a systematic review and meta-analysis of epilepsy. J. Neurosurg. 2018, 130, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falco-Walter, J.J.; Scheffer, I.E.; Fisher, R.S. The new definition and classification of seizures and epilepsy. Epilepsy Res. 2018, 139, 73–79. [Google Scholar] [CrossRef]
- Engel, J. Mechanisms of Neuronal Excitation and Synchronization. In Seizures and Epilepsy, 2nd ed.; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus—Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, S.I.; Landmark, C.J. Antiepileptic drug interactions—principles and clinical implications. Curr. Neuropharmacol. 2010, 8, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, T.; Bonaccorso, A.; Puglisi, G. Epilepsy Disease and Nose-to-Brain Delivery of Polymeric Nanoparticles: An Overview. Pharmaceutics 2019, 11, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samia, O.; Hanan, R.; Kamal, E.T. Carbamazepine mucoadhesive nanoemulgel (MNEG) as brain targeting delivery system via the olfactory mucosa. Drug Deliv. 2012, 19, 58–67. [Google Scholar] [CrossRef]
- Sharma, D.; Sharma, R.K.; Sharma, N.; Gabrani, R.; Sharma, S.K.; Ali, J.; Dang, S. Nose-To-Brain Delivery of PLGA-Diazepam Nanoparticles. AAPS PharmSciTech 2015, 16, 1108–1121. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, T.; Serapide, M.F.; Pellitteri, R.; Dalpiaz, A.; Ferraro, L.; Dal Magro, R.; Bonaccorso, A.; Carbone, C.; Veiga, F.; Sancini, G.; et al. Oxcarbazepine free or loaded PLGA nanoparticles as effective intranasal approach to control epileptic seizures in rodents. Eur. J. Pharm. Biopharm. 2018, 133, 309–320. [Google Scholar] [CrossRef]
- El-Zaafarany, G.M.; Soliman, M.E.; Mansour, S.; Awad, G.A. Identifying lipidic emulsomes for improved oxcarbazepine brain targeting: In vitro and rat in vivo studies. Int. J. Pharm. 2016, 503, 127–140. [Google Scholar] [CrossRef]
- Ahmad, N.; Ahmad, R.; Alam, M.A.; Ahmad, F.J.; Amir, M. Impact of ultrasonication techniques on the preparation of novel Amiloride-nanoemulsion used for intranasal delivery in the treatment of epilepsy. Artif. Cells Nanomed. Biotechnol. 2018, 46, S192–S207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubek, M.J.; Domb, A.J.; Veronesi, M.C. Attenuation of kindled seizures by intranasal delivery of neuropeptide-loaded nanoparticles. Neurotherapeutics 2009, 6, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Maheshwari, D.; Philip, G.; Rana, R.; Bhatia, S.; Singh, M.; Gabrani, R.; Sharma, S.K.; Ali, J.; Sharma, R.K.; et al. Formulation and optimization of polymeric nanoparticles for intranasal delivery of lorazepam using Box-Behnken design: In vitro and in vivo evaluation. Biomed. Res. Int. 2014, 2014, 156010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praveen, A.; Aqil, M.; Imam, S.S.; Ahad, A.; Moolakkadath, T.; Ahmad, F.J. Lamotrigine encapsulated intra-nasal nanoliposome formulation for epilepsy treatment: Formulation design, characterization and nasal toxicity study. Colloids Surf. B Biointerfaces 2019, 174, 553–562. [Google Scholar] [CrossRef]
- Aithal, G.C.; Narayan, R.; Nayak, U.Y. Nanoemulgel: A Promising Phase in Drug Delivery. Curr. Pharm. Des. 2020, 26, 279–291. [Google Scholar] [CrossRef]
- Newell, B.D.; Moinfar, M.; Mancini, A.J.; Nopper, A.J. Retrospective analysis of 32 pediatric patients with anticonvulsant hypersensitivity syndrome (ACHSS). Pediatr. Dermatol. 2009, 26, 536–546. [Google Scholar] [CrossRef]
- Mehta, M.; Shah, J.; Khakhkhar, T.; Shah, R.; Hemavathi, K.G. Anticonvulsant hypersensitivity syndrome associated with carbamazepine administration: Case series. J. Pharmacol. Pharmacother. 2014, 5, 59–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, Y.M.; Argin-Soysal, S.; Hsu, C.-H. Chapter 22—Bioconversion of Whey Lactose into Microbial Exopolysaccharides. In Bioprocessing for Value-Added Products from Renewable Resources; Yang, S.-T., Ed.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 559–583. [Google Scholar]
- Dhaliwal, J.S.; Rosani, A.; Saadabadi, A. Diazepam; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Abou-Khalil, B.W. Oxcarbazepine and carbamazepine: Expected and unexpected differences and similarities. Epilepsy Curr. 2007, 7, 74–76. [Google Scholar] [CrossRef] [Green Version]
- Ucisik, M.H.; Sleytr, U.B.; Schuster, B. Emulsomes meet S-layer proteins: An emerging targeted drug delivery system. Curr. Pharm. Biotechnol. 2015, 16, 392–405. [Google Scholar] [CrossRef] [Green Version]
- Neugroschl, J.; Wang, S. Alzheimer’s disease: Diagnosis and treatment across the spectrum of disease severity. Mt. Sinai J. Med. 2011, 78, 596–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mebane-Sims, I. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunnström, H.R.; Englund, E.M. Cause of death in patients with dementia disorders. Eur. J. Neurol. 2009, 16, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.; Goldman, J.; Marder, K.S. Genetic aspects of Alzheimer disease. Neurologist 2009, 15, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peron, E.P.; Zimmerman, K.M.; Crouse, E.L.; Slattum, P.W.; Hobgood, S.E. Alzheimer Disease. In Pharmacotherapy: A Pathophysiologic Approach, 11e; DiPiro, J.T., Yee, G.C., Posey, L., Haines, S.T., Nolin, T.D., Ellingrod, V., Eds.; McGraw Hill: New York, NY, USA, 2020. [Google Scholar]
- Sadowsky, C.H.; Galvin, J.E. Guidelines for the management of cognitive and behavioral problems in dementia. J. Am. Board Fam. Med. 2012, 25, 350–366. [Google Scholar] [CrossRef]
- Md, S.; Ali, M.; Ali, R.; Bhatnagar, A.; Baboota, S.; Ali, J. Donepezil nanosuspension intended for nose to brain targeting: In vitro and in vivo safety evaluation. Int. J. Biol. Macromol. 2014, 67, 418–425. [Google Scholar] [CrossRef]
- Md, S.; Ali, M.; Baboota, S.; Sahni, J.K.; Bhatnagar, A.; Ali, J. Preparation, characterization, in vivo biodistribution and pharmacokinetic studies of donepezil-loaded PLGA nanoparticles for brain targeting. Drug Dev. Ind. Pharm. 2014, 40, 278–287. [Google Scholar] [CrossRef]
- Singh, S.K.; Mishra, D.N. Nose to Brain Delivery of Galantamine Loaded Nanoparticles: In-vivo Pharmacodynamic and Biochemical Study in Mice. Curr. Drug Deliv. 2019, 16, 51–58. [Google Scholar] [CrossRef]
- Li, W.; Zhou, Y.; Zhao, N.; Hao, B.; Wang, X.; Kong, P. Pharmacokinetic behavior and efficiency of acetylcholinesterase inhibition in rat brain after intranasal administration of galanthamine hydrobromide loaded flexible liposomes. Environ. Toxicol. Pharmacol. 2012, 34, 272–279. [Google Scholar] [CrossRef]
- Misra, S.; Chopra, K.; Sinha, V.R.; Medhi, B. Galantamine-loaded solid-lipid nanoparticles for enhanced brain delivery: Preparation, characterization, in vitro and in vivo evaluations. Drug Deliv. 2016, 23, 1434–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, A.; Nigam, K.; Srivastava, S.; Tyagi, A.; Dang, S. Memantine nanoemulsion: A new approach to treat Alzheimer’s disease. J. Microencapsul. 2020, 37, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Jojo, G.M.; Kuppusamy, G.; De, A.; Karri, V. Formulation and optimization of intranasal nanolipid carriers of pioglitazone for the repurposing in Alzheimer’s disease using Box-Behnken design. Drug Dev. Ind. Pharm. 2019, 45, 1061–1072. [Google Scholar] [CrossRef]
- Meng, Q.; Wang, A.; Hua, H.; Jiang, Y.; Wang, Y.; Mu, H.; Wu, Z.; Sun, K. Intranasal delivery of Huperzine A to the brain using lactoferrin-conjugated N-trimethylated chitosan surface-modified PLGA nanoparticles for treatment of Alzheimer’s disease. Int. J. Nanomedicine 2018, 13, 705–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazil, M.; Md, S.; Haque, S.; Kumar, M.; Baboota, S.; Sahni, J.K.; Ali, J. Development and evaluation of rivastigmine loaded chitosan nanoparticles for brain targeting. Eur. J. Pharm. Sci. 2012, 47, 6–15. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Zhang, Y.Q.; Wang, Z.Z.; Wu, K.; Lou, J.N.; Qi, X.R. Enhanced brain distribution and pharmacodynamics of rivastigmine by liposomes following intranasal administration. Int. J. Pharm. 2013, 452, 344–354. [Google Scholar] [CrossRef]
- Rassu, G.; Soddu, E.; Posadino, A.M.; Pintus, G.; Sarmento, B.; Giunchedi, P.; Gavini, E. Nose-to-brain delivery of BACE1 siRNA loaded in solid lipid nanoparticles for Alzheimer’s therapy. Colloids Surf. B Biointerfaces 2017, 152, 296–301. [Google Scholar] [CrossRef]
- Wong, L.R.; Ho, P.C. Role of serum albumin as a nanoparticulate carrier for nose-to-brain delivery of R-flurbiprofen: Implications for the treatment of Alzheimer’s disease. J. Pharm. Pharmacol. 2018, 70, 59–69. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 97–101. [Google Scholar] [CrossRef]
- Hu, S.H.; Jiang, T.; Yang, S.S.; Yang, Y. Pioglitazone ameliorates intracerebral insulin resistance and tau-protein hyperphosphorylation in rats with type 2 diabetes. Exp. Clin. Endocrinol. Diabetes 2013, 121, 220–224. [Google Scholar] [CrossRef] [Green Version]
- Seok, H.; Lee, M.; Shin, E.; Yun, M.R.; Lee, Y.-h.; Moon, J.H.; Kim, E.; Lee, P.H.; Lee, B.-W.; Kang, E.S.; et al. Low-dose pioglitazone can ameliorate learning and memory impairment in a mouse model of dementia by increasing LRP1 expression in the hippocampus. Sci. Rep. 2019, 9, 4414. [Google Scholar] [CrossRef]
- Alam, F.; Islam, M.A.; Mohamed, M.; Ahmad, I.; Kamal, M.A.; Donnelly, R.; Idris, I.; Gan, S.H. Efficacy and Safety of Pioglitazone Monotherapy in Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. Sci. Rep. 2019, 9, 5389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Song, Y.; Lu, C.; Zhang, J.; Yuan, J.; Wang, T.; Fu, F. The effects of huperzine A on gastrointestinal acetylcholinesterase activity and motility after single and multiple dosing in mice. Exp. Ther. Med. 2013, 5, 793–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orozco, J.L.; Valderrama-Chaparro, J.A.; Pinilla-Monsalve, G.D.; Molina-Echeverry, M.I.; Pérez Castaño, A.M.; Ariza-Araújo, Y.; Prada, S.I.; Takeuchi, Y. Parkinson’s disease prevalence, age distribution and staging in Colombia. Neurol. Int. 2020, 12, 8401. [Google Scholar] [CrossRef]
- Chen, J.J.; Dashtipour, K. Parkinson Disease. In Pharmacotherapy: A Pathophysiologic Approach, 11e; DiPiro, J.T., Yee, G.C., Posey, L., Haines, S.T., Nolin, T.D., Ellingrod, V., Eds.; McGraw Hill: New York, NY, USA, 2020. [Google Scholar]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Aguilar, L.G. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr. Dis. Treat 2008, 4, 743–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, D.; Md, S.; Hasan, Q.; Fazil, M.; Ali, A.; Baboota, S.; Ali, J. Brain targeted nanoparticulate drug delivery system of rasagiline via intranasal route. Drug Deliv. 2016, 23, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, A.; Yan, X.; Chu, L.; Yang, X.; Song, Y.; Sun, K.; Yu, X.; Liu, R.; Wu, Z.; et al. Brain-targeted intranasal delivery of dopamine with borneol and lactoferrin co-modified nanoparticles for treating Parkinson’s disease. Drug Deliv. 2019, 26, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Md, S.; Khan, R.A.; Mustafa, G.; Chuttani, K.; Baboota, S.; Sahni, J.K.; Ali, J. Bromocriptine loaded chitosan nanoparticles intended for direct nose to brain delivery: Pharmacodynamic, pharmacokinetic and scintigraphy study in mice model. Eur. J. Pharm. Sci. 2013, 48, 393–405. [Google Scholar] [CrossRef]
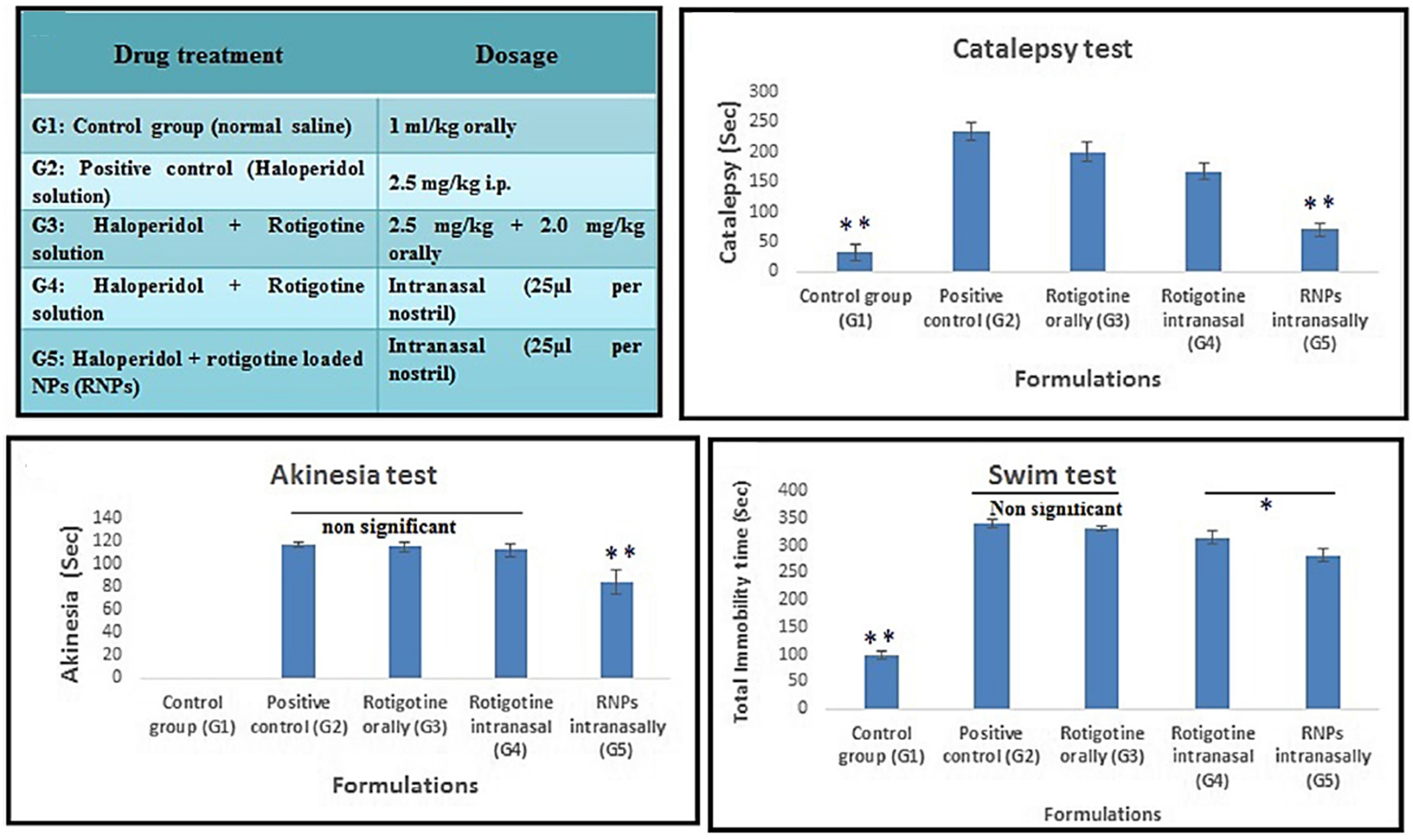
- Bhattamisra, S.K.; Shak, A.T.; Xi, L.W.; Safian, N.H.; Choudhury, H.; Lim, W.M.; Shahzad, N.; Alhakamy, N.A.; Anwer, M.K.; Radhakrishnan, A.K.; et al. Nose to brain delivery of rotigotine loaded chitosan nanoparticles in human SH-SY5Y neuroblastoma cells and animal model of Parkinson’s disease. Int. J. Pharm. 2020, 579, 119148. [Google Scholar] [CrossRef]
- Bi, C.; Wang, A.; Chu, Y.; Liu, S.; Mu, H.; Liu, W.; Wu, Z.; Sun, K.; Li, Y. Intranasal delivery of rotigotine to the brain with lactoferrin-modified PEG-PLGA nanoparticles for Parkinson’s disease treatment. Int. J. Nanomedicine 2016, 11, 6547–6559. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.; Sharma, S.; Deshmukh, R.; Kumar, A.; Sharma, R. Development and Characterization of Nasal Delivery of Selegiline Hydrochloride Loaded Nanolipid Carriers for the Management of Parkinson’s Disease. Cent. Nerv. Syst. Agents Med. Chem. 2019, 19, 46–56. [Google Scholar] [CrossRef]
- Kumar, S.; Dang, S.; Nigam, K.; Ali, J.; Baboota, S. Selegiline Nanoformulation in Attenuation of Oxidative Stress and Upregulation of Dopamine in the Brain for the Treatment of Parkinson’s Disease. Rejuvenation Res. 2018, 21, 464–476. [Google Scholar] [CrossRef]
- De Oliveira Junior, E.R.; Truzzi, E.; Ferraro, L.; Fogagnolo, M.; Pavan, B.; Beggiato, S.; Rustichelli, C.; Maretti, E.; Lima, E.M.; Leo, E.; et al. Nasal administration of nanoencapsulated geraniol/ursodeoxycholic acid conjugate: Towards a new approach for the management of Parkinson’s disease. J. Control. Release 2020, 321, 540–552. [Google Scholar] [CrossRef]
- Pardeshi, C.V.; Rajput, P.V.; Belgamwar, V.S.; Tekade, A.R.; Surana, S.J. Novel surface modified solid lipid nanoparticles as intranasal carriers for ropinirole hydrochloride: Application of factorial design approach. Drug Deliv. 2013, 20, 47–56. [Google Scholar] [CrossRef]
- Jafarieh, O.; Md, S.; Ali, M.; Baboota, S.; Sahni, J.K.; Kumari, B.; Bhatnagar, A.; Ali, J. Design, characterization, and evaluation of intranasal delivery of ropinirole-loaded mucoadhesive nanoparticles for brain targeting. Drug Dev. Ind. Pharm. 2015, 41, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Patil, G.B.; Surana, S.J. Fabrication and statistical optimization of surface engineered PLGA nanoparticles for naso-brain delivery of ropinirole hydrochloride: In-vitro-ex-vivo studies. J. Biomater. Sci. Polym. Ed. 2013, 24, 1740–1756. [Google Scholar] [CrossRef] [PubMed]
- Raj, R.; Wairkar, S.; Sridhar, V.; Gaud, R. Pramipexole dihydrochloride loaded chitosan nanoparticles for nose to brain delivery: Development, characterization and in vivo anti-Parkinson activity. Int. J. Biol. Macromol. 2018, 109, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Gaba, B.; Khan, T.; Haider, M.F.; Alam, T.; Baboota, S.; Parvez, S.; Ali, J. Vitamin E Loaded Naringenin Nanoemulsion via Intranasal Delivery for the Management of Oxidative Stress in a 6-OHDA Parkinson’s Disease Model. Biomed. Res. Int. 2019, 2019, 2382563. [Google Scholar] [CrossRef]
- Wen, Z.; Yan, Z.; Hu, K.; Pang, Z.; Cheng, X.; Guo, L.; Zhang, Q.; Jiang, X.; Fang, L.; Lai, R. Odorranalectin-conjugated nanoparticles: Preparation, brain delivery and pharmacodynamic study on Parkinson’s disease following intranasal administration. J. Control. Release 2011, 151, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Negro, S.; Slowing, K.; Fernández-Carballido, A.; Barcia, E. An effective novel delivery strategy of rasagiline for Parkinson’s disease. Int. J. Pharm. 2011, 419, 271–280. [Google Scholar] [CrossRef]
- Gavini, E.; Rassu, G.; Ferraro, L.; Generosi, A.; Rau, J.V.; Brunetti, A.; Giunchedi, P.; Dalpiaz, A. Influence of chitosan glutamate on the in vivo intranasal absorption of rokitamycin from microspheres. J. Pharm. Sci. 2011, 100, 1488–1502. [Google Scholar] [CrossRef]
- Qi, H.P.; Gao, X.C.; Zhang, L.Q.; Wei, S.Q.; Bi, S.; Yang, Z.C.; Cui, H. In vitro evaluation of enhancing effect of borneol on transcorneal permeation of compounds with different hydrophilicities and molecular sizes. Eur. J. Pharmacol. 2013, 705, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Vautier, S.; Lacomblez, L.; Chacun, H.; Picard, V.; Gimenez, F.; Farinotti, R.; Fernandez, C. Interactions between the dopamine agonist, bromocriptine and the efflux protein, P-glycoprotein at the blood-brain barrier in the mouse. Eur. J. Pharm. Sci. 2006, 27, 167–174. [Google Scholar] [CrossRef]
- Chaudhuri, K.R. Crystallisation within transdermal rotigotine patch: Is there cause for concern? Expert Opin. Drug Deliv. 2008, 5, 1169–1171. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease With the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef]
- Johnson, W.; Onuma, O.; Owolabi, M.; Sachdev, S. Stroke: A global response is needed. Bull. World Health Organ. 2016, 94, 634. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418. [Google Scholar] [CrossRef]
- Hemphill, J.C., III; Greenberg, S.M.; Anderson, C.S.; Becker, K.; Bendok, B.R.; Cushman, M.; Fung, G.L.; Goldstein, J.N.; Macdonald, R.L.; Mitchell, P.H.; et al. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2015, 46, 2032–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.Y.; Zhu, Y.X.; Bu, J.Y.; Li, G.W.; Zhou, J.H.; Zhou, S.P. Evaluation of Neuroprotective Effect of Thymoquinone Nanoformulation in the Rodent Cerebral Ischemia-Reperfusion Model. Biomed. Res. Int. 2016, 2016, 2571060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
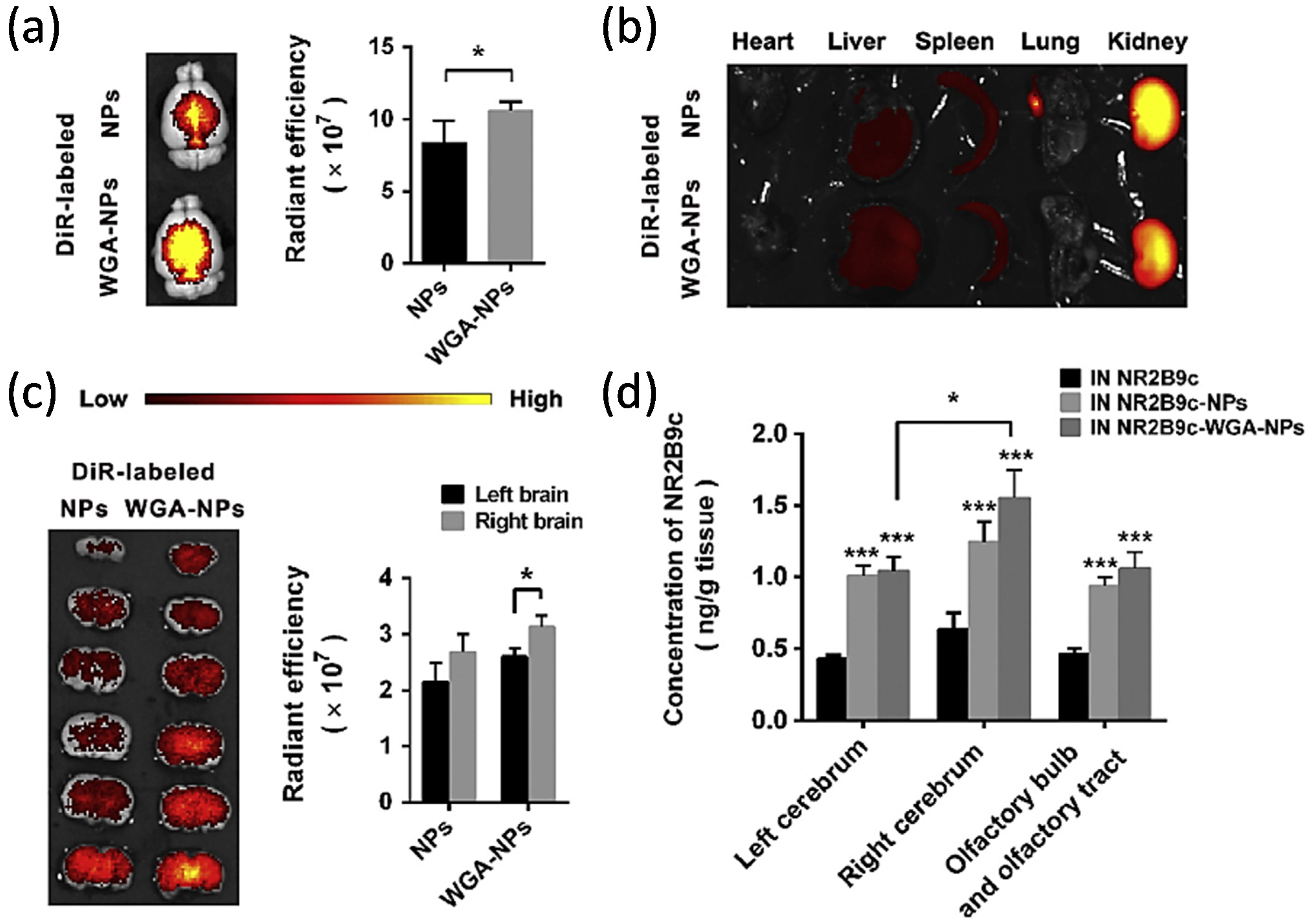
- Li, R.; Huang, Y.; Chen, L.; Zhou, H.; Zhang, M.; Chang, L.; Shen, H.; Zhou, M.; Su, P.; Zhu, D. Targeted delivery of intranasally administered nanoparticles-mediated neuroprotective peptide NR2B9c to brain and neuron for treatment of ischemic stroke. Nanomedicine 2019, 18, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Ahmad, I.; Umar, S.; Iqbal, Z.; Samim, M.; Ahmad, F.J. PNIPAM nanoparticles for targeted and enhanced nose-to-brain delivery of curcuminoids: UPLC/ESI-Q-ToF-MS/MS-based pharmacokinetics and pharmacodynamic evaluation in cerebral ischemia model. Drug Deliv. 2016, 23, 2095–2114. [Google Scholar] [CrossRef]
- Goyal, S.N.; Prajapati, C.P.; Gore, P.R.; Patil, C.R.; Mahajan, U.B.; Sharma, C.; Talla, S.P.; Ojha, S.K. Therapeutic Potential and Pharmaceutical Development of Thymoquinone: A Multitargeted Molecule of Natural Origin. Front. Pharmacol. 2017, 8, 656. [Google Scholar] [CrossRef]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Baker, H.; Spencer, R.F. Transneuronal transport of peroxidase-conjugated wheat germ agglutinin (WGA-HRP) from the olfactory epithelium to the brain of the adult rat. Exp. Brain Res. 1986, 63, 461–473. [Google Scholar] [CrossRef]
- McGrath, J.; Saha, S.; Chant, D.; Welham, J. Schizophrenia: A concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 2008, 30, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Crismon, M.; Smith, T.; Buckley, P.F. Schizophrenia. In Pharmacotherapy: A Pathophysiologic Approach, 11e; DiPiro, J.T., Yee, G.C., Posey, L., Haines, S.T., Nolin, T.D., Ellingrod, V., Eds.; McGraw Hill: New York, NY, USA, 2020. [Google Scholar]
- Chokhawala, K.; Stevens, L. Antipsychotic Medications. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- De Araújo, A.N.; de Sena, E.P.; de Oliveira, I.R.; Juruena, M.F. Antipsychotic agents: Efficacy and safety in schizophrenia. Drug Healthc. Patient Saf. 2012, 4, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroney, M. An update on current treatment strategies and emerging agents for the management of schizophrenia. Am. J. Manag. Care 2020, 26, S55–S61. [Google Scholar] [CrossRef]
- Tan, M.S.A.; Parekh, H.S.; Pandey, P.; Siskind, D.J.; Falconer, J.R. Nose-to-brain delivery of antipsychotics using nanotechnology: A review. Expert Opin. Drug Deliv. 2020, 17, 839–853. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Khunt, D.; Misra, M.; Padh, H. Application of Box-Behnken design for optimization and development of quetiapine fumarate loaded chitosan nanoparticles for brain delivery via intranasal route*. Int. J. Biol. Macromol. 2016, 89, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Boche, M.; Pokharkar, V. Quetiapine Nanoemulsion for Intranasal Drug Delivery: Evaluation of Brain-Targeting Efficiency. AAPS PharmSciTech 2017, 18, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Sawant, K.; Pandey, A.; Patel, S. Aripiprazole loaded poly(caprolactone) nanoparticles: Optimization and in vivo pharmacokinetics. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 66, 230–243. [Google Scholar] [CrossRef]
- Fonseca, F.N.; Betti, A.H.; Carvalho, F.C.; Gremião, M.P.; Dimer, F.A.; Guterres, S.S.; Tebaldi, M.L.; Rates, S.M.; Pohlmann, A.R. Mucoadhesive Amphiphilic Methacrylic Copolymer-Functionalized Poly(ε-caprolactone) Nanocapsules for Nose-to-Brain Delivery of Olanzapine. J. Biomed. Nanotechnol. 2015, 11, 1472–1481. [Google Scholar] [CrossRef]
- Seju, U.; Kumar, A.; Sawant, K.K. Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: In vitro and in vivo studies. Acta Biomater. 2011, 7, 4169–4176. [Google Scholar] [CrossRef]
- Kumar, M.; Misra, A.; Babbar, A.K.; Mishra, A.K.; Mishra, P.; Pathak, K. Intranasal nanoemulsion based brain targeting drug delivery system of risperidone. Int. J. Pharm. 2008, 358, 285–291. [Google Scholar] [CrossRef]
- Bahadur, S.; Pathak, K. Buffered nanoemulsion for nose to brain delivery of ziprasidone hydrochloride: Preformulation and pharmacodynamic evaluation. Curr. Drug Deliv. 2012, 9, 596–607. [Google Scholar] [CrossRef]
- Horacek, J.; Bubenikova-Valesova, V.; Kopecek, M.; Palenicek, T.; Dockery, C.; Mohr, P.; Höschl, C. Mechanism of action of atypical antipsychotic drugs and the neurobiology of schizophrenia. CNS Drugs 2006, 20, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Narala, A.; Veerabrahma, K. Preparation, Characterization and Evaluation of Quetiapine Fumarate Solid Lipid Nanoparticles to Improve the Oral Bioavailability. J. Pharm. 2013, 2013, 265741. [Google Scholar] [CrossRef] [PubMed]
- Emmert, D.; Campos, C.R.; Ward, D.; Lu, P.; Namanja, H.A.; Bohn, K.; Miller, D.S.; Sharom, F.J.; Chmielewski, J.; Hrycyna, C.A. Reversible dimers of the atypical antipsychotic quetiapine inhibit p-glycoprotein-mediated efflux in vitro with increased binding affinity and in situ at the blood-brain barrier. ACS Chem. Neurosci. 2014, 5, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Silki; Sinha, V.R. Enhancement of In Vivo Efficacy and Oral Bioavailability of Aripiprazole with Solid Lipid Nanoparticles. AAPS PharmSciTech 2018, 19, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Bassi, A.K.; Gough, J.E.; Zakikhani, M.; Downes, S. The Chemical and Physical Properties of Poly(ε-caprolactone) Scaffolds Functionalised with Poly(vinyl phosphonic acid-co-acrylic acid). J. Tissue Eng. 2011, 2011, 615328. [Google Scholar] [CrossRef]
- Mena, A.; Ruiz-Salas, J.C.; Puentes, A.; Dorado, I.; Ruiz-Veguilla, M.; De la Casa, L.G. Reduced Prepulse Inhibition as a Biomarker of Schizophrenia. Front. Behav. Neurosci. 2016, 10, 202. [Google Scholar] [CrossRef] [Green Version]
- Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [CrossRef] [Green Version]
- Shadrina, M.; Bondarenko, E.A.; Slominsky, P.A. Genetics Factors in Major Depression Disease. Front. Psychiatry 2018, 9, 334. [Google Scholar] [CrossRef] [Green Version]
- VandenBerg, A.M. Major Depressive Disorder. In Pharmacotherapy: A Pathophysiologic Approach, 11e; DiPiro, J.T., Yee, G.C., Posey, L.M., Haines, S.T., Nolin, T.D., Ellingrod, V., Eds.; McGraw-Hill Education: New York, NY, USA, 2020. [Google Scholar]
- Al-Harbi, K.S. Treatment-resistant depression: Therapeutic trends, challenges, and future directions. Patient Prefer. Adherence 2012, 6, 369–388. [Google Scholar] [CrossRef] [Green Version]
- Demeule, M.; Régina, A.; Jodoin, J.; Laplante, A.; Dagenais, C.; Berthelet, F.; Moghrabi, A.; Béliveau, R. Drug transport to the brain: Key roles for the efflux pump P-glycoprotein in the blood–brain barrier. Vasc. Pharmacol. 2002, 38, 339–348. [Google Scholar] [CrossRef]
- Vitorino, C.; Silva, S.; Bicker, J.; Falcão, A.; Fortuna, A. Antidepressants and nose-to-brain delivery: Drivers, restraints, opportunities and challenges. Drug Discov. Today 2019, 24, 1911–1923. [Google Scholar] [CrossRef]
- Haque, S.; Md, S.; Fazil, M.; Kumar, M.; Sahni, J.K.; Ali, J.; Baboota, S. Venlafaxine loaded chitosan NPs for brain targeting: Pharmacokinetic and pharmacodynamic evaluation. Carbohydr. Polym. 2012, 89, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Tong, G.F.; Qin, N.; Sun, L.W. Development and evaluation of Desvenlafaxine loaded PLGA-chitosan nanoparticles for brain delivery. Saudi Pharm. J. 2017, 25, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Bari, N.K.; Fazil, M.; Hassan, M.Q.; Haider, M.R.; Gaba, B.; Narang, J.K.; Baboota, S.; Ali, J. Brain delivery of buspirone hydrochloride chitosan nanoparticles for the treatment of general anxiety disorder. Int. J. Biol. Macromol. 2015, 81, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Fatouh, A.M.; Elshafeey, A.H.; Abdelbary, A. Intranasal agomelatine solid lipid nanoparticles to enhance brain delivery: Formulation, optimization and in vivo pharmacokinetics. Drug Des. Devel. Ther. 2017, 11, 1815–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, S.M.; Grady, M.M.; Moret, C.; Briley, M. SNRIs: Their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005, 10, 732–747. [Google Scholar] [CrossRef] [PubMed]
- Bronowska, A.; Chilmonczyk, Z.; Leś, A.; Edvardsen, O.; Ostensen, R.; Sylte, I. Molecular dynamics of 5-HT1A and 5-HT2A serotonin receptors with methylated buspirone analogues. J. Comput. Aided. Mol. Des. 2001, 15, 1005–1023. [Google Scholar] [CrossRef]
- Mahmood, I.; Sahajwalla, C. Clinical pharmacokinetics and pharmacodynamics of buspirone, an anxiolytic drug. Clin. Pharmacokinet. 1999, 36, 277–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javia, A.; Thakkar, H. Intranasal delivery of tapentadol hydrochloride-loaded chitosan nanoparticles: Formulation, characterisation and its in vivo evaluation. J. Microencapsul. 2017, 34, 644–658. [Google Scholar] [CrossRef]
- Nigam, K.; Kaur, A.; Tyagi, A.; Manda, K.; Gabrani, R.; Dang, S. Baclofen-Loaded Poly (D,L-Lactide-Co-Glycolic Acid) Nanoparticles for Neuropathic Pain Management: In Vitro and In Vivo Evaluation. Rejuvenation Res. 2019, 22, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Gadhave, D.G.; Kokare, C.R. Nanostructured lipid carriers engineered for intranasal delivery of teriflunomide in multiple sclerosis: Optimization and in vivo studies. Drug Dev. Ind. Pharm. 2019, 45, 839–851. [Google Scholar] [CrossRef]
- Bhatt, R.; Singh, D.; Prakash, A.; Mishra, N. Development, characterization and nasal delivery of rosmarinic acid-loaded solid lipid nanoparticles for the effective management of Huntington’s disease. Drug Deliv. 2015, 22, 931–939. [Google Scholar] [CrossRef]
- Jain, K.; Sood, S.; Gowthamarajan, K. Optimization of artemether-loaded NLC for intranasal delivery using central composite design. Drug Deliv. 2015, 22, 940–954. [Google Scholar] [CrossRef] [Green Version]
- Nigam, K.; Kaur, A.; Tyagi, A.; Nematullah, M.; Khan, F.; Gabrani, R.; Dang, S. Nose-to-brain delivery of lamotrigine-loaded PLGA nanoparticles. Drug Deliv. Transl. Res. 2019, 9, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.S.; Patel, H.S.; Belgamwar, V.S.; Agrawal, A.; Tekade, A.R. Solid lipid nanoparticles of ondansetron HCl for intranasal delivery: Development, optimization and evaluation. J. Mater. Sci. Mater. Med. 2012, 23, 2163–2175. [Google Scholar] [CrossRef] [PubMed]
- Patil, G.B.; Surana, S.J. Bio-fabrication and statistical optimization of polysorbate 80 coated chitosan nanoparticles of tapentadol hydrochloride for central antinociceptive effect: In vitro-in vivo studies. Artif. Cells Nanomed. Biotechnol. 2017, 45, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.R.; Nag, K.; Shetti, A.N.; Krishnaveni, N. Tapentadol hydrochloride: A novel analgesic. Saudi J. Anaesth. 2013, 7, 322–326. [Google Scholar] [CrossRef]
- Ghanavatian, S.; Derian, A. Baclofen. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Peterson, L.K.; Fujinami, R.S. Inflammation, demyelination, neurodegeneration and neuroprotection in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2007, 184, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanasescu, R.; Evangelou, N.; Constantinescu, C.S. Role of oral teriflunomide in the management of multiple sclerosis. Neuropsychiatr. Dis. Treat 2013, 9, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Diamond, M.I. Huntington disease and the huntingtin protein. Prog. Mol. Biol. Transl. Sci. 2012, 107, 189–214. [Google Scholar] [CrossRef] [PubMed]
- Ghasemzadeh Rahbardar, M.; Hosseinzadeh, H. Effects of rosmarinic acid on nervous system disorders: An updated review. Naunyn Schmiedeberg Arch. Pharmacol. 2020, 393, 1779–1795. [Google Scholar] [CrossRef] [PubMed]
- Idro, R.; Marsh, K.; John, C.C.; Newton, C.R. Cerebral malaria: Mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr. Res. 2010, 68, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Padmakumar, S.; Jones, G.; Khorkova, O.; Hsiao, J.; Kim, J.; Bleier, B.S.; Amiji, M.M. Osmotic core-shell polymeric implant for sustained BDNF AntagoNAT delivery in CNS using minimally invasive nasal depot (MIND) approach. Biomaterials 2021, 276, 120989. [Google Scholar] [CrossRef]
- Padmakumar, S.; Jones, G.; Pawar, G.; Khorkova, O.; Hsiao, J.; Kim, J.; Amiji, M.M.; Bleier, B.S. Minimally Invasive Nasal Depot (MIND) technique for direct BDNF AntagoNAT delivery to the brain. J. Control. Release 2021, 331, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Majumder, J.; Taratula, O.; Minko, T. Nanocarrier-based systems for targeted and site specific therapeutic delivery. Adv. Drug Deliv. Rev. 2019, 144, 57–77. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Nanocarrier | Lipids | Surfactant/Co-Surfactant | Surface Modification | Size, nm | Z-Potential, mV | PDI | EE, % | DTE, % | DTP, % | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Letrozole | Nanoemulsion | Triacetin | Tween 80, | PEG-400 | 95.59 ± 2.34 | 0.162 ±0.012 | 97.37 ± 1.13 | - | - | [120] | |
| Carbamazepine | Nanoemulgel | Oleic acid | Labrasol | Xanthan gum | 45–146 | - | - | - | - | - | [155] |
| Diazepam | PLGA Nanoparticle | - | Poloxamer 407 | 183.2 | - | - | 87.8 | 258 | 61.3 | [156] | |
| Oxcarbazepine | PLGA Nanoparticle | - | Tween 80 | - | 256.16 ± 2.94 | −15.12 ± 0.36 | 0.144 ± 0.024 | 85.1 ± 2.1 | - | - | [157] |
| Emulsome | Triolein | Tween 80 | 120.4 ± 1.45 * 101.5 † | −34.1 ± 1.27 * −6.72 † | 0.36 * - | 81.19 ± 2.3 * - | 265.7 | 62.3 | [158] | ||
| Amiloride | Nanoemulsion | Oleic acid | Tween-80/Carbitol | 89.36 ± 11.18 | −9.83 ± 0.12 | 0.231 ± 0.018 | 98.28 ± 0.29 | 1993 ± 46 | 586.2 ± 11.6 | [159] | |
| Thyrotropin-release hormone | PLA Nanoparticle | - | PVA | - | 108 ± 12 | - | - | - | - | - | [160] |
| Lorazepam | PLGA Nanoparticle | - | Poloxamer 407 | 168.2 | −18.4 | 0.08 | 83.8 | [161] | |||
| Lamotrigine | Liposome | Phospholipon 90G/Cholesterol | Tween 80 | - | 88.90 ± 1.56 | - | 0.247 ± 0.04 | 68.75 ± 0.02 | - | - | [162] |
| Drug | Nano-Carrier | Lipids | Surfactant/Co-Surfactant | Surface Modification | Size, nm | Z-Potential, mV | PDI | EE, % | DTE, % | DTP, % | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tarenflurbil | PLGA Nanoparticle | - | PF-68 | - | 133.13 ± 7.82 | −30.25 ± 2.11 | 0.21 ± 0.02 | 64.11 ± 2.21 | 287.24 | 65.18 | [139] | ||
| SLN | Glyceryl monostearate/Stearic acid/Soy lecithin | Tween 20 | - | 169.87 ± 10.98 | −23.13 ± 2.32 | 0.24 ± 0.04 | 57.81 ± 5.32 | 183.15 | 45.41 | ||||
| Donepezil | Liposome | DSPC/Cholesterol | - | PEG | 102 ± 3.3 | -28.31 ± 0.85 | 0.28 ± 0.03 | 84.91 ± 3.31 | - | - | [99] | ||
| Chitosan Nanosuspension | - | - | - | 150–200 | - | 0.341 | 92–96 | - | - | [178] | |||
| PLGA Nanoparticle | - | PVA/Tween 80 | - | 89.67 ± 6.43 | −36 ± 1.05 | 0.013 ± 0.002 | 88.65 ± 2.51 | - | - | [179] | |||
| Galantamine | Thiolated Chitosan Nanoparticle | - | - | - | 148.2–150 | +27.2 | 0.216–0.225 | 87.65–89 | - | - | [180] | ||
| Liposome | SPC/Cholesterol | - | Propylene glycol | 112 ± 8 | −49.2 ± 0.7 | - | 83.6 ± 1.8 | - | - | [181] | |||
| SLN | Compritol 888 ATO | PF-127/Tween 80 | - | 92.0 ± 3.51 | −17.22 ± 1.1 | 0.380 ± 0.16 | 83.42 ± 0.63 | - | - | [182] | |||
| Memantine | Nanoemulsion | Labrasol | Tween 20/Propylene glycol | ~11 | -19.6 | 0.080 | - | 158.78 | 37.05 | [183] | |||
| Pioglitazone | NLC | Capmul MCM/Tripalmitin | PF 68/Tween 80 | - | 211.4 ± 3.54 | +14.9 ± 1.09 | 0.257 ± 0.108 | 70.18 ± 4.5 | - | - | [184] | ||
| Huperzine A | PLGA Nanoparticle | - | PVA | TMC/Lactoferrin | 153.2 ± 13.7 | +35.6 ± 5.2 | 0.229 ± 0.078 | 73.8 ± 5.7 | - | - | [185] | ||
| Rivastigmine | Chitosan Nanoparticle | - | - | - | 185.4 ± 8.4 | +38.4 ± 2.85 | 0.391 ± 0.065 | 85.3 ± 3.5 | 355 ± 13.5 | 71.80 ± 6.7 | [186] | ||
| Liposome | EPC/cholesterol | - | DSPE-PEG/CPP | 178.9 ± 11.7 | −8.6 ± 2.4 | 0.333 ± 0.032 | 30.5 ± 8.0 | - | - | [187] | |||
| BACE1 SiRNA | SLN | Witepsol E 85 | PVA | RVG-9R/Chitosan | 335.76 ± 34.81 * 358.44 ± 25.89 # | −17.31 ± 0.68 * + 10.54 ± 0.75 # | 0.013 ± 0.00 * 0.028 ± 0.02 # | - - | - | - | [188] | ||
| R-flurbiprofen | Albumin Nanoparticle | - | - | - | 284.4 ± 14.9 | - | 0.404 ± 0.065 | - | - | - | [189] | ||
| Drug | Nanocarrier | Lipids | Surfactant/Co-Surfactant | Surface Modification | Size, nm | Z-Potential, mV | PDI | EE, % | DTE, % | DTP, % | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Rasagiline | Chitosan glutamate Nanoparticle | - | - | - | 151.1 ± 10.31 | - | 0.380 ± 0.01 | 96.43 ± 4.23 | 325 ± 40 | 69.27 ± 2.1 | [200] |
| Dopamine | PLGA Nanoparticle | - | PVA | PEG/Borneol/Lactoferrin | 175.3 ± 9.6 | −15.7 ± 0.86 | 0.129± 0.011 | 25.43 ± 5.32 | - | - | [201] |
| Bromocriptine | Chitosan Nanoparticle | - | - | - | 161.3 ± 4.7 | +40.32 ± 2.78 | 0.44 ± 0.03 | 84.2 ± 3.5 | 633 ± 86.1 | 84.2 ± 1.9 | [202] |
| Rotigotine | Chitosan Nanoparticle | - | - | - | 75.37 ±3.37 | +25.53 ± 0.45 | 0.368 ± 0.02 | 96.08 ± 0.01 | 258.10 ±17.13 | 53.87 ± 10.14 | [203] |
| PLGA Nanoparticle | - | - | PEG/Lactoferrin | 122.0 ± 19.3 | −21.28 ± 2.15 | 0.194 ± 0.023 | 92.57 ± 9.41 | - | - | [204] | |
| Selegiline | NLC | Stearylamine/Olive oil | PF 68 Tween 80 | - | 133 ± 6.08 | - | 0.357 ± 0.06 | 93 ± 5.25 | - | - | [205] |
| Nanoemulsion | Grape seed oil/Sefsol 218 | Tween 80/Lauroglycol 90 | - | 61.43 ± 4.10 | -34 | - | - | - | - | [206] | |
| Basic fibroblast growth factor | NLC | Gelatin | Poloxamer 188 | - | 172 ± 1.31 | −27.6 ± 1.1 | 0.105 ± 0.011 | 86.7 ± 1.1 | - | - | [91] |
| Geraniol/ursodeoxycholic acid conjugate | SLN | Compritol ATO 888 | Span 85 | - | 121 ± 8.4 | −22.5 ± 7.7 | 0.164 ± 0.03 | 94.5 ± 2.6 | - | - | [207] |
| Ropinirole | SLN | Dynasan 114/Stearylamine | PF 68/Soy lecithin | - | 66.22 ± 6.22 | +28.19 ± 3.02 | 0.023 ± 0.21 | 61.90 ± 0.18 | - | - | [208] |
| Chitosan Nanoparticle | - | - | - | 173.7 ± 2.32 | +32.7 ± 1.5 | 0.39 ± 0.03 | 69.6 ± 3.3 | - | - | [209] | |
| PLGA Nanoparticle | - | TPGS | - | 279.4 ± 1.8 | -29.4 ± 2.6 | 0.329 ± 0.09 | 72.3 ± 6.1 | - | - | [210] | |
| Pramipexole | Chitosan Nanoparticle | - | - | - | 292.5 ± 8.80 | +14.0 ± 2.89 | 0.292 | 91.25 ± 0.95 | - | - | [211] |
| Naringenin | Nanoemulsion | Vitamin E Capryol 90 | Tween 80/Transcutol-HP | - | 38.70 ± 3.11 | − 27.4 ± 0.14 | 0.14 ± 0.0024 | - | 822.71 ± 9.14 | 72.14 ± 5.87 | [212] |
| Urocortin | PLGA Nanoparticle | - | Sodium cholate | OL/PEG | 114.8 ± 5.6 | −24.7 ± 1.5 | 0.193 | 75.5 ± 0.8 | - | - | [213] |
| Drug | Nanocarrier | Surfactant/Co-Surfactant | Surface Modification | Size, nm | Z-potential, mV | PDI | EE, % | DTE, % | DTP, % | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Thymoquinone | PLGA Nanoparticle | - | Chitosan | 183.5 ± 8.2 | +33.63 ± 2.25 | 0.257 ± 0.02 | 73.2 ± 2.6 | 524.17 | 80.47 | [225] |
| NR2B9c | PLGA Nanoparticle | Sodium cholate | WGA/PEG | ~139 | −23.3 | <0.2 | ~50 | ~150 | 78.58 | [226] |
| Curcumin | PNIPAM Nanoparticle | - | - | 92.46 ± 2.8 | -16.2 ± 1.42 | 0.191 ± 0.052 | 84.63 ± 4.2 | 659.23 ± 83.59 | 84.03 ± 1.81 | [227] |
| Demethoxycurcumin | - | - | 91.23 ± 4.2 | -15.6 ± 1.33 | 0.183 ± 0.063 | 84.71 ± 3.99 | 667.84 ± 85.12 | 85.23 ± 2.19 | ||
| Bisdemethoxycurcumin | - | - | 94.28 ± 1.91 | −16.6 ± 1.21 | 0.142 ± 0.046 | 85.73 ± 4.31 | 677.12 ± 289.99 | 85.47 ± 2.49 |
| Drug | Nanocarrier | Lipids | Surfactant/Co-Surfactant | Surface Modification | Size, nm | Z-Potential, mV | PDI | EE, % | DTE, % | DTP, % | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Quetiapine | Chitosan Nanoparticle | - | - | - | 131.08 ± 7.45 | +34.4 ± 1.87 | 0.252 ± 0.064 | 89.93 ± 3.85 | 374.93 ± 15.02 | 73.33 ± 4.14 | [237] |
| Nanoemulsion | Capmul MCM | Tween 80/Transcutol | - | 144 ± 0.5 | −8.131 ± 1.8 | 0.193 ± 0.04 | 91 ± 0.3 | 267.98 ± 3.06 | 63.63 | [238] | |
| Aripiprazole | PCL Nanoparticle | - | Poloxamer 188/Poloxamer 407 | - | 199.2 ± 5.65 | −21.4 ± 4.6 | - | 69.2 ± 2.34 | 64.11 ± 4.68 | 74.34 ± 3.76 | [239] |
| Olanzapine | PCL Nanoparticle | - | - | MMA/DMAEMA | 254.9 ± 12.1 | +22.2 ± 1.2 | 0.03 ± 0.01 | 99.00 ± 0.05 | - | - | [240] |
| PLGA Nanoparticle | - | Poloxamer 407 | - | 91.2 ± 5.2 | −23.7 ± 2.1 | 0.120 ± 0.018 | 68.91 ± 2.31 | - | - | [241] | |
| Risperidone | SLN | Compritol 888 ATO | PF-127 | - | 148.05 ± 0.85 | −25.35 ± 0.45 | 0.148 ± 0.028 | 59.65 ± 1.18 | - | - | [111] |
| Nanoemulsion | Capmul MCM | Tween 80/Transcutol/Propylene glycol | - | 16.7 ± 1.21 | −9.15 ± 2.14 | 0.19 ± 0.04 | 98.86 ± 1.21 | 476 ± 0.14 | 78 ± 1.31 | [242] | |
| Ziprasidone | Nanoemulsion | Capmul MCM | Labrasol/Transcutol | - | 145.24 ± 4.75 | −30.2 ± 3.21 | 0.186 ± 0.40 | - | - | - | [243] |
| Drug | Nanocarrier | Lipids | Surfactant/Co-Surfactant | Surface Modification | Size, nm | Z-Potential, mV | PDI | EE, % | DTE, % | DTP, % | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Venlafaxine | Chitosan Nanoparticle | - | - | - | 167 ± 6.5 | +23.83± 1.76 | 0.367 ± 0.045 | 79.3 ± 2.6 | 508.59 | 80.34 | [256] |
| Alginate Nanoparticle | - | - | - | 173.7 ± 2.5 | +37.40 ± 1.74 | 0.391 ± 0.045 | 81.3 ± 1.9 | 425.77 | 76.52 | [134] | |
| Desvenlafaxine | PLGA Nanoparticle | - | - | Chitosan | 172.5 ± 10.2 | +35.63 ± 8.25 | 0.254 ± 0.02 | 76.4 ± 4.2 | 544.23 | 81.62 | [257] |
| Buspirone | TC Nanoparticle | - | - | - | 226.7 ± 2.52 | - | 0.483 ± 0.031 | 81.13 ± 2.8 | 78.94 ± 15.31 | 95.97 ± 11.31 | [258] |
| Agomelatine | SLN | Gelucire 43/01 | PVA/SDC | - | 167.70 ± 0.42 | −17.90 ± 2.70 | 0.12 ± 0.1 | 91.25 ± 1.7 | 190.02 | 47.37 | [259] |
| Selegiline | TC Nanoparticle | - | - | - | 215 ± 34.71 | +17.06 | 0.214 ± 0.0421 | 70 ± 2.71 | - | - | [131] |
| Drug | Nanocarrier | Lipids | Surfactant/Co-Surfactant | Surface Modification | Size, nm | Z-Potential, mV | PDI | EE, % | DTE, % | DTP, % | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tapentadol | Chitosan Nanoparticle | - | - | - | 201.2 ± 1.5 | +49.3 ± 1.2 | 0.201 ± 0.01 | 63.49 ± 1.61 | 321 ± 60 | 68.85 ± 0.49 | [263] |
| Baclofen | PLGA Nanoparticle | - | Poloxamer 407 | - | 124.8 | −20.4 | 0.225 | 86.45 ± 1.65 | 183.85 | 45.92 | [264] |
| Teriflunomide | NLC | Compritol 888 ATO/Maisine 35-1/ | Gelucire 44/14/Tween 20 | - | 99.82 ± 1.36 | −22.29 ± 1.8 | 0.35 ± 0.01 | 83.39 ± 1.24 | - | - | [265] |
| Rosmarinic acid | SLN | HSPC | Soya lecithin/Tween 80 | - | 149.2 ± 18.2 | −38.27 | 0.290 ± 0.021 | 61.9 ± 2.2 | - | - | [266] |
| Artemether | NLC | TM/MCT | PF 68 | - | 123.4 ± 3.6 | −34.4 ± 1.2 | - | 91.2 ± 2.5 | 278.16 | 64.02 | [267] |
| Leucine-enkephalin | TMC Nanoparticle | - | - | - | 443 ± 23 | +15 ± 2 | 0.317 ± 0.17 | 78.28 ±3.8 | - | - | [130] |
| Cyclobenzaprine | TC Nanoparticle | - | SDC | - | 282.9 ± 15.6 | +27.7 ± 1.2 | - | 80.20 ± 3.2 | 2101 | 95.24 | [132] |
| Lamotrigine | PLGA Nanoparticle | - | Poloxamer 407 | - | 184.6 | −18.8 | 0.082 | 84.87 ± 1.2 | 129.81 | 22.96 | [268] |
| Ondansetron | SLN | Glycerol monostearate | Lecithin/Poloxamer 188 | - | 299.67 | −16.5 | 0.296 | 49.82 | - | - | [269] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.; Minko, T. Nanotherapeutics for Nose-to-Brain Drug Delivery: An Approach to Bypass the Blood Brain Barrier. Pharmaceutics 2021, 13, 2049. https://doi.org/10.3390/pharmaceutics13122049
Lee D, Minko T. Nanotherapeutics for Nose-to-Brain Drug Delivery: An Approach to Bypass the Blood Brain Barrier. Pharmaceutics. 2021; 13(12):2049. https://doi.org/10.3390/pharmaceutics13122049
Chicago/Turabian StyleLee, David, and Tamara Minko. 2021. "Nanotherapeutics for Nose-to-Brain Drug Delivery: An Approach to Bypass the Blood Brain Barrier" Pharmaceutics 13, no. 12: 2049. https://doi.org/10.3390/pharmaceutics13122049