
A Novel Sustained Anti-Inflammatory Effect of Atorvastatin—Calcium PLGA Nanoparticles: In Vitro Optimization and In Vivo Evaluation

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Atorvastatin Calcium (At-Ca) PLGA NPs Fabrication
2.3. Particle Size and Zeta Potential Measurements
2.4. Determination of Percent Entrapment Efficiency (EE)
2.5. Morphological Imaging
2.6. Analysis of Drug—Polymer Interaction
2.6.1. Fourier-Transform Infrared Spectroscopy (FTIR)
2.6.2. Differential Scanning Colorimetry (DSC)
2.7. In Vitro Drug Release
2.8. In Vivo Study
2.8.1. Induction of Carrageenan Induced Inflammation
2.8.2. Experimental Groups
2.8.3. Histological Examination
2.9. Statistical Analysis
3. Results
3.1. Effect of Formulation Variables
3.1.1. PLGA Concentration
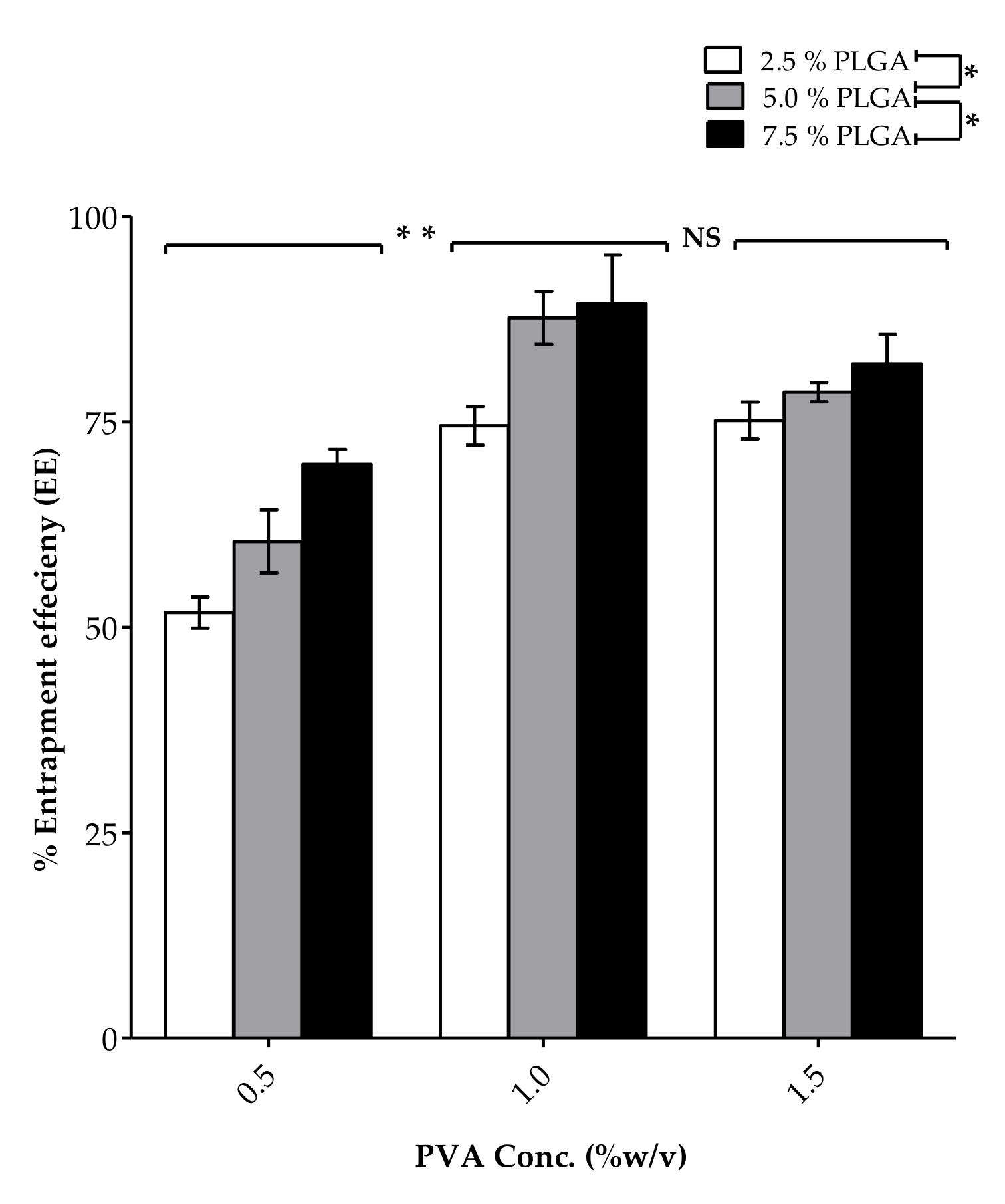
3.1.2. PVA Concentration
3.2. Morphological Imaging
3.3. In Vitro Drug Stability
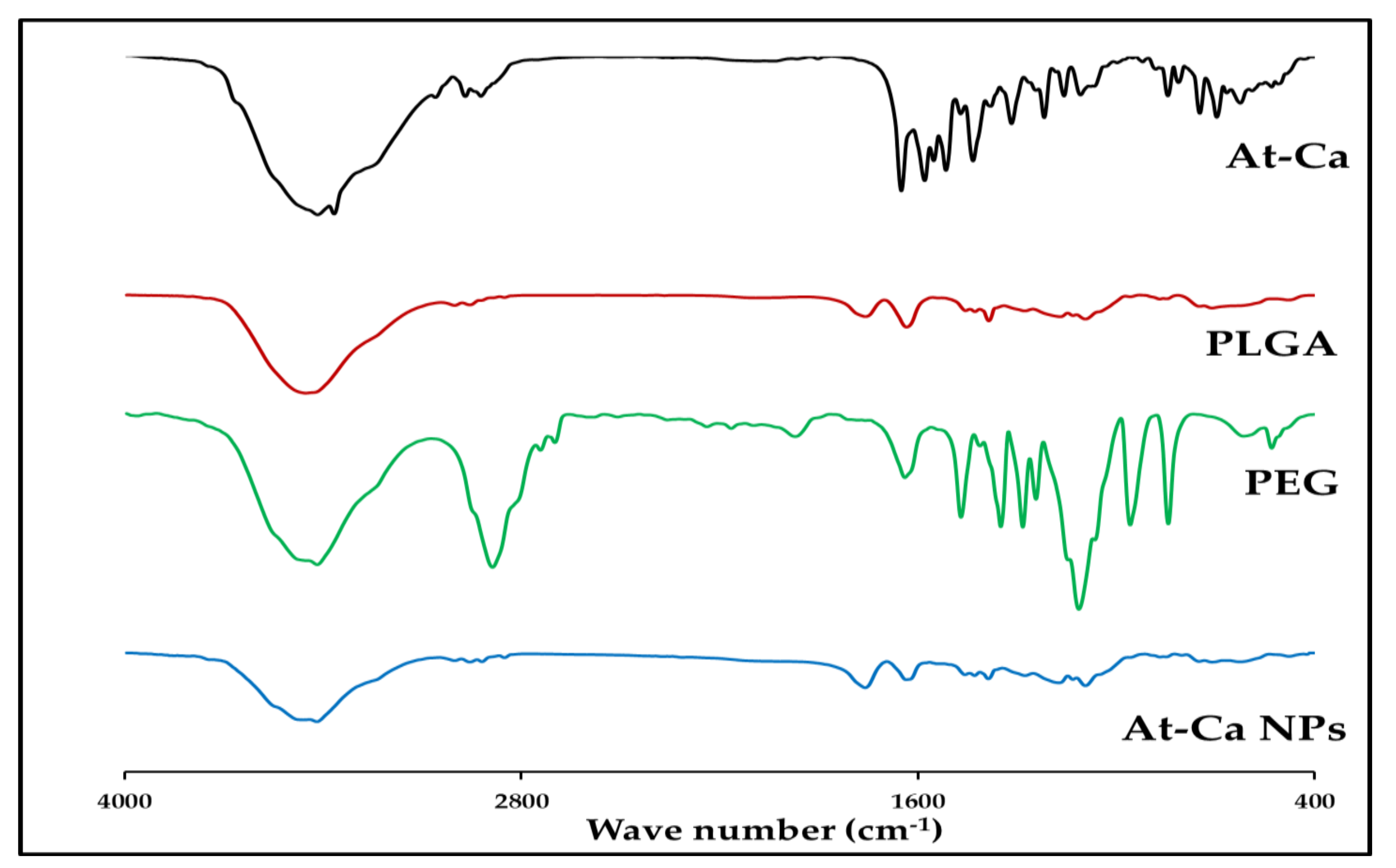
3.3.1. Fourier-Transform Infrared Spectroscopy (FTIR)
3.3.2. Differential Scanning Colorimetry (DSC)
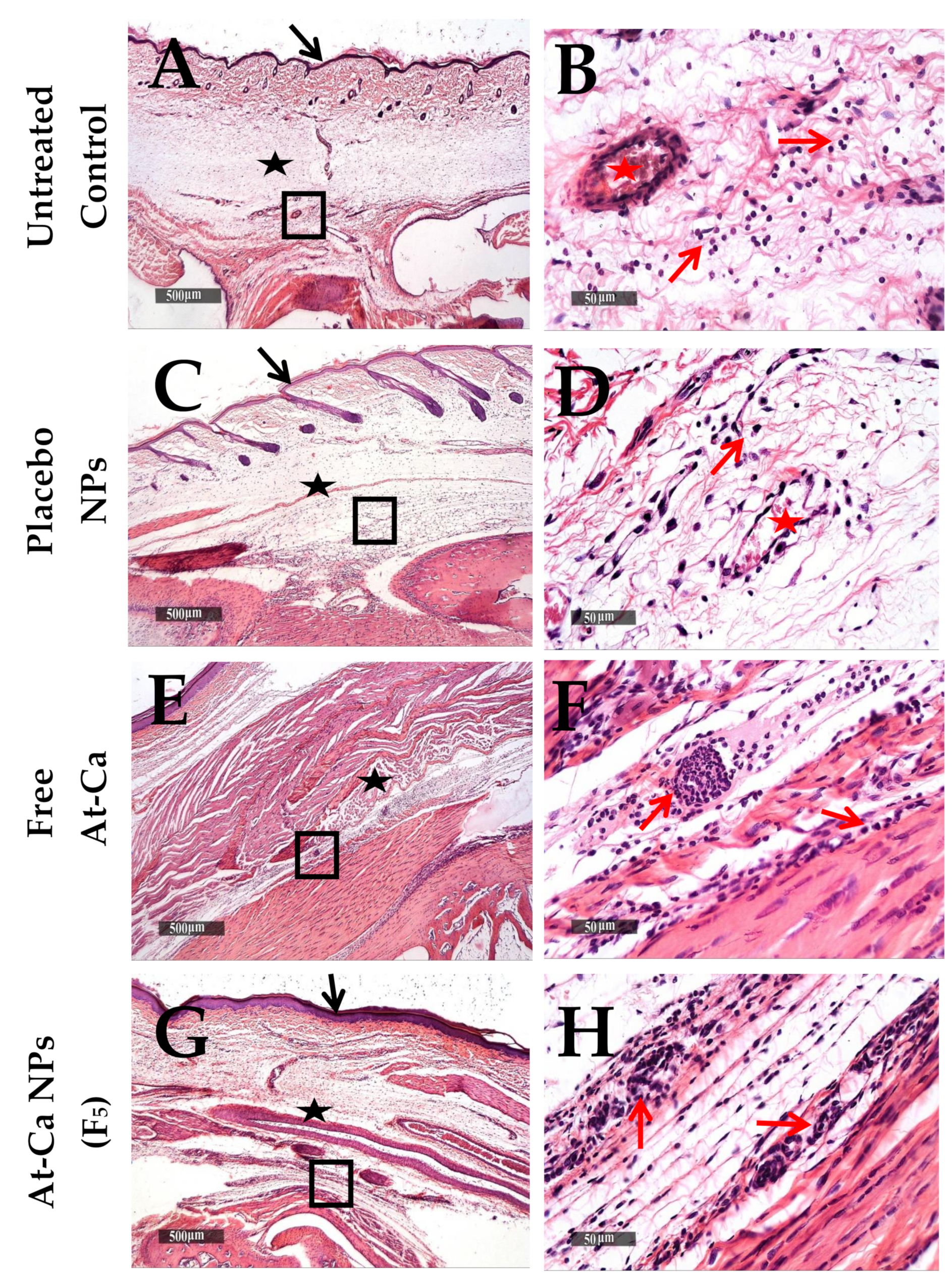
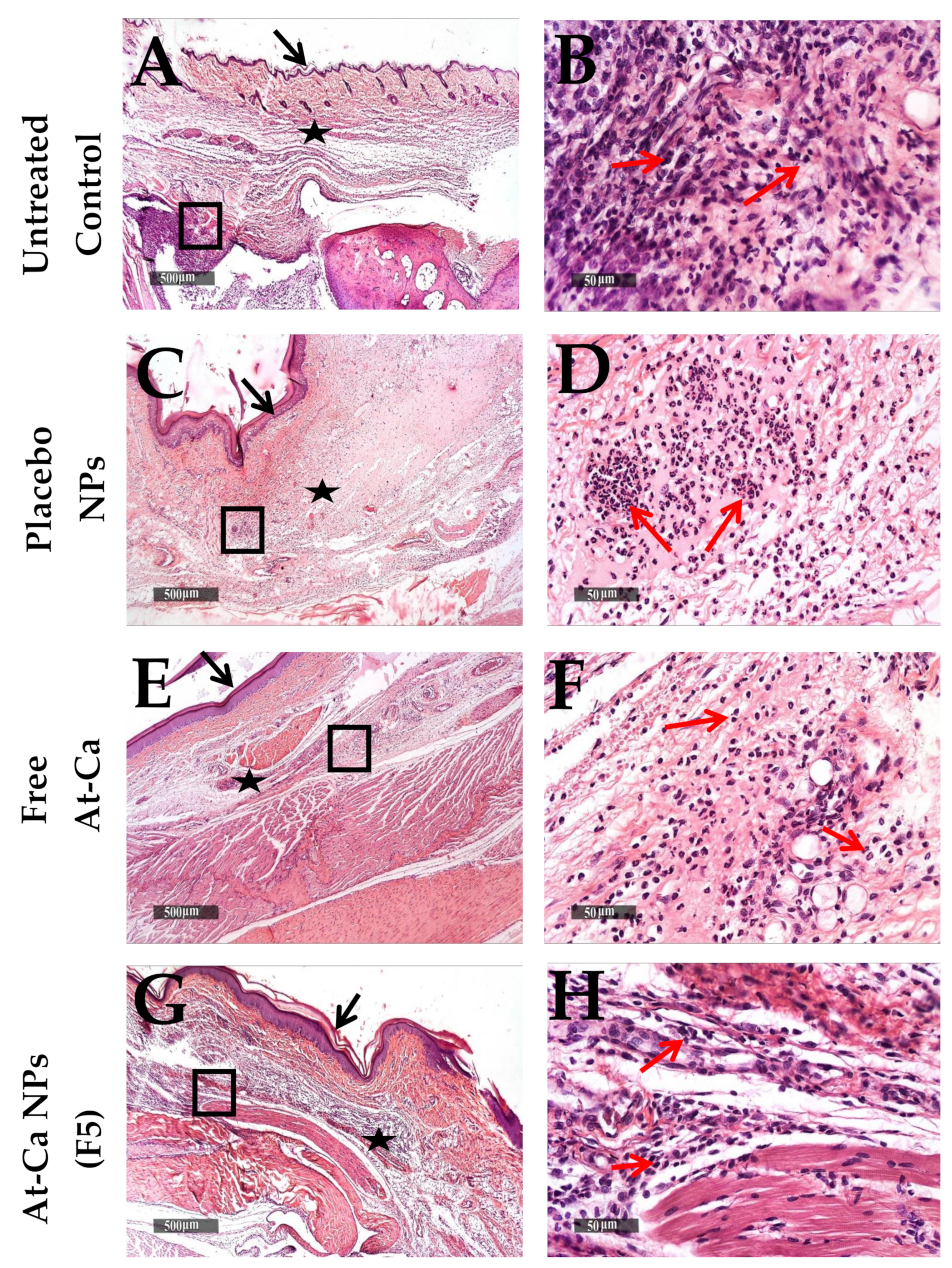
3.4. In Vivo Anti-Inflammatory Study
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clin. Rev. Allergy Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef]
- Gilbert, R.; Al-Janabi, A.; Tomkins-Netzer, O.; Lightman, S. Statins as anti-inflammatory agents: A potential therapeutic role in sight-threatening non-infectious uveitis. Porto Biomed. J. 2017, 2, 33–39. [Google Scholar] [CrossRef]
- Macin, S.M.; Perna, E.R.; Farías, E.F.; Franciosi, V.; Cialzeta, J.R.; Brizuela, M.; Medina, F.; Tajer, C.; Doval, H.; Badaracco, R. Atorvastatin has an important acute anti-inflammatory effect in patients with acute coronary syndrome: Results of a randomized, double-blind, placebo-controlled study. Am. Heart J. 2005, 149, 451–457. [Google Scholar] [CrossRef]
- Bu, D.X.; Griffin, G.; Lichtman, A.H. Mechanisms for the anti-inflammatory effects of statins. Curr. Opin. Lipidol. 2011, 22, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J.; Zhang, Y.; Sun, J.; Yan, K.; Liu, A. Oxidized Low-Density Lipoprotein (OxLDL)-Treated Dendritic Cells Promote Activation of T Cells in Human Atherosclerotic Plaque and Blood, Which Is Repressed by Statins: MicroRNA let-7c Is Integral to the Effect. J. Am. Heart Assoc. 2016, 5, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönbeck, U.; Libby, P. Inflammation, immunity, and HMG-CoA reductase inhibitors: Statins as antiinflammatory agents? Circulation 2004, 109. [Google Scholar] [CrossRef] [Green Version]
- Shaker, M.A.; Elbadawy, H.M.; Al Thagfan, S.S.; Shaker, M.A. Enhancement of atorvastatin oral bioavailability via encapsulation in polymeric nanoparticles. Int. J. Pharm. 2021, 592. [Google Scholar] [CrossRef]
- Mahmoud, M.O.; Aboud, H.M.; Hassan, A.H.; Ali, A.A.; Johnston, T.P. Transdermal delivery of atorvastatin calcium from novel nanovesicular systems using polyethylene glycol fatty acid esters: Ameliorated effect without liver toxicity in poloxamer 407-induced hyperlipidemic rats. J. Control. Release 2017, 254, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Hossain, M.S.; Shamim, M.A.; Islam, M.; Siddique, M.A.T. Formulation, in vitro evaluation and characterization of atorvastatin solid dispersion. Trop. J. Pharm. Res. 2020, 19, 1131–1138. [Google Scholar] [CrossRef]
- Al-kazemi, R.; Al-basarah, Y.; Nada, A. Atorvastatin Cocrystals: Tablet Formulation and Stability. Asian J. Pharm. 2020, 14, 578–595. [Google Scholar]
- Gardouh, A.R.; Nasef, A.M.; Mostafa, Y.; Gad, S. Design and evaluation of combined atorvastatin and ezetimibe optimized self- nano emulsifying drug delivery system. J. Drug Deliv. Sci. Technol. 2020, 60, 102093. [Google Scholar] [CrossRef]
- Abdelkader, D.H.; El-Gizawy, S.A.; Faheem, A.M.; McCarron, P.A.; Osman, M.A. Effect of process variables on formulation, in-vitro characterisation and subcutaneous delivery of insulin PLGA nanoparticles: An optimisation study. J. Drug Deliv. Sci. Technol. 2018, 43, 160–171. [Google Scholar] [CrossRef]
- Faheem, A.M.; Abdelkader, D.H. Novel drug delivery systems. In Engineering Drug Delivery Systems; Woodhead Publishing: Sawston, UK, 2020; ISBN 9780081025482. [Google Scholar]
- Zhang, Y.; Zhang, R.; Illangakoon, U.E.; Harker, A.H.; Thrasivoulou, C.; Parhizkar, M.; Edirisinghe, M.; Luo, C.J. Copolymer composition and nanoparticle configuration enhance in vitro drug release behavior of poorly water-soluble progesterone for oral formulations. Int. J. Nanomed. 2020, 15, 5389–5403. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tao, W.; Zhang, D.; Wu, C. The studies of PLGA nanoparticles loading atorvastatin calcium for oral administration in vitro and in vivo. Asian J. Pharm. Sci. 2017, 12, 285–291. [Google Scholar] [CrossRef]
- Kurakula, M.A.T. Co-Delivery of Atorvastatin Nanocrystals in PLGA based in situ Gel for Anti-Hyperlipidemic Efficacy. Curr. Drug Deliv. 2016, 13, 211–220. [Google Scholar] [CrossRef]
- Şimşek, S.; Eroğlu, H.; Kurum, B.; Ulubayram, K. Brain targeting of Atorvastatin loaded amphiphilic PLGA-b-PEG nanoparticles. J. Microencapsul. 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kizilbey, K. Optimization of Rutin-Loaded PLGA Nanoparticles Synthesized by Single-Emulsion Solvent Evaporation Method. ACS Omega 2019, 4, 555–562. [Google Scholar] [CrossRef]
- Abdelkader, D.H.; Osman, M.A.; El-Gizawy, S.A.; Hawthorne, S.J.; Faheem, A.M.; McCarron, P.A. Effect of poly(ethylene glycol) on insulin stability and cutaneous cell proliferation in vitro following cytoplasmic delivery of insulin-loaded nanoparticulate carriers—A potential topical wound management approach. Eur. J. Pharm. Sci. 2018, 114, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Tong, H.H.Y.; Chow, S.F. In vitro release study of the polymeric drug nanoparticles: Development and validation of a novel method. Pharmaceutics 2020, 12, 732. [Google Scholar] [CrossRef]
- Rivat, C.; Laulin, J.-P.; Corcuff, J.-B.; Célèrier, E.; Pain, L.; Simonnet, G. Fentanyl Enhancement of Carrageenan-induced Long-lasting Hyperalgesia in Rats. Anesthesiology 2002, 96, 381–391. [Google Scholar] [CrossRef]
- Whiteley, P.E.; Dalrymple, S.A. Models of Inflammation: Carrageenan-Induced Paw Edema in the Rat. Curr. Protoc. Pharmacol. 2001, 4–6. [Google Scholar] [CrossRef]
- Culling, C.F.A. Handbook of Histopathological and Histochemical Techniques, 3rd ed.; Elsevier: London, UK, 2013; ISBN 9781483164793. [Google Scholar]
- Abdelkader, D.H.; Osman, M.A.; El-Gizawy, S.A.; Faheem, A.M.; McCarron, P.A. Characterisation and in vitro stability of low-dose, lidocaine-loaded poly(vinyl alcohol)-tetrahydroxyborate hydrogels. Int. J. Pharm. 2016, 500, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, D.H.; Osman, M.A.; El-Gizawy, S.A.; Faheem, A.M.; McCarron, P.A. Adhesiveness of poly(vinyl alcohol) tetrahydroxyborate hydrogels to the stratum corneum and their applicability as topical drug delivery systems. Int. J. Cosmet. Sci. 2015, 37, 141–163. [Google Scholar]
- Singh, G.; Kaur, T.; Kaur, R.; Kaur, A. Recent biomedical applications and patents on biodegradable polymer-PLGA. Int. J. Pharmacol. 2014, 1, 30–42. [Google Scholar]
- Singh, R.; Kesharwani, P.; Mehra, N.K.; Singh, S.; Banerjee, S.; Jain, N.K. Development and characterization of folate anchored Saquinavir entrapped PLGA nanoparticles for anti-tumor activity. Drug Dev. Ind. Pharm. 2015, 41, 1888–1901. [Google Scholar] [CrossRef]
- Xiao, H.; Wang, L. Effects of X-shaped reduction-sensitive amphiphilic block copolymer on drug delivery. Int. J. Nanomed. 2015, 10, 5309–5325. [Google Scholar] [CrossRef] [Green Version]
- Asadi, H.; Rostamizadeh, K.; Salari, D.; Hamidi, M. Preparation of biodegradable nanoparticles of tri-block PLA-PEG-PLA copolymer and determination of factors controlling the particle size using artificial neural network. J. Microencapsul. 2011, 28, 406–416. [Google Scholar] [CrossRef]
- Xiang, H.; Wang, S.; Wang, R.; Zhou, Z.; Peng, C.; Zhu, M. Synthesis and characterization of an environmentally friendly PHBV/PEG copolymer network as a phase change material. Sci. China Chem. 2013, 56, 716–723. [Google Scholar] [CrossRef]
- Zhang, X.; Li, N.; Liu, Y.; Ji, B.; Wang, Q.; Wang, M.; Dai, K.; Gao, D. On-demand drug release of ICG-liposomal wedelolactone combined photothermal therapy for tumor. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 2019–2029. [Google Scholar] [CrossRef]
- Shaker, M.A.; Elbadawy, H.M.; Shaker, M.A. Improved solubility, dissolution, and oral bioavailability for atorvastatin-Pluronic® solid dispersions. Int. J. Pharm. 2020, 574, 118891. [Google Scholar] [CrossRef]
- Shaker, M.A. Dissolution and bioavailability enhancement of Atorvastatin: Gelucire semi-solid binary system. J. Drug Deliv. Sci. Technol. 2018, 43, 178–184. [Google Scholar] [CrossRef]
- Sun, S.B.; Liu, P.; Shao, F.M.; Miao, Q.L. Formulation and evaluation of PLGA nanoparticles loaded capecitabine for prostate cancer. Int. J. Clin. Exp. Med. 2015, 8, 19670–19681. [Google Scholar] [PubMed]
- Valor, D.; Montes, A.; Monteiro, M.; García-Casas, I.; Pereyra, C.; de la Ossa, E.M. Determining the optimal conditions for the production by supercritical co2 of biodegradable plga foams for the controlled release of rutin as a medical treatment. Polymers 2021, 13, 1645. [Google Scholar] [CrossRef]
- Dabbagh, A.; Mahmoodian, R.; Abdullah, B.J.J.; Abdullah, H.; Hamdi, M.; Abu Kasim, N.H. Low-melting-point polymeric nanoshells for thermal-triggered drug release under hyperthermia condition. Int. J. Hyperth. 2015, 31, 920–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuru, A.; Aksoy, S.A. Cellulose–PEG grafts from cotton waste in thermo-regulating textiles. Text. Res. J. 2014, 84, 337–346. [Google Scholar] [CrossRef]
- Öztürk, A.A.; Kırımlıoğlu, G.Y. Preparation and in vitro characterization of lamivudine loaded nanoparticles prepared by acid and/or ester terminated PLGA for effective oral anti-retroviral therapy. J. Res. Pharm. 2019, 23, 897–913. [Google Scholar] [CrossRef] [Green Version]
- Gebreel, R.M.; Edris, N.A.; Elmofty, H.M.; Tadros, M.I.; El-Nabarawi, M.A.; Hassan, D.H. Development and Characterization of PLGA Nanoparticle-Laden Hydrogels for Sustained Ocular Delivery of Norfloxacin in the Treatment of Pseudomonas Keratitis: An Experimental Study. Drug Des. Devel. Ther. 2021, 15, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.R.; Chaurasia, S.; Khan, G.; Chaubey, P.; Kumar, N.; Mishra, B. Cromolyn sodium encapsulated PLGA nanoparticles: An attempt to improve intestinal permeation. Int. J. Biol. Macromol. 2016, 83, 249–258. [Google Scholar] [CrossRef]
- Sharma, D.; Maheshwari, D.; Philip, G.; Rana, R.; Bhatia, S.; Singh, M.; Gabrani, R.; Sharma, S.K.; Ali, J.; Sharma, R.K.; et al. Formulation and Optimization of Polymeric Nanoparticles for Intranasal Delivery of Lorazepam Using Box-Behnken Design:In Vitro and In Vivo Evaluation. Biomed. Res. Int. 2014, 2014, 1–14. [Google Scholar]
- Sharma, N.; Madan, P.; Lin, S. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study. Asian J. Pharm. Sci. 2016, 11, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Mohan, L.J.; McDonald, L.; Daly, J.S.; Ramtoola, Z. Optimising PLGA-PEG nanoparticle size and distribution for enhanced drug targeting to the inflamed intestinal barrier. Pharmaceutics 2020, 12, 1114. [Google Scholar] [CrossRef]
- Seju, U.; Kumar, A.; Sawant, K.K. Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: In vitro and in vivo studies. Acta Biomater. 2011, 7, 4169–4176. [Google Scholar] [CrossRef]
- Mares, A.G.; Pacassoni, G.; Marti, J.S.; Pujals, S.; Albertazzi, L. Formulation of tunable size PLGA-PEG nanoparticles for drug delivery using microfluidic technology. PLoS ONE 2021, 16, e0251821. [Google Scholar] [CrossRef] [PubMed]
- Beletsi, A.; Panagi, Z.; Avgoustakis, K. Biodistribution properties of nanoparticles based on mixtures of PLGA with PLGA–PEG diblock copolymers. Int. J. Pharm. 2005, 298, 233–241. [Google Scholar] [CrossRef]
- Samkange, T.; D’Souza, S.; Obikeze, K.; Dube, A. Influence of PEGylation on PLGA nanoparticle properties,hydrophobic drug release and interactions with humanserum albumin.pdf. J. Pharm. Pharmacol. 2019, 71, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.A.; Kazarian, S.G. Detection of trace materials with Fourier transform infrared spectroscopy using a multi-channel detector. Analyst 2006, 131, 126–131. [Google Scholar] [CrossRef]
- Panda, B.P.; Wei, M.X.; Shivashekaregowda, N.K.H.; Patnaik, S. Design, Fabrication and Characterization of PVA/PLGA Electrospun Nanofibers Carriers for Improvement of Drug Delivery of Gliclazide in Type-2 Diabetes. Proceedings 2020, 78, 14. [Google Scholar] [CrossRef]
- Bachhav, S.S.; Dighe, V.D.; Devarajan, P. V Exploring Peyer’s Patch Uptake as a Strategy for Targeted Lung Delivery of Polymeric Rifampicin Nanoparticles Exploring Peyer ’ s Patch Uptake as a Strategy for Targeted Lung Delivery of Polymeric Rifampicin Nanoparticles National Center for Preclinical. Mol. Pharm. 2018, 1–38. [Google Scholar]
- Abdelkader, D.H.; Murtaza, T.; Mitchell, C.A.; Osman, M.A.; El-Gizawy, S.A.; Faheem, A.M.; Mohamed, E.-T.; McCarron, P.A. Enhanced cutaneous wound healing in rats following topical delivery of insulin-loaded nanoparticles embedded in poly(vinyl alcohol)-borate hydrogels. J. Drug Deliv. Transl. Res. 2018, 8, 1053–1065. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula Code | PLGA Concentration (% w/v) | PEG Content in Primary Emulsion (% w/w) | PVA Concentration in Aqueous External Phase (% w/v) | Drug Loading (mg) | Organic/Aqueous Phase Volume Ratio |
|---|---|---|---|---|---|
| F1 | 2.50 | ||||
| F2 | 5.00 | 5.00 | 0.50 | 10 | 1:100 |
| F3 | 7.50 | ||||
| F4 F5 F6 | 2.50 5.00 7.50 | 5.00 | 1.00 | 10 | 1:100 |
| F7 F8 F9 | 2.50 5.00 7.50 | 5.00 | 1.50 | 10 | 1:100 |
| Formula Code | Z-Average | Polydispersity Index (PDI) | Zeta Potential (mV) | % EE |
|---|---|---|---|---|
| (nm) | ||||
| F1 | 200.7 ± 18.85 | 0.096 ± 0.011 | −5.57 ± 0.88 | 51.78 ± 1.89 |
| F2 | 238.3 ± 22.43 | 0.134 ± 0.025 | −8.23 ± 0.82 | 60.42 ± 3.84 |
| F3 | 257.9 ± 19.53 | 0.145 ± 0.012 | −10.77 ± 0.36 | 69.81 ± 1.84 |
| F4 | 217.5 ± 20.87 | 0.107 ± 0.066 | −9.02 ± 3.22 | 74.52 ± 2.35 |
| F5 | 248.2 ± 15.13 | 0.126 ± 0.048 | −12.41 ± 4.80 | 87.63 ± 3.21 |
| F6 | 280.8 ± 13.55 | 0.151 ± 0.039 | −17.18 ± 0.87 | 89.36 ± 5.89 |
| F7 | 238.1 ± 22.58 | 0.076 ± 0.047 | −11.71 ± 0.78 | 75.15 ± 2.24 |
| F8 | 289.5 ± 17.89 | 0.102 ± 0.029 | −14.55 ± 2.95 | 78.95 ± 1.17 |
| F9 | 326.9 ± 16.74 | 0.110 ± 0.012 | −19.28 ± 0.77 | 82.02 ± 3.59 |
| Assignment | Approximate Frequency (cm−1) | Description | |
|---|---|---|---|
| PLGA 50:50 [26,27,28] | O-H (very broad) | 3452.21 | Stretching |
| C-O-C | 1093.27 | ||
| =C-O | 1169.23 | Asymmetric Stretching | |
| C-H | 3001.64 (CH3) | Stretching | |
| 2955.70 (CH2) | |||
| 2851.21 (CH2) | |||
| 1455.76 (CH3) | Bending | ||
| 1385.24 (CH3) | |||
| 1276.49 (CH2) | |||
| C=O | 1758.52 | Stretching | |
| 1427.20 1385.24 | Bending | ||
| PEG [29,30,31] | O-H (very broad) | 3414.43 | Stretching |
| C-H | 2886.97 | ||
| C-O | 1112.8 | ||
| C-C | 842.13 | ||
| At-Ca [7,32,33] | O-H (very broad) | 3416.25 | Stretching |
| N-H | 3366.44 | ||
| C-H | 3059.59 2969.41 29922.27 | ||
| C=O | 1649.89 1579.24 | ||
| C-C | 1515.04 1433.1 | ||
| CH3 and -CH2 | 1316.51 | Deformation Bending | |
| C-N | 1217.72 | Stretching | |
| C-O | 1157.55 | ||
| aromatic C-H | 843.52 747.00 | Out plane Bending | |
| C-H | 695.58 623.99 | Deformation Bending |
| A Inflammatory cells infiltrates Subcutaneous oedema Congested BVs | Untreated Control | Placebo NPs | Free AT-Ca | AT-Ca NPs (F5) |
| +++ +++ ++ | ++ +++ ++ | ++ ++ ++ | ++ ++ ++ | |
| B Inflammatory cells infiltrates Subcutaneous oedema Congested BVs | Untreated Control | Placebo NPs | Free AT-Ca | AT-Ca NPs (F5) |
| +++ +++ ++ | ++ +++ ++ | ++ ++ + | ++ + + | |
| C Inflammatory cells infiltrates Subcutaneous oedema Congested BVs | Untreated Control | Placebo NPs | Free AT-Ca | AT-Ca NPs (F5) |
| +++ | +++ | ++ | + | |
| ++ | ++ | ++ | + | |
| ++ | + | + | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelkader, D.H.; Abosalha, A.K.; Khattab, M.A.; Aldosari, B.N.; Almurshedi, A.S. A Novel Sustained Anti-Inflammatory Effect of Atorvastatin—Calcium PLGA Nanoparticles: In Vitro Optimization and In Vivo Evaluation. Pharmaceutics 2021, 13, 1658. https://doi.org/10.3390/pharmaceutics13101658
Abdelkader DH, Abosalha AK, Khattab MA, Aldosari BN, Almurshedi AS. A Novel Sustained Anti-Inflammatory Effect of Atorvastatin—Calcium PLGA Nanoparticles: In Vitro Optimization and In Vivo Evaluation. Pharmaceutics. 2021; 13(10):1658. https://doi.org/10.3390/pharmaceutics13101658
Chicago/Turabian StyleAbdelkader, Dalia H., Ahmed Kh. Abosalha, Mohamed A. Khattab, Basmah N. Aldosari, and Alanood S. Almurshedi. 2021. "A Novel Sustained Anti-Inflammatory Effect of Atorvastatin—Calcium PLGA Nanoparticles: In Vitro Optimization and In Vivo Evaluation" Pharmaceutics 13, no. 10: 1658. https://doi.org/10.3390/pharmaceutics13101658