
Direct Delivery of ANA-TA9, a Peptide Capable of Aβ Hydrolysis, to the Brain by Intranasal Administration

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Purification of ANA-TA9
2.3. Evaluation of the Disposition and Nasal Absorption of ANA-TA9
2.3.1. Preparation of Dosing Solutions
2.3.2. Animal Study
2.3.3. Degradation of ANA-TA9 in the Rat Nasal Cavity
2.4. In Vitro Stability in the Plasma, Whole Blood, and Cerebrospinal Fluid
2.4.1. Preparation of the Whole Blood, Plasma, and Cerebrospinal Fluid
2.4.2. Stability of ANA-TA9 in the Plasma, Whole Blood, and Cerebrospinal Fluid
2.5. Uptake of ANA-TA9 to the Cerebrospinal Fluid after Nasal Administration
2.6. In Vivo Brain Distribution Study
2.6.1. Preparation of Dosing Solutions
2.6.2. Animal Study
2.7. Assays of ANA-TA9
2.8. Calculation of the Pharmacokinetic Parameters
2.9. Calculation of Direct Transport Percentage (DTP)
2.10. Data Analysis
3. Results
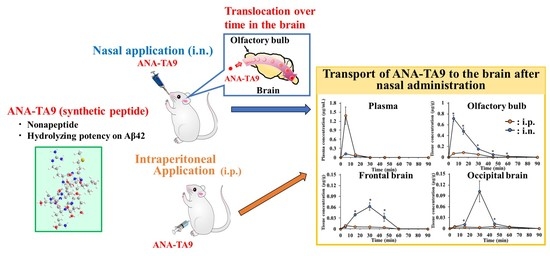
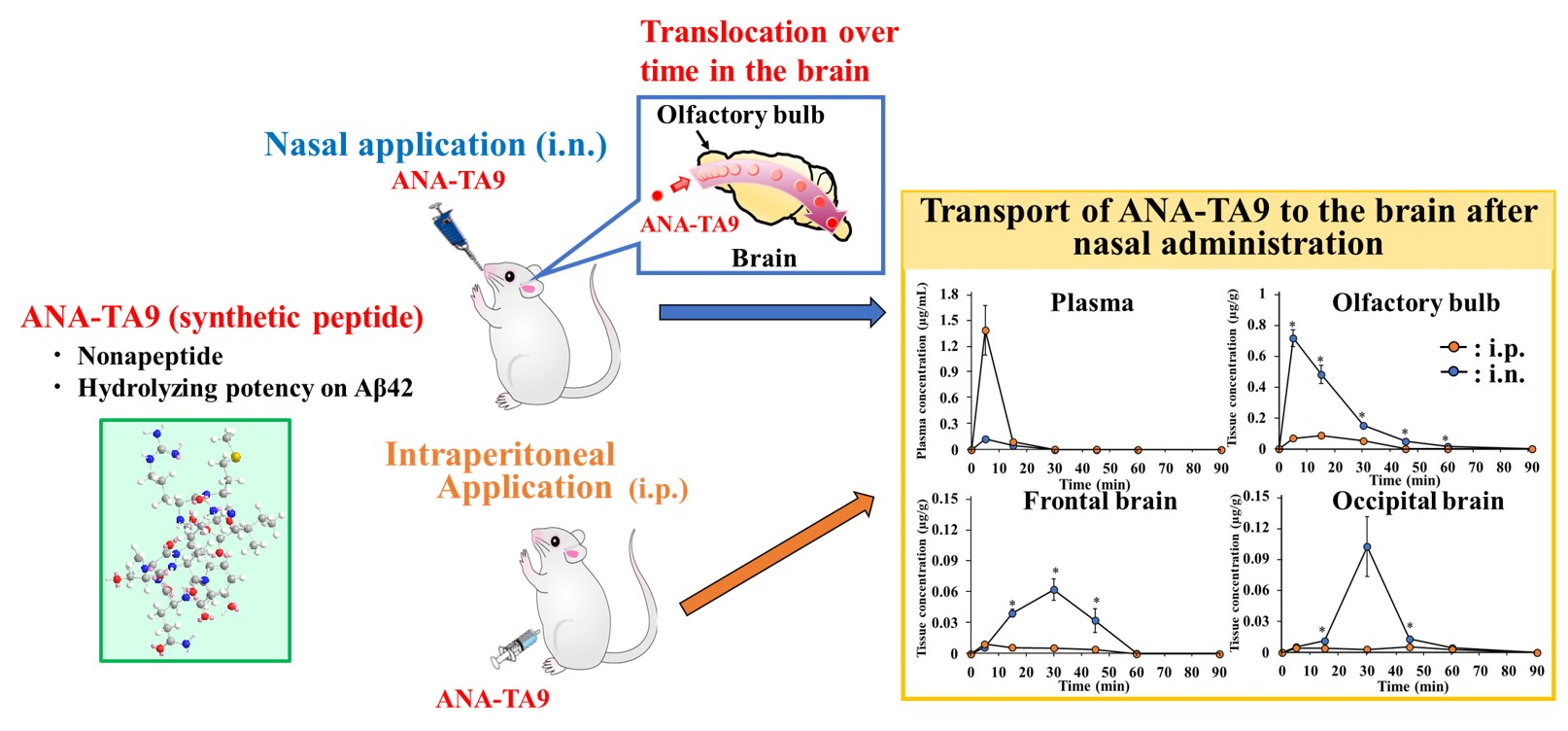
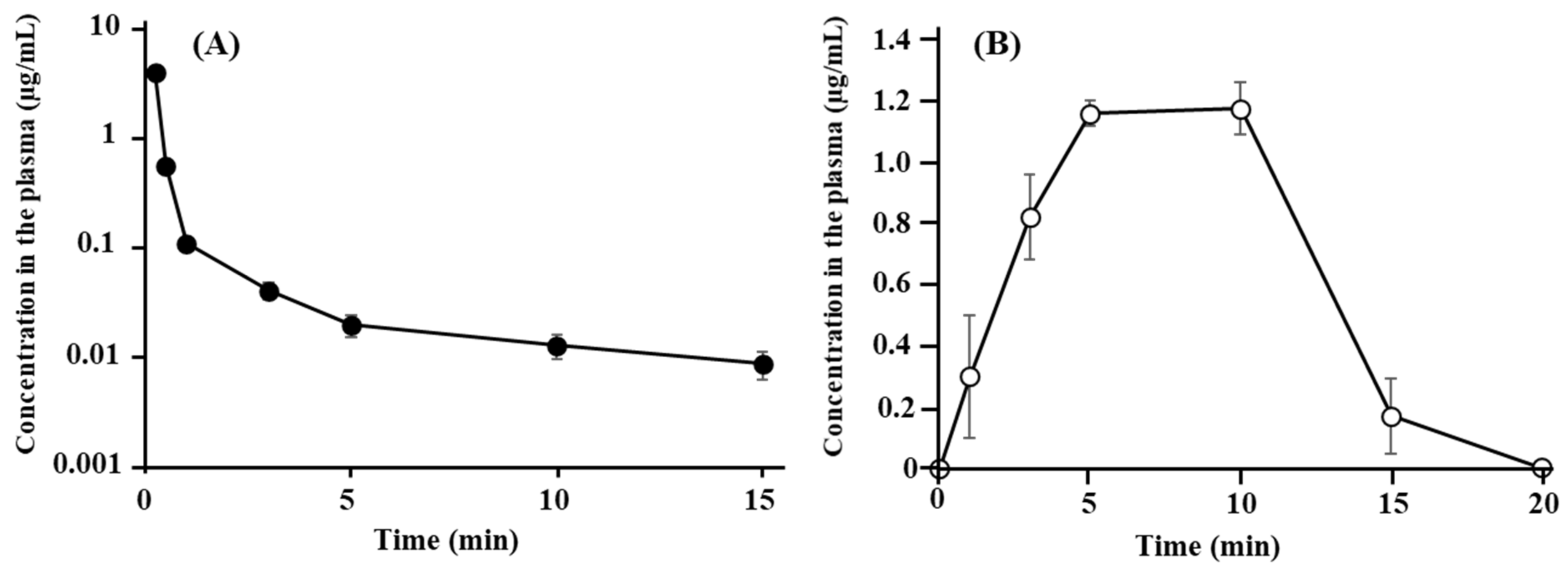
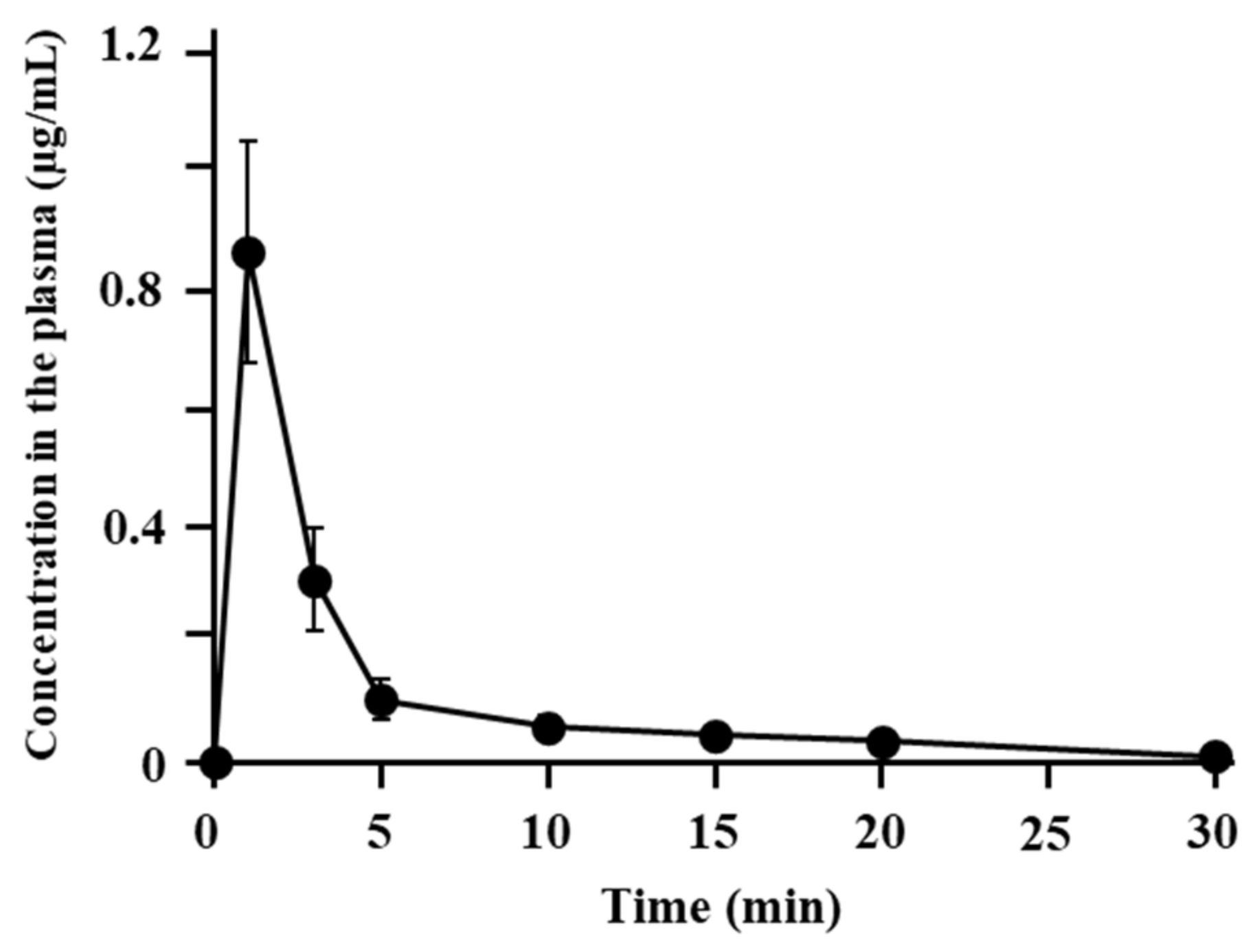
3.1. Disposition and Nasal Absorption of ANA-TA9 in Rats
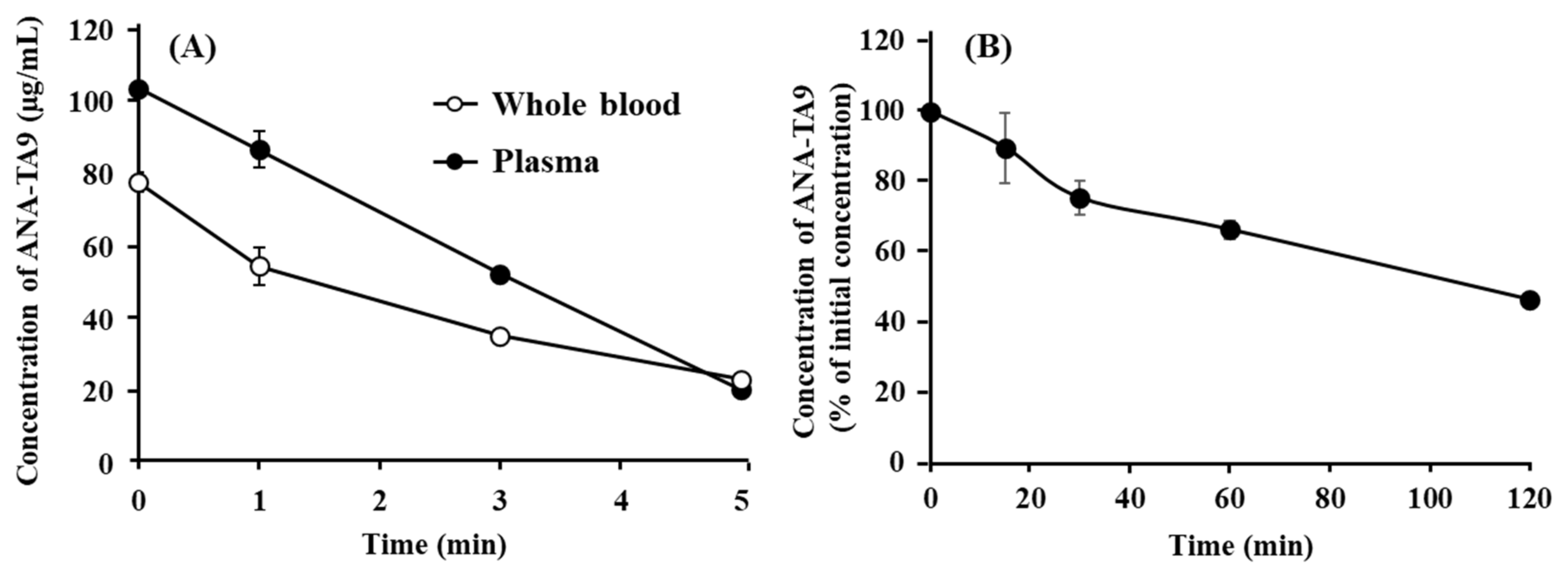
3.2. Degradation of ANA-TA9 in the Plasma, Whole Blood, and Cerebrospinal Fluid
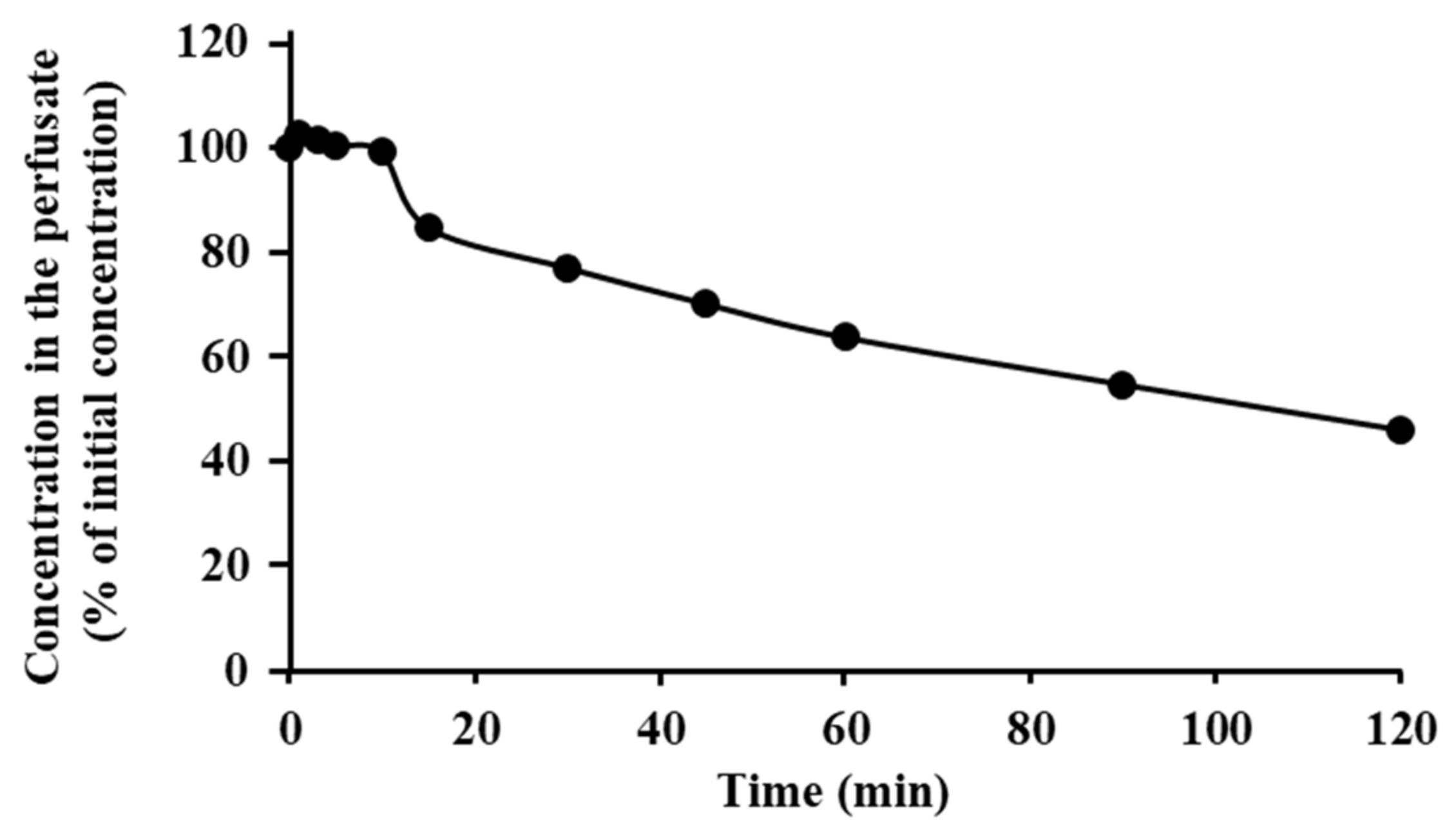
3.3. Degradation of ANA-TA9 on the Surface of Nasal Epithelial Cells
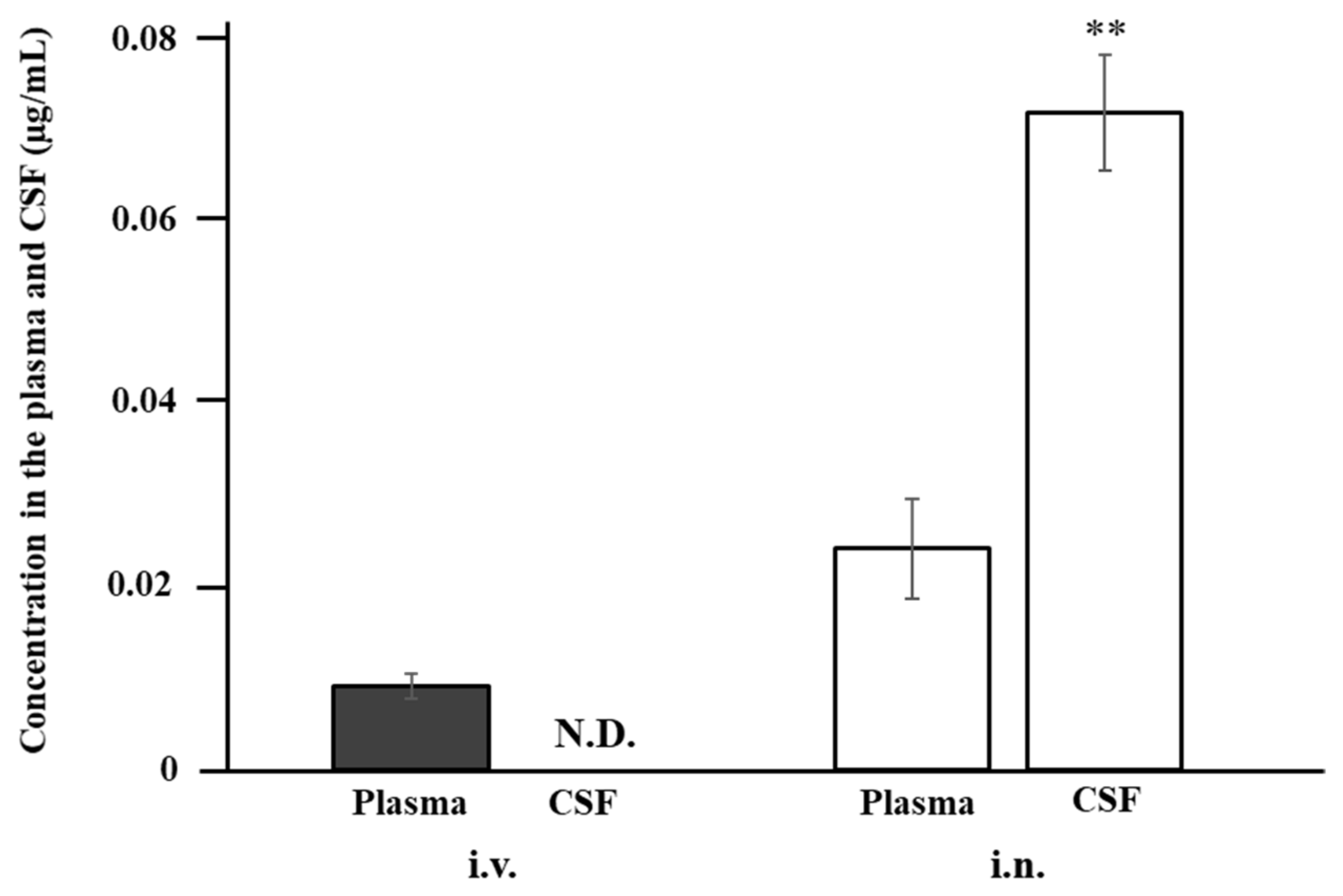
3.4. Uptake of ANA-TA9 by the Cerebrospinal Fluid after Nasal Administration to Rats
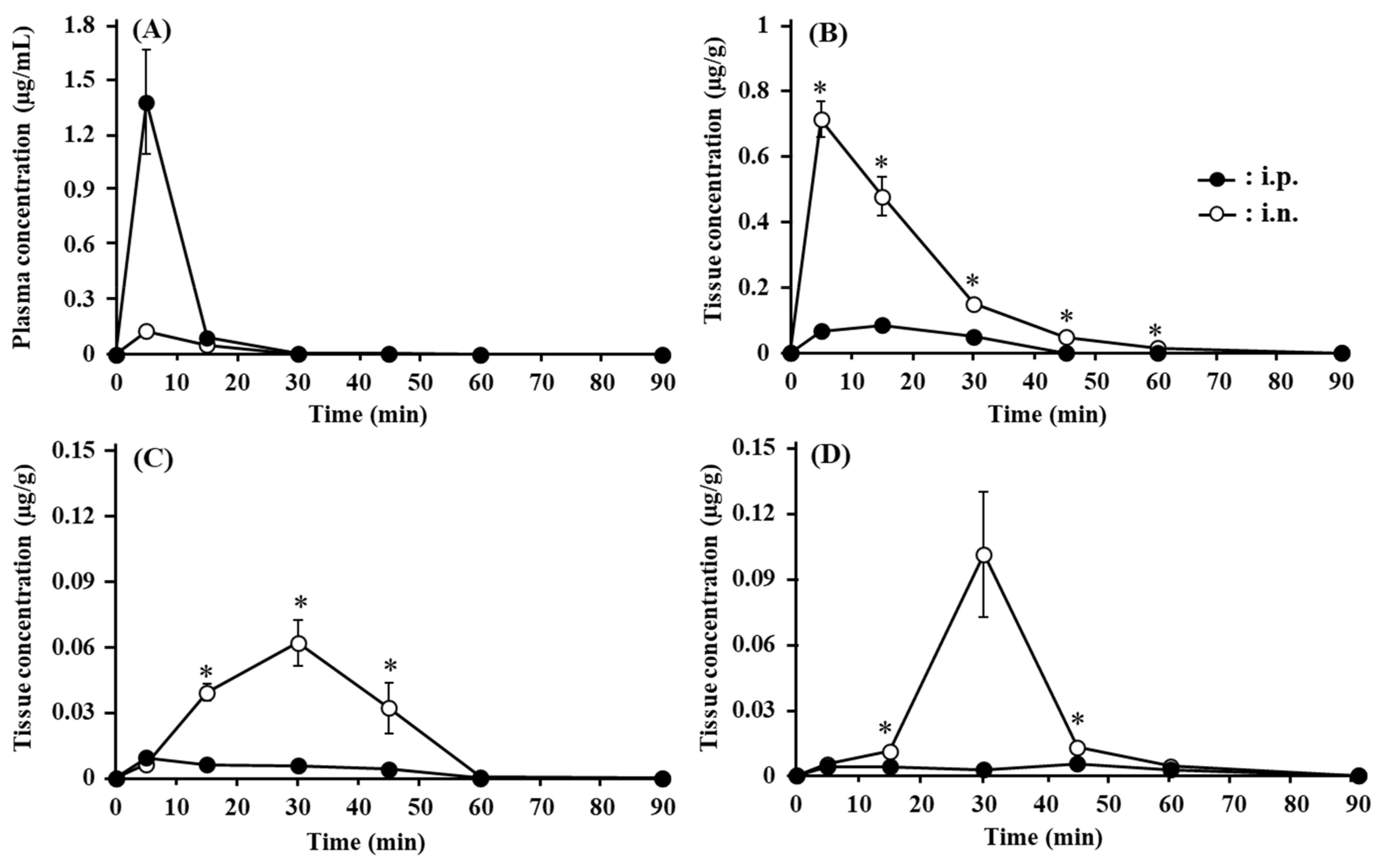
3.5. Transport of ANA-TA9 to the Brain after Nasal Administration to Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crous-Bou, M.; Minguillon, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer‘s disease prevention: From risk factors to early intervention. Alzheimers Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef]
- Jason, W.; Andrew, B. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar]
- Martin, P.; Renata, B.; Emiliano, A.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 2013, 9, 63–75. [Google Scholar]
- Benilova, I.; Karran, E.; Strooper, B.D. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H. Alzheimer’s Disease and the β-Amyloid Peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Chen, W.D.; Wang, Y.D. β-Amyloid: The key peptide in the pathogenesis of Alzheimer’s disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef] [Green Version]
- Karran, E.; Mercken, M.; Strooper, B.D. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Smith, S.O. Structural conversion of neurotoxic amyloid-β (1-42) oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Hamley, I.W. The amyloid beta peptide: A chemist’s perspective. role in Alzheimer’s and fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef]
- Luhrs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Riek, R. 3D structure of Alzheimer’s amyloid-β (1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esler, W.P.; Wolfe, M.S. A portrait of Alzheimer secretases--New features and familiar faces. Science 2001, 293, 1449–1454. [Google Scholar] [CrossRef]
- Kotler, S.A.; Brender, J.R.; Vivekanandan, S.; Suzuki, Y.; Yamamoto, K.; Monette, M.; Ramamoorthy, A. High-resolution NMR characterization of low abundance oligomers of amyloid-β without purification. Sci. Rep. 2015, 5, 11811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotler, S.A.; Walsh, P.; Brender, J.R.; Ramamoorthy, A. Differences between amyloid-β aggregation in solution and on the membrane: Insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6692–6700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maia, M.A.; Sousa, E. BACE-1 and γ-Secretase as Therapeutic Targets for Alzheimer’s Disease. Pharmaceuticals 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Mohs, R. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Eng. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef]
- Sato, T.; Kienlen-Campard, P.; Ahmed, M.; Liu, W.; Li, H.; Elliott, J.I.; Smith, S.O. Inhibitors of amyloid toxicity based on β-sheet and reduce cytotoxicity. Biochemistry 2006, 45, 5503–5516. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.; Namsechi, R.; Sim, L.V. Structure-Based Peptide Design to Modulate Amyloid Beta Aggregation and Reduce Cytotoxicity. PLoS ONE 2015, 10, e0129087. [Google Scholar] [CrossRef] [Green Version]
- Young, L.M.; Saunders, J.C.; Mahood, R.A.; Revill, C.H.; Foster, R.J.; Tu, L.H.; Ashcroft, A.E. Screening and classifying small-molecule inhibitors of amyloid formation using ion mobility spectrometry-mass spectrometry. Nat. Chem. 2015, 7, 73–81. [Google Scholar] [CrossRef]
- Crouch, P.J.; Tew, D.J.; Du, T.; Nguyen, D.N.; Caragounis, A.; Filiz, G.; White, A.R. Restored degradation of the Alzheimer’s amyloid-β peptide by targeting amyloid formation. J. Neurochem. 2009, 108, 1198–1207. [Google Scholar] [CrossRef]
- Storr, T.; Merkel, M.; Song-Zhao, G.X.; Scott, L.E.; Green, D.E.; Bowen, M.L.; Orvig, C. Synthesis, characterization, and metal coordinating ability of multifunctional carbohydrate-containing compounds for Alzheimer’s therapy. J. Am. Chem. Soc. 2007, 129, 7453–7463. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Sandrock, A. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Konishi, M.; Taniguchi, M.; Hatakawa, Y.; Akizawa, T. The discovery of shorter synthetic proteolytic peptides derived from Tob1 protein. Peptides 2019, 116, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Konishi, M.; Hatakawa, Y.; Saito, M.; Akizawa, T. The novel catalytic peptide, a synthetic nona-peptide (JAL-TA9) derived from Tob1 protein, digests the amyloid-β peptide. J. Royal Sci. 2019, 1, 30–35. [Google Scholar]
- Hatakawa, Y.; Nakamura, R.; Konishi, M.; Sakane, T.; Saito, M.; Akizawa, T. Catalytides derived from the Box A region in the ANA/BTG3 protein cleave amyloid-β fragment peptide. Heliyon 2019, 5, e02454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Furubayashi, T.; Arai, M.; Inoue, D.; Kimura, S.; Kiriyama, A.; Yamamoto, A. Delivery of Oxytocin to the Brain for the Treatment of Autism Spectrum Disorder by Nasal Application. Mol. Pharm. 2018, 15, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Takayama, K.; Furubayashi, T.; Mori, K.; Takemura, Y.; Amano, M.; Yamamoto, A. Transnasal Delivery of the Peptide Agonist Specific to Neuromedin-U Receptor 2 to the Brain for the Treatment of Obesity. Mol. Pharm. 2020, 17, 32–39. [Google Scholar] [CrossRef]
- Kojima, A.; Konishi, M.; Akizawa, T. Prion fragment peptides are digested with membrane type matrix metalloproteinases and acquire enzyme resistance through Cu2+-binding. Biomolecules 2014, 4, 510–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinichiro, H.; Takatsuka, Y.; Tai, M.; Hiroyuki, M. Absorption of drugs from the nasal mucosa of rat. Int. J. Pharm. 1981, 7, 317–325. [Google Scholar] [CrossRef]
- Chou, R.C.; Levy, G. Effect of heparin or salicylate infusion on serum protein binding and on concentrations of phenytoin in serum, brain and cerebrospinal fluid of rats. J. Pharmacol. Exp. Ther. 1981, 219, 42–49. [Google Scholar]
- Fukuda, M.; Kanazawa, T.; Iioka, S.; Oguma, T.; Iwasa, R.; Masuoka, S.; Suzuki, T. Quantitative analysis of inulin distribution in the brain focused on nose-to-brain route via olfactory epithelium by reverse esophageal cannulation. J. Control. Release 2021, 332, 493–501. [Google Scholar] [CrossRef]
- Sasaki, Y.; Wagner, H.N.J. Measurement of the distribution of cardiac output in unanesthetized rats. J. Appl. Physiol. 1971, 30, 879–884. [Google Scholar] [CrossRef]
- Gartziandia, O.; Egusquiaguirre, S.P.; Bianco, J.; Pedraz, J.L.; Igartua, M.; Hernandez, R.M.; Beloqui, A. Nanoparticle transport across in vitro olfactory cell monolayers. Int. J. Pharm. 2016, 499, 81–89. [Google Scholar] [CrossRef]
- Singh, K.; Ahmad, Z.; Shakya, P.; Ansari, V.A.; Kumar, A.; Zishan, M.; Arif, M. Nano formulation: A novel approach for nose to brain drug delivery. J. Chem. Pharm. Res. 2016, 8, 208–215. [Google Scholar]
- Kulkarni, A.D.; Vanjari, Y.H.; Sancheti, K.H.; Belgamwar, V.S.; Surana, S.J.; Pardeshi, C.V. Nanotechnology-mediated nose to brain drug delivery for Parkinson’s disease: A mini review. J. Drug Target. 2015, 23, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Sakane, T.; Akizuki, M.; Yamashita, S.; Nadai, T.; Hashida, M.; Sezaki, H. Transport of cephalexin to the cerebrospinal fluid directly from the nasal cavity. J. Pharm. Pharmacol. 1991, 43, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Frey II, W.H.; Liu, J.; Chen, X.; Thorne, R.G.; Fawcett, J.R.; Ala, T.A.; Rahman, Y.E. Delivery of 125 I-NGF to the Brain via the Olfactory Route. Drug Deliv. 1997, 4, 87–92. [Google Scholar] [CrossRef]
- Zada, M.H.; Kubek, M.; Khan, W.; Kumar, A.; Domb, A. Dispersible hydrolytically sensitive nanoparticles for nasal delivery of thyrotropin releasing hormone (TRH). J. Control. Release 2019, 295, 278–289. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, J.C.; Sosa-Teste, I. The nasal route as a potential pathway for delivery of erythropoietin in the treatment of acute ischemic stroke in humans. Sci. World J. 2009, 9, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, N.; Farr, S.A.; Kageyama, H.; Shioda, S.; Banks, W.A. Delivery of galanin-like peptide to the brain: Targeting with intranasal delivery and cyclodextrins. J. Pharmacol. Exp. Ther. 2008, 325, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Thorne, R.G.; Hanson, L.R.; Ross, T.M.; Tung, D.; Frey Ii, W.H. Delivery of interferon-beta to the monkey nervous system following intranasal administration. Neuroscience 2008, 152, 785–797. [Google Scholar] [CrossRef]
- Yang, J.P.; Liu, H.J.; Cheng, S.M.; Wang, Z.L.; Cheng, X.; Yu, H.X.; Liu, X.F. Direct transport of VEGF from the nasal cavity to brain. Neurosci. Lett. 2009, 449, 108–111. [Google Scholar] [CrossRef]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H., II. Intranasal drug targeting of hypocretin-1 (orexin-A) to the central nervous system. J. Pharm. Sci. 2009, 98, 2501–2515. [Google Scholar] [CrossRef]
- Veening, J.G.; Olivier, B. Intranasal administration of oxytocin: Behavioral and clinical effects, a review. Neurosci. Biobehav. Rev. 2013, 37, 1445–1465. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I.; Giladi, E.; Pinhasov, A.; Bardea, A.; Brenneman, D.E. Activity-dependent neurotrophic factor: Intranasal administration of femtomolar-acting peptides improve performance in a water maze. J. Pharmacol. Exp. Ther. 2000, 293, 1091–1098. [Google Scholar] [PubMed]
- Alcalay, R.N.; Giladi, E.; Pick, C.G.; Gozes, I. Intranasal administration of NAP, a neuroprotective peptide, decreases anxiety-like behavior in aging mice in the elevated plus maze. Neurosci. Lett. 2004, 361, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Erdo, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef]
- Iliff, J.J.; Lee, H.; Yu, M.; Feng, T.; Logan, J.; Nedergaard, M.; Benveniste, H. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J. Clin. Investig. 2013, 123, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The glymphatic system: A beginner’s guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infusion | i.n. | |
|---|---|---|
| AUC (min·µg/mL) | 12.9 ± 1.10 | 3.10 ± 0.782 |
| CL (mL/min) | 260.2 ± 13.8 | − |
| F (%) | − | 36.0 ± 4.65 |
| Olfactory Bulb | Frontal Brain | Occipital Brain | |
|---|---|---|---|
| DTP (%) | 96.3 ± 0.9 | 96.6 ± 0.8 | 94.2 ± 1.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatakawa, Y.; Tanaka, A.; Furubayashi, T.; Nakamura, R.; Konishi, M.; Akizawa, T.; Sakane, T. Direct Delivery of ANA-TA9, a Peptide Capable of Aβ Hydrolysis, to the Brain by Intranasal Administration. Pharmaceutics 2021, 13, 1673. https://doi.org/10.3390/pharmaceutics13101673
Hatakawa Y, Tanaka A, Furubayashi T, Nakamura R, Konishi M, Akizawa T, Sakane T. Direct Delivery of ANA-TA9, a Peptide Capable of Aβ Hydrolysis, to the Brain by Intranasal Administration. Pharmaceutics. 2021; 13(10):1673. https://doi.org/10.3390/pharmaceutics13101673
Chicago/Turabian StyleHatakawa, Yusuke, Akiko Tanaka, Tomoyuki Furubayashi, Rina Nakamura, Motomi Konishi, Toshifumi Akizawa, and Toshiyasu Sakane. 2021. "Direct Delivery of ANA-TA9, a Peptide Capable of Aβ Hydrolysis, to the Brain by Intranasal Administration" Pharmaceutics 13, no. 10: 1673. https://doi.org/10.3390/pharmaceutics13101673