Targeting Activated Hepatic Stellate Cells Using Collagen-Binding Chitosan Nanoparticles for siRNA Delivery to Fibrotic Livers


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
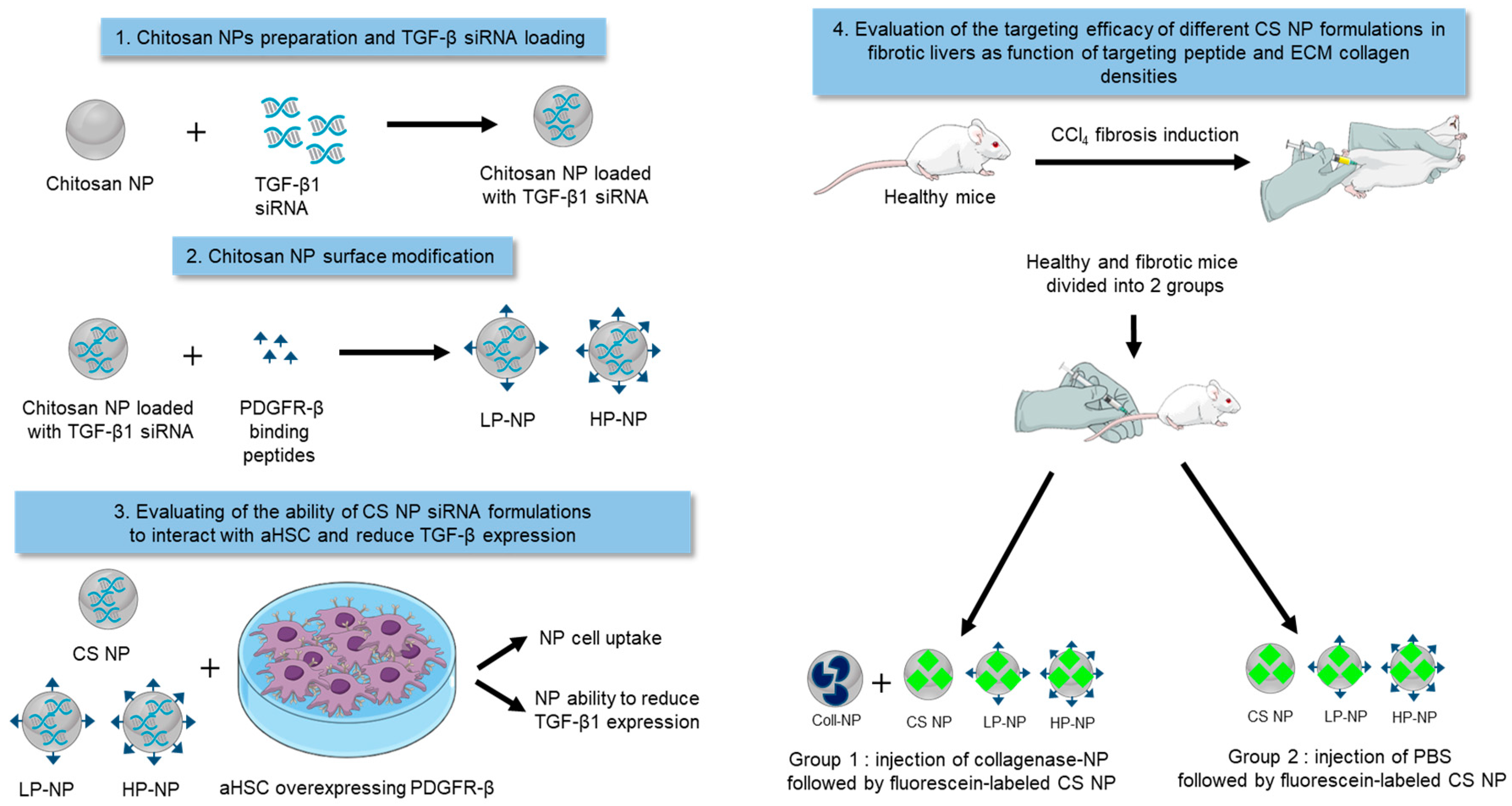
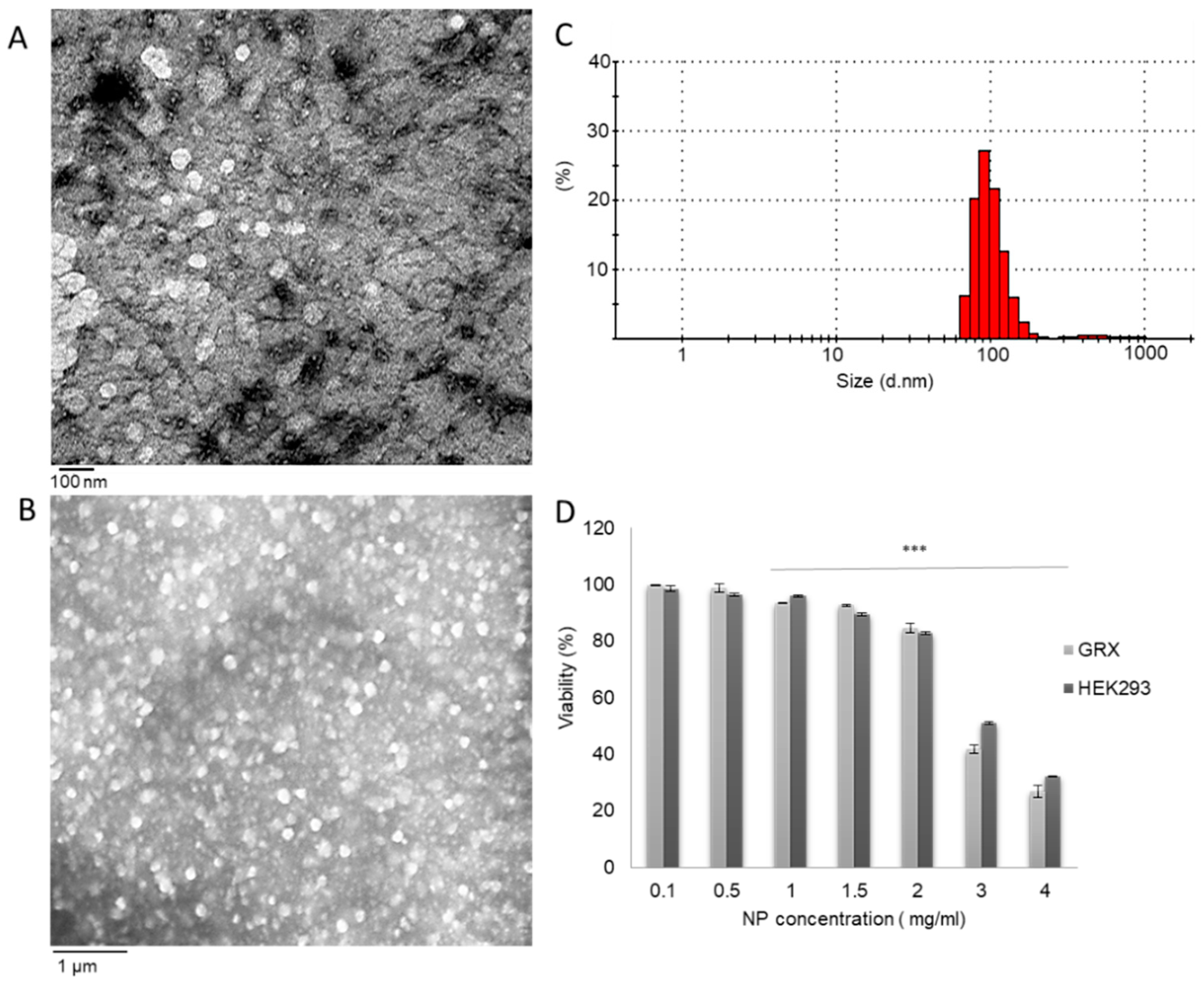
2.1. Nanoparticle Preparation and Characterization
2.2. In-Vitro CS-NPs Cytotoxicity Studies
2.3. Nanoparticle Modification with PDGFR-β Binding Peptide
2.4. Nanoparticle Loading and Determination of Encapsulation Efficiency
2.5. In-Vitro CS-NPs Association in GRX and HEK293 Cells
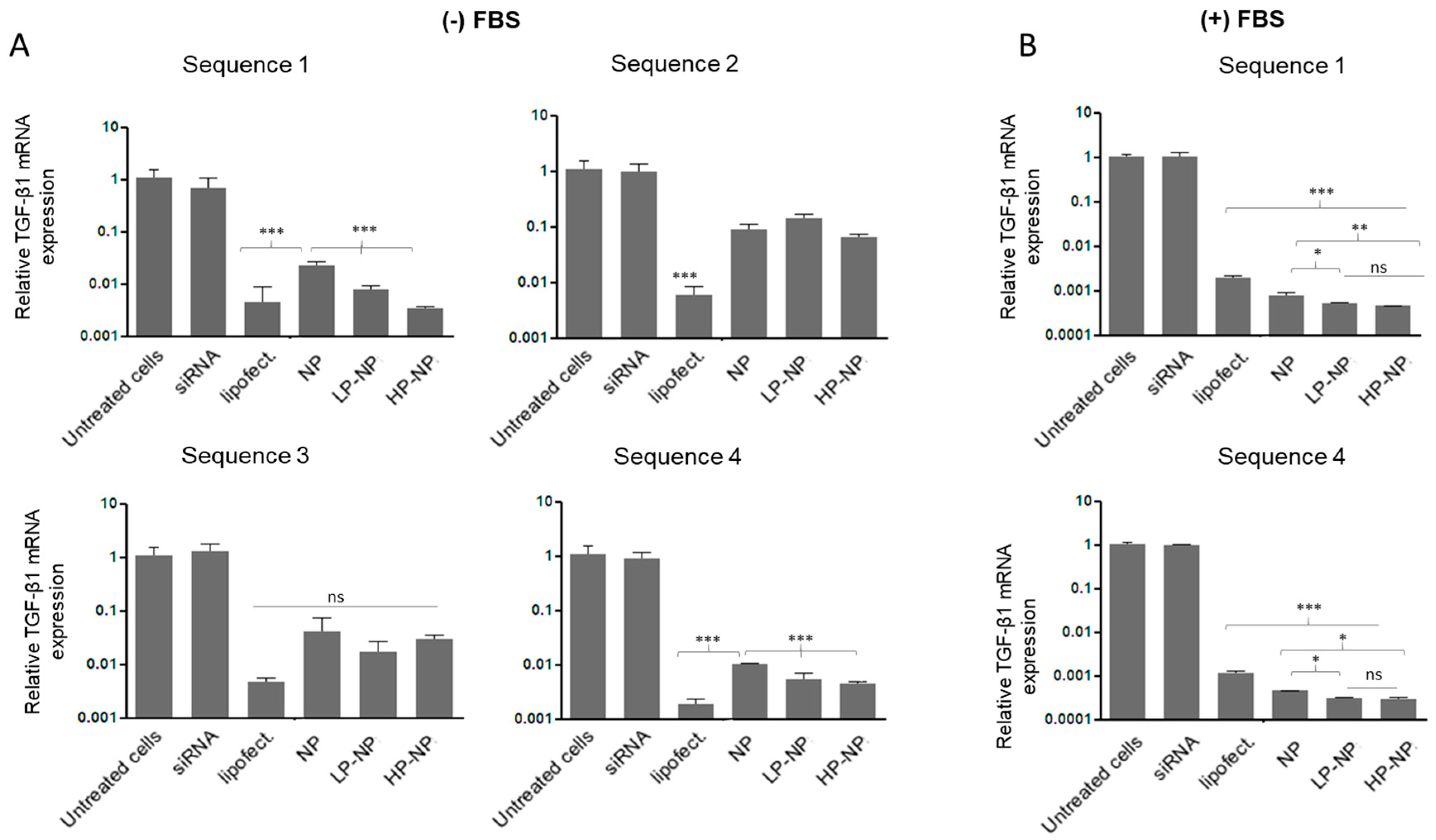
2.6. Quantitative Real-Time PCR Analysis
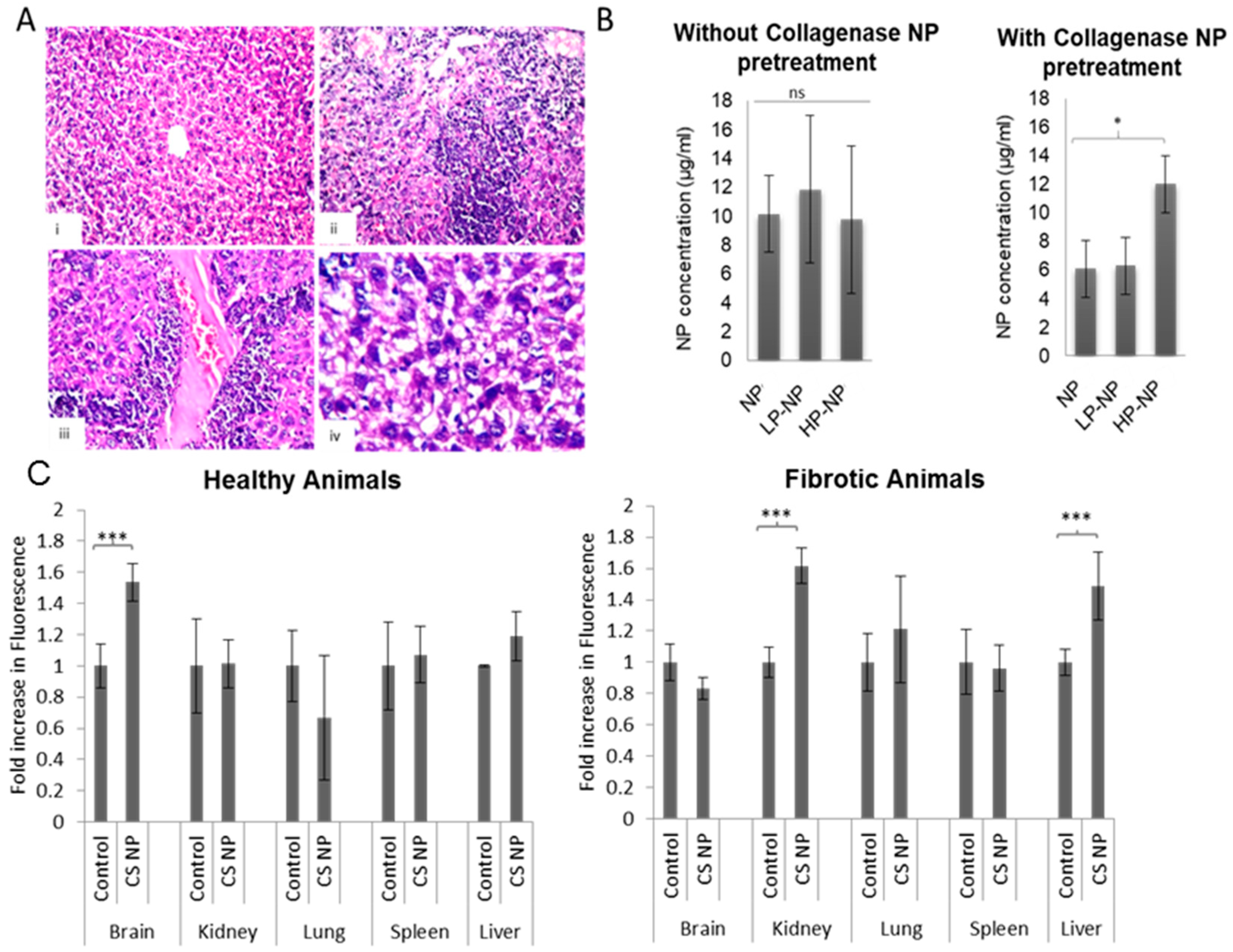
2.7. In-Vivo Nanoparticle Biodistribution
2.8. Statistical Analysis
3. Results and Discussion
3.1. Nanoparticle Synthesis and Characterization
3.2. In-Vitro Association of Chitosan Nanoparticles by HEK293 and GRX Cells
3.3. siRNA-Containing Chitosan Nanoparticles Reduce Profibrogenic Gene Expression
3.4. In-Vivo Nanoparticle Biodistribution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, C.-Y.; Yuan, W.-G.; He, P.; Lei, J.-H.; Wang, C.-X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Tammam, S.N.; El Safy, S.; Abdel-Halim, M.; Asimakopoulou, A.; Weiskirchen, R.; Mansour, S. Prevention of hepatic stellate cell activation using JQ1- and atorvastatin-loaded chitosan nanoparticles as a promising approach in therapy of liver fibrosis. Eur. J. Pharm. Biopharm. 2019, 134, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2011, 3, 1473–1492. [Google Scholar]
- Lepreux, S.; Desmoulière, A. Human liver myofibroblasts during development and diseases with a focus on portal (myo)fibroblasts. Front. Physiol. 2015, 6, 173. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; He, L.; Guo, H.; Chen, H.; Shan, H. Targeting activated hepatic stellate cells (aHSCs) for liver fibrosis imaging. EJNMMI Res. 2015, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Jain, A.; Liu, H.; Zhao, Z.; Cheng, K. Targeted drug delivery to hepatic stellate cells for the treatment of liver fibrosis. J. Pharmacol. Exp. Ther. 2019, 370, 695–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.-Y.; Hu, J.-J.; Shen, J.; Wang, M.; Zhang, Q.-Q.; Qu, Y.; Lu, L.-G. Stat3 signaling activation crosslinking of TGF-β1 in hepatic stellate cell exacerbates liver injury and fibrosis. Biochim. Biophys. Acta (BBA)–Mol. Basis Dis. 2014, 1842, 2237–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, S.; Morris, J.C. Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccines Immunother. 2017, 13, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The role of transforming growth factor (TGF)-β in the infarcted myocardium. J. Thorac. Dis. 2017, 9, S52–S63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, D.; Boyle, G.M.; Parsons, P.G.; Coman, W.B. What is transforming growth factor-beta (TGF-β)? Br. J. Plast. Surg. 2004, 57, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Schon, H.-T.; Bartneck, M.; Borkham-Kamphorst, E.; Nattermann, J.; Lammers, T.; Tacke, F.; Weiskirchen, R. Pharmacological intervention in hepatic stellate cell activation and hepatic fibrosis. Front. Pharmacol. 2016, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammam, S.; Malak, P.; Correa, D.; Rothfuss, O.; M.E., A.H.; Lamprecht, A.; Schulze-Osthoff, K. Nuclear delivery of recombinant OCT4 by chitosan nanoparticles for Transgene-Free generation of Protein-Induced pluripotent stem cells. Oncotarget 2016, 7, 37728–37739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammam, S.N.; Lamprecht, A.; Cornier, J.; Owen, A.; Kwade, A.; Van De Voorde, M. Nanostructures in Drug Delivery. Pharm. Nanotechnol. Innov. Prod. 2016, 101–134. [Google Scholar]
- Iwakiri, Y.; Shah, V.; Rockey, D.C. Vascular pathobiology in chronic liver disease and Cirrhosis–Current status and future directions. J. Hepatol. 2014, 61, 912–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkham-Kamphorst, E.; Weiskirchen, R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016, 28, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Maass, T.; Thieringer, F.R.; Mann, A.; Longerich, T.; Schirmacher, P.; Strand, D.; Hansen, T.; Galle, P.R.; Teufel, A.; Kanzler, S. Liver specific overexpression of Platelet-Derived growth Factor-B accelerates liver cancer development in chemically induced liver carcinogenesis. Int. J. Cancer 2011, 128, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Beljaars, L.; Weert, B.; Geerts, A.; Meijer, D.K.F.; Poelstra, K. The preferential homing of a platelet derived growth factor receptor-recognizing macromolecule to Fibroblast-Like cells in fibrotic tissue. Biochem. Pharmacol. 2003, 66, 1307–1317. [Google Scholar] [CrossRef]
- Askoxylakis, V.; Marr, A.; Altmann, A.; Markert, A.; Mier, W.; Debus, J.; Huber, P.E.; Haberkorn, U. Peptide-Based targeting of the Platelet-Derived growth factor receptor beta. Mol. Imaging Boil. 2012, 15, 212–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donalisio, M.; Leone, F.; Civra, A.; Spagnolo, R.; Ozer, O.; Lembo, D.; Cavalli, R. Acyclovir-Loaded Chitosan Nanospheres from Nano-Emulsion Templating for the Topical Treatment of Herpesviruses Infections. Pharmaceutics 2018, 10, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammam, S.N.; Azzazy, H.M.; Breitinger, H.G.; Lamprecht, A. Chitosan nanoparticles for nuclear targeting: The effect of nanoparticle size and nuclear localization sequence density. Mol. Pharm. 2015, 12, 4277–4289. [Google Scholar] [CrossRef] [PubMed]
- Tammam, S.N.; Azzazy, H.M.; Lamprecht, A. Nuclear and cytoplasmic delivery of lactoferrin in glioma using chitosan nanoparticles: Cellular location dependent-action of lactoferrin. Eur. J. Pharm. Biopharm. 2018, 129, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, C.; Negre, J.M.S.; Pérez-Lozano, P.; García-Montoya, E.; Sarrate, R.; Fàbregas, A.; Miñarro, M.; Ticó, J.R. Chitosan nanoparticles as Non-Viral gene delivery systems: Determination of loading efficiency. Biomed. Pharmacother. 2014, 68, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.-K.; Chen, L.; Lu, X.; Cao, D.; Guo, L.; Zhang, Y.-S.; Li, L.-B.; Zhang, L.; Kuang, Y.-T.; Wang, S.-L.; et al. Optimization of transforming growth Factor-β1 siRNA loaded Chitosan-Tripolyphosphate nanoparticles for the treatment of colorectal cancer hepatic metastasis in a mouse model. J. Biomed. Nanotechnol. 2016, 12, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Wary, R.; Sivaraj, S.; Gurukarthikeyana, R.K.P.; MARI, S.L.; Suraja, G.D.; Kannayiram, G. Chitosan gallic acid microsphere incorporated collagen matrix for chronic wounds: Biophysical and biochemical characterization. Int. J. Pharm. Pharm. Sci. 2014, 6, 94–100. [Google Scholar]
- El-Safy, S.; Tammam, S.N.; Abdel-Halim, M.; Ali, M.E.; Youshia, J.; Boushehri, M.A.S.; Lamprecht, A.; Mansour, S. Collagenase loaded chitosan nanoparticles for digestion of the collagenous scar in liver fibrosis: The effect of chitosan intrinsic collagen binding on the success of targeting. Eur. J. Pharm. Biopharm. 2020, 148, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Borojevic, R.; Monteiro, A.N.; Vinhas, S.A.; Domont, G.B.; Mourão, P.A.S.; Emonard, H.; Grimaldi, G.; Grimaud, J.-A. Establishment of a continuous cell line from fibrotic schistosomal granulomas in mice livers. Vitr. Cell. Dev. Boil.–Anim. 1985, 21, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Graham, F.L.; Russell, W.C.; Smiley, J.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Buch, K.; Peters, T.; Nawroth, T.; Sänger, M.; Schmidberger, H.; Langguth, P. Determination of cell survival after irradiation via clonogenic assay versus multiple MTT Assay–A comparative study. Radiat. Oncol. 2012, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; Bossche, B.V.D.; De Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2015, 90, 1025–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bancroft, J.D.; Gamble, M. Theory and Practice of Histological Techniques; Elsevier Health Sciences: London, UK, 2008. [Google Scholar]
- Tammam, S.N.; Azzazy, H.M.E.; Lamprecht, A. Biodegradable particulate carrier formulation and tuning for targeted drug delivery. J. Biomed. Nanotechnol. 2015, 11, 555–577. [Google Scholar] [CrossRef] [PubMed]
- Bazak, R.; Houri, M.; El Achy, S.; Kamel, S.; Refaat, T. Cancer active targeting by nanoparticles: A comprehensive review of literature. J. Cancer Res. Clin. Oncol. 2014, 141, 769–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, R.; Prakash, J.; Post, E.; Beljaars, L.; Schuppan, D.; Poelstra, K. Novel engineered targeted Interferon-Gamma blocks hepatic fibrogenesis in mice. Hepatology 2011, 54, 586–596. [Google Scholar] [CrossRef] [PubMed]
- SPDP Crosslinker; Thermo Scientific: Rockford, IL, USA, 2011.
- Pinheiro-Margis, M.; Margis, R.; Borojevic, R. Collagen synthesis in an established liver connective tissue cell line (GRX) during induction of the Fat-Storing phenotype. Exp. Mol. Pathol. 1992, 56, 108–118. [Google Scholar] [CrossRef]
- Tammam, S.N.; Azzazy, H.M.; Lamprecht, A. A high throughput method for quantification of cell surface bound and internalized chitosan nanoparticles. Int. J. Boil. Macromol. 2015, 81, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Shingleton, W.D.; Cawston, T.E.; Hodges, D.J.; Brick, P. Collagenase: A key enzyme in collagen turnover. Biochem. Cell Boil. 1996, 74, 759–775. [Google Scholar] [CrossRef] [PubMed]
- Muhamad, N.; Plengsuriyakarn, T.; Na-Bangchang, K. Application of active targeting nanoparticle delivery system for chemotherapeutic drugs and traditional/herbal medicines in cancer therapy: A systematic review. Int. J. Nanomed. 2018, 13, 3921–3935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boushehri, M.A.S.; Lamprecht, A. Nanoparticles as drug carriers: Current issues within vitrotesting. Nanomedicine 2015, 10, 3213–3230. [Google Scholar] [CrossRef] [PubMed]
- Scientific, T.F. Factors Influencing Transfection Efficiency. Available online: https://www.thermofisher.com/se/en/home/references/gibco-cell-culture-basics/transfection-basics/factors-influencing-transfection-efficiencyhtml (accessed on 29 October 2019).
- Lipofectamine® 2000 Reagent Protocol; Life Technologies: Carlsbad, CA, USA, 2013.
- Tammam, S.N.; Azzazy, H.M.; Lamprecht, A. How successful is nuclear targeting by nanocarriers? J. Control. Release 2016, 229, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-W.; Chiang, I.-N.; Wang, J.-H.; Young, T. Chitosan delaying human fibroblast senescence through downregulation of TGF-β signaling pathway. Artif. Cells Nanomed. Biotechnol. 2017, 46, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tammam, S.; Mathur, S.; Afifi, N. Preparation and biopharmaceutical evaluation of tacrolimus loaded biodegradable nanoparticles for liver targeting. J. Biomed. Nanotechnol. 2012, 8, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C.W. Nanoparticle–liver interactions: Cellular uptake and hepatobiliary elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-Y.; Hu, Y.-L.; Gao, J.-Q. Brain Localization and neurotoxicity evaluation of polysorbate 80-Modified chitosan nanoparticles in rats. PLoS ONE 2015, 10, e0134722. [Google Scholar] [CrossRef] [PubMed]
- Shadab, M.; Rashid, A.K.; Gulam, M.; Krishna, C.; Sanjula, B.; Jaseet, K.S.; Ali, J. Bromocriptine loaded chitosan nanoparticles intended for direct nose to brain delviery: Pharmacodynamic, pharmacokinetic and scintigraphy study in mice model. Eur. J. Pharm. Sci. 2013, 48, 393–405. [Google Scholar]
- Ogawa, M.; Mori, T.; Mori, Y.; Ueda, S.; Azemoto, R.; Makino, Y.; Wakashin, Y.; Ohto, M.; Wakashin, M.; Yoshida, H.; et al. Study on chronic renal injuries induced by carbon tetrachloride: Selective inhibtion of nephrotoxcity by irradiation. Nephron 1992, 60, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, K.; Anantharaman, P.; Chidambaram, N.; Balasubramanian, T.; Somasundaram, S.T. Padina boergessenii ameliorates carbon tetrachloride induced nephrotoxcity in wistar rats. J. King Saud. Univ.-Sci. 2012, 24, 227–232. [Google Scholar]
- Aly, M.S.; Galaly, S.R.; Moustafa, N.; Mohammed, H.M.; Khadrawy, S.M.; Mahmoud, A.M. Hesperidin protects against diethylnitrosamine/carbon Tetrachloride-Induced renal repercussions via Up-Regulation of Nrf2/HO-1 signaling and attenuation of oxidative stress. J. Appl. Pharm. Sci. 2017, 7, 7–14. [Google Scholar]
- Bülow, R.D.; Boor, P. Extracellular matrix in kidney fibrosis: More than just a scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demoy, M.; Andreux, J.; Weingarten, C.; Gouritin, B.; Guilloux, V.; Couvreur, P. Spleen capture of nanoparticles: Influence of animal species and surface characteristics. Pharm. Res. 1999, 16, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, M.; Vigliotti, C.; Mosca, T.; Cammarota, M.; Capone, D. Emerging role of the spleen in the pharmacokinetics of monoclonal antibodies, nanoparticles and exosomes. Int. J. Mol. Sci. 2017, 18, 1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghimi, S.M.; Porter, C.; Muir, I.; Illum, L.; Davis, S. Non-Phagocytic uptake of intravenously injected microspheres in rat spleen: Influence of particle size and hydrophilic coating. Biochem. Biophys. Res. Commun. 1991, 177, 861–866. [Google Scholar] [CrossRef]
- Amoozgar, Z.; Park, J.; Lin, Q.; Yeo, Y. Low Molecular-Weight chitosan as a pH-Sensitive stealth coating for Tumor-Specific drug delivery. Mol. Pharm. 2012, 9, 1262–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abouelmagd, S.A.; Ku, Y.J.; Yeo, Y. Low molecular weight Chitosan-Coated polymeric nanoparticles for sustained and pH-Sensitive delivery of paclitaxel. J. Drug Target. 2015, 23, 725–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastin, A.J. Handbook of Biologically Active Peptides, 2nd ed.; Elsevier: San Diego, CA, USA, 2013. [Google Scholar]
- Axel, M.G. Roles of TGF-Beta in hepatic fibrosis. Front. Biosci. 2002, 7, 793–807. [Google Scholar]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-Beta targted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.C.; Shapiro, G.I.; Tan, A.R.; Lawrence, D.P.; Olencki, T.E.; Dezube, B.J.; Hsu, F.J.; Reiss, M.; Berzofsky, J.A. Phase I/II study of GC1008: A human Anti-Transforming growth Factor-Beta (TGFβ) monoclonal antibody (MAb) in patients with advanced malignant melanoma (MM) or renal cell carcinoma (RCC). J. Clin. Oncol. 2008, 26, 9028. [Google Scholar] [CrossRef]
- Rice, L.M.; Padilla, C.M.; McLaughlin, S.R.; Mathes, A.; Ziemek, J.; Goummih, S.; Nakerakanti, S.; York, M.; Farina, G.; Whitfield, M.L.; et al. Fresolimumab treatmnet decreases biomarkers and improves clinical symptoms in systemic patients. J. Clin. Investig. 2015, 125, 2795–2807. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzam, M.; El Safy, S.; Abdelgelil, S.A.; Weiskirchen, R.; Asimakopoulou, A.; de Lorenzi, F.; Lammers, T.; Mansour, S.; Tammam, S. Targeting Activated Hepatic Stellate Cells Using Collagen-Binding Chitosan Nanoparticles for siRNA Delivery to Fibrotic Livers. Pharmaceutics 2020, 12, 590. https://doi.org/10.3390/pharmaceutics12060590
Azzam M, El Safy S, Abdelgelil SA, Weiskirchen R, Asimakopoulou A, de Lorenzi F, Lammers T, Mansour S, Tammam S. Targeting Activated Hepatic Stellate Cells Using Collagen-Binding Chitosan Nanoparticles for siRNA Delivery to Fibrotic Livers. Pharmaceutics. 2020; 12(6):590. https://doi.org/10.3390/pharmaceutics12060590
Chicago/Turabian StyleAzzam, Menna, Sara El Safy, Sarah A. Abdelgelil, Ralf Weiskirchen, Anastasia Asimakopoulou, Federica de Lorenzi, Twan Lammers, Samar Mansour, and Salma Tammam. 2020. "Targeting Activated Hepatic Stellate Cells Using Collagen-Binding Chitosan Nanoparticles for siRNA Delivery to Fibrotic Livers" Pharmaceutics 12, no. 6: 590. https://doi.org/10.3390/pharmaceutics12060590