Importance of Mesoporous Silica Particle Size in the Stabilization of Amorphous Pharmaceuticals—The Case of Simvastatin

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
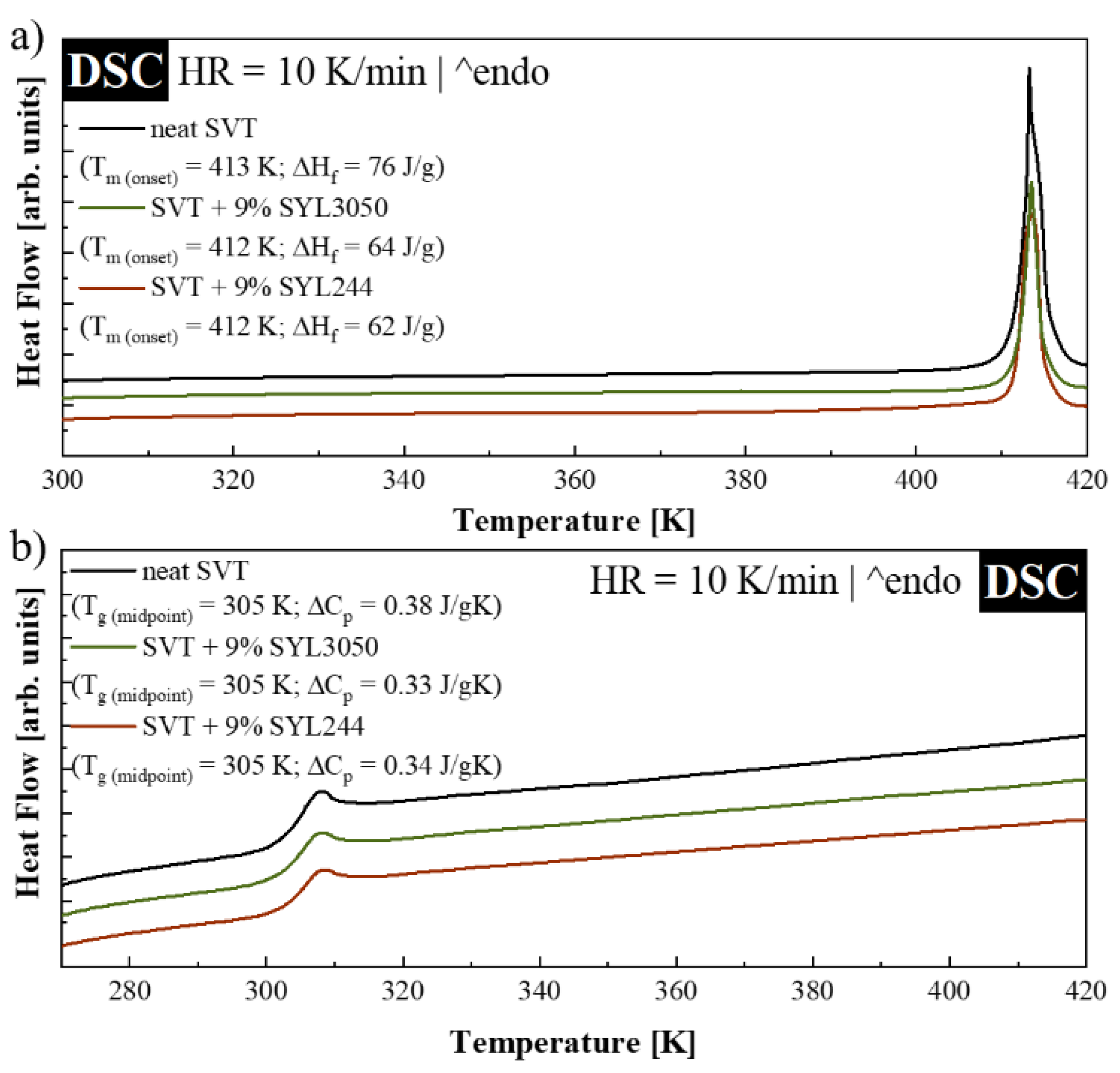
2.3. Differential Scanning Calorimetry (DSC)
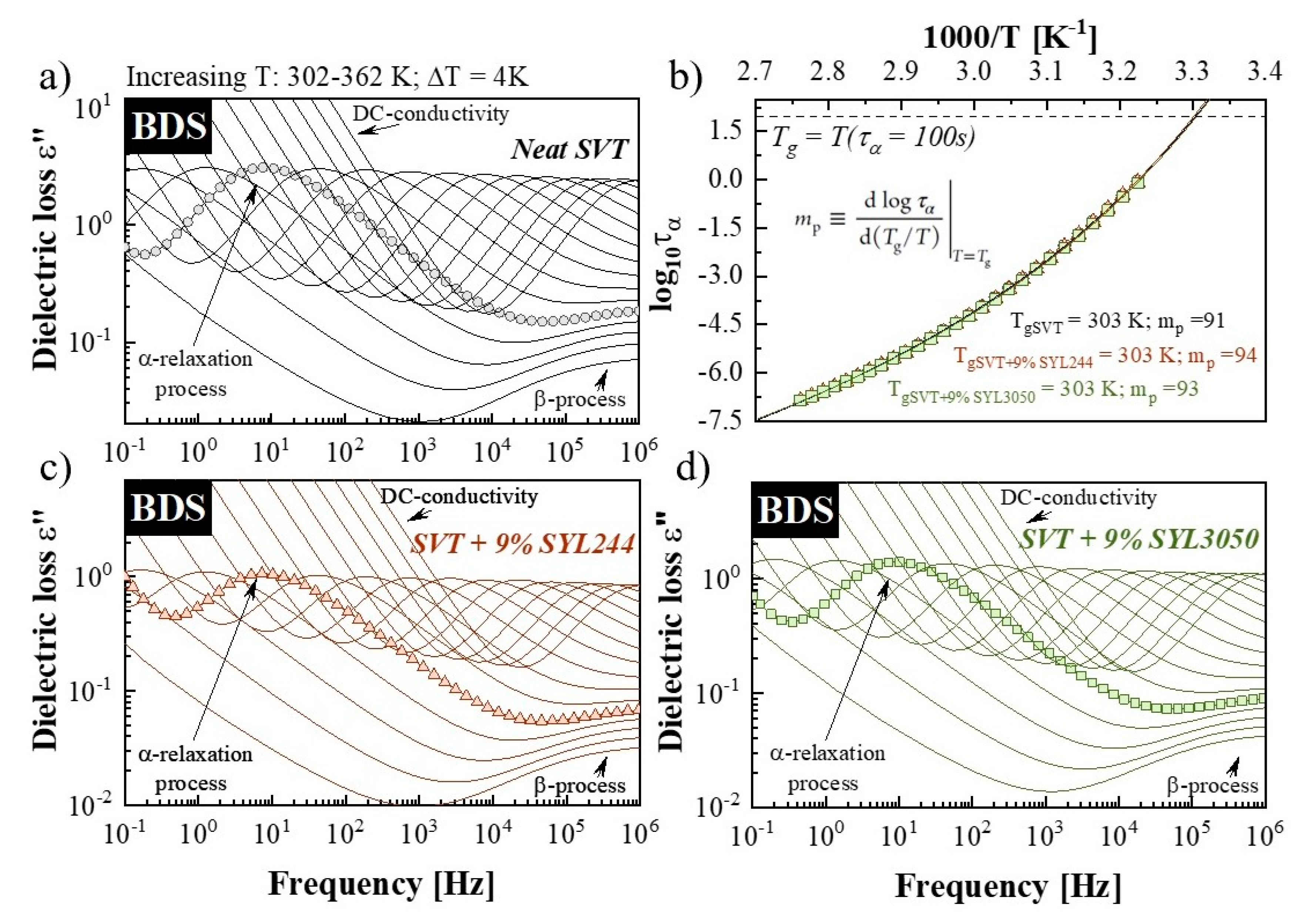
2.4. Broadband Dielectric Spectroscopy (BDS)
2.5. Optical Microscope
3. Results and Discussion
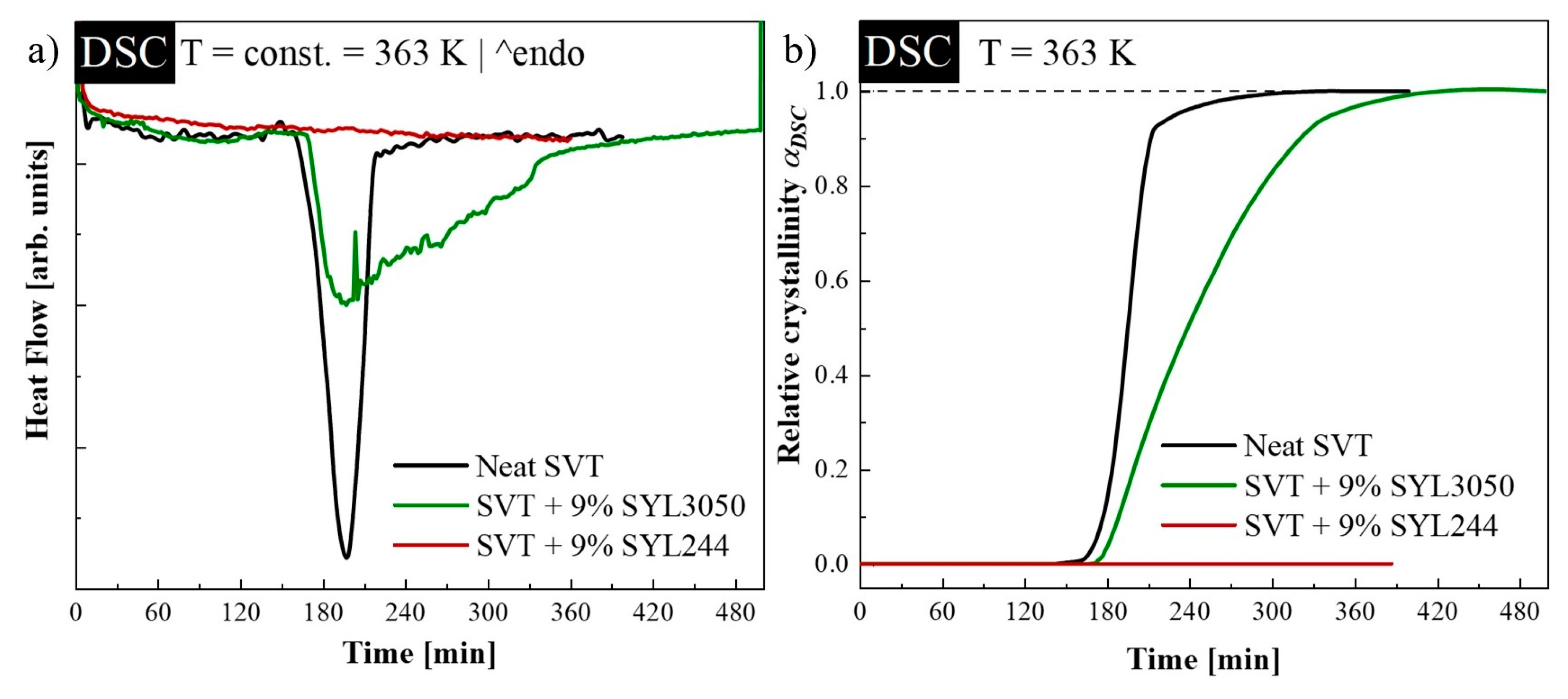
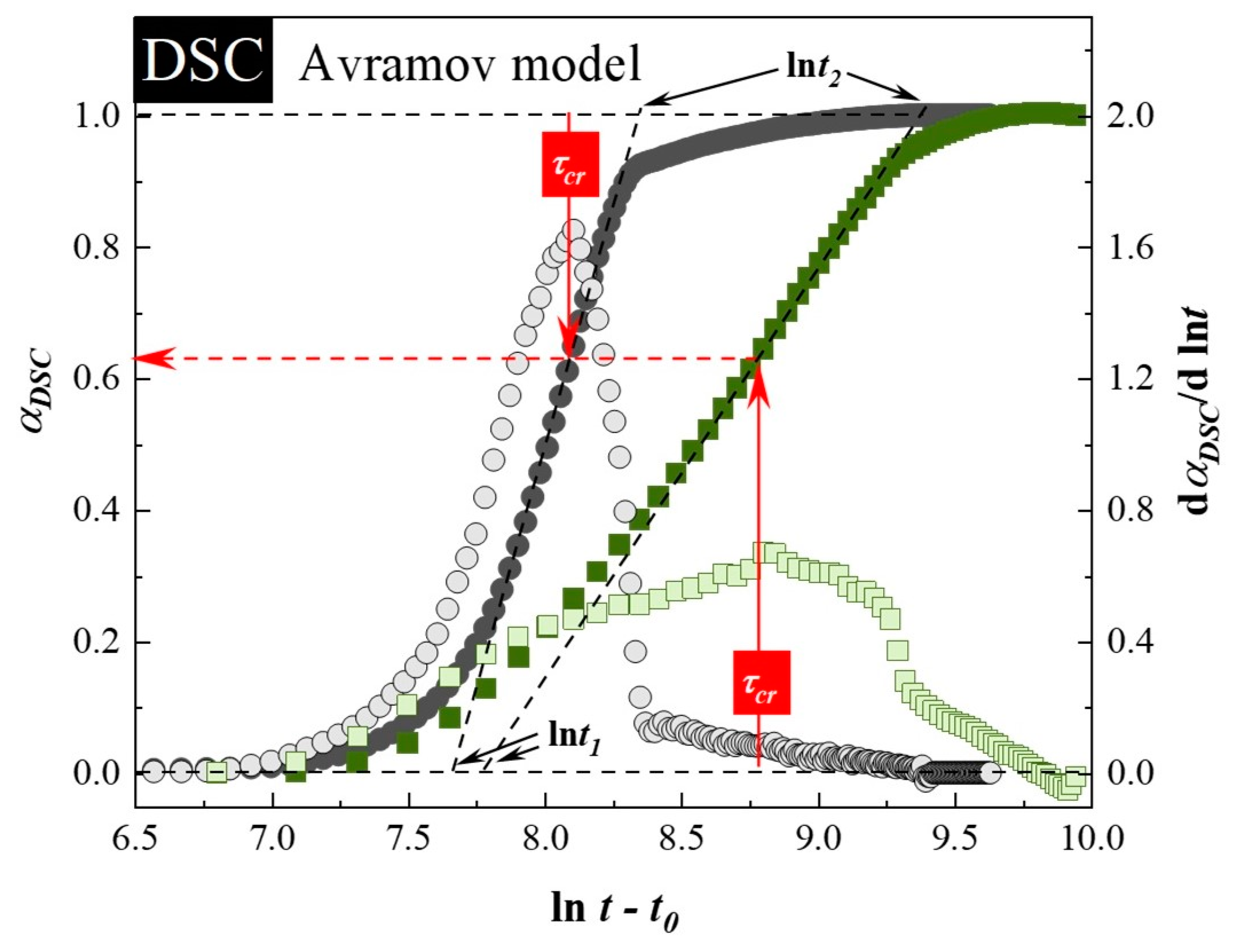
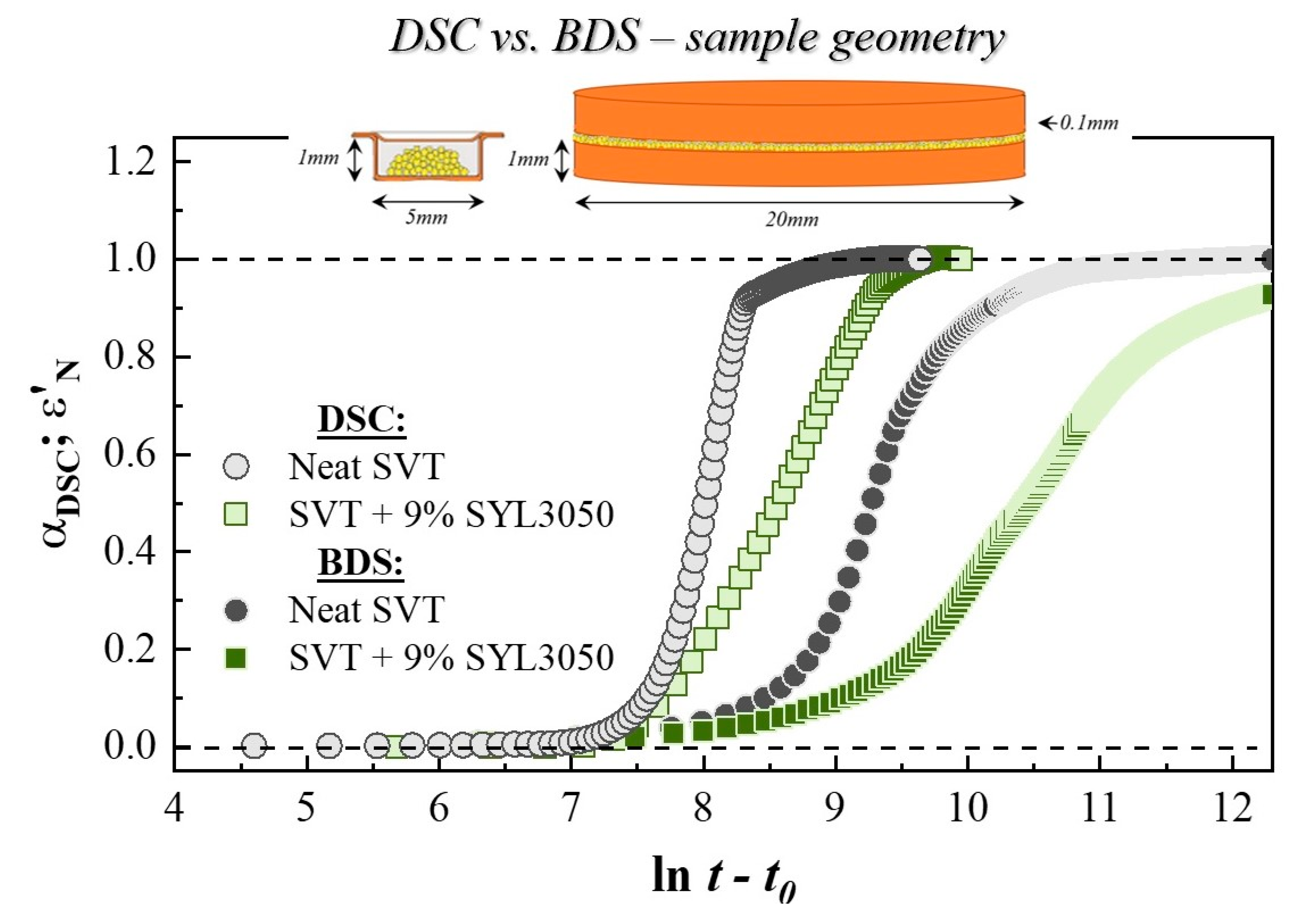
3.1. Isothermal Crystallization Studies Performed by DSC
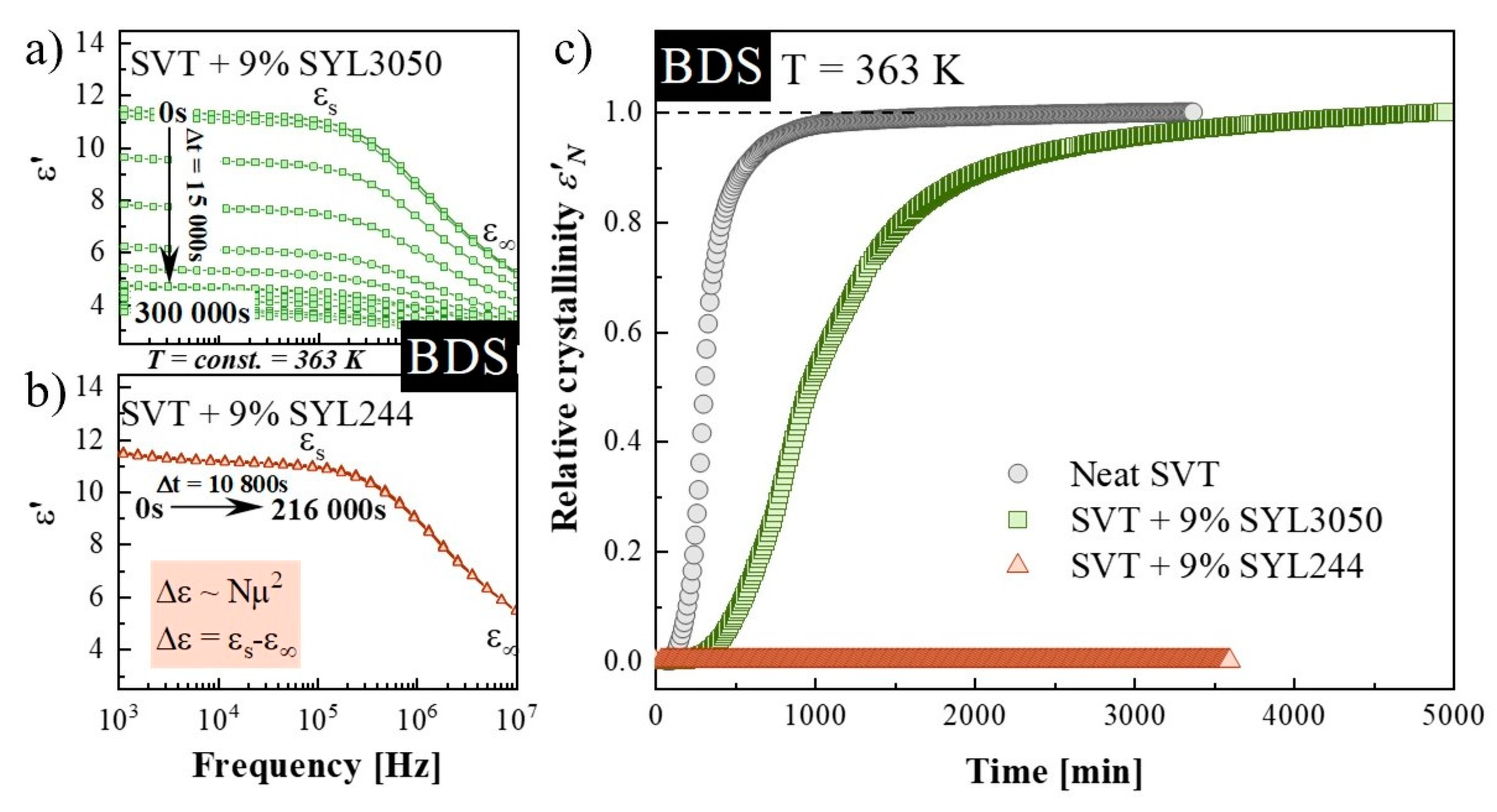
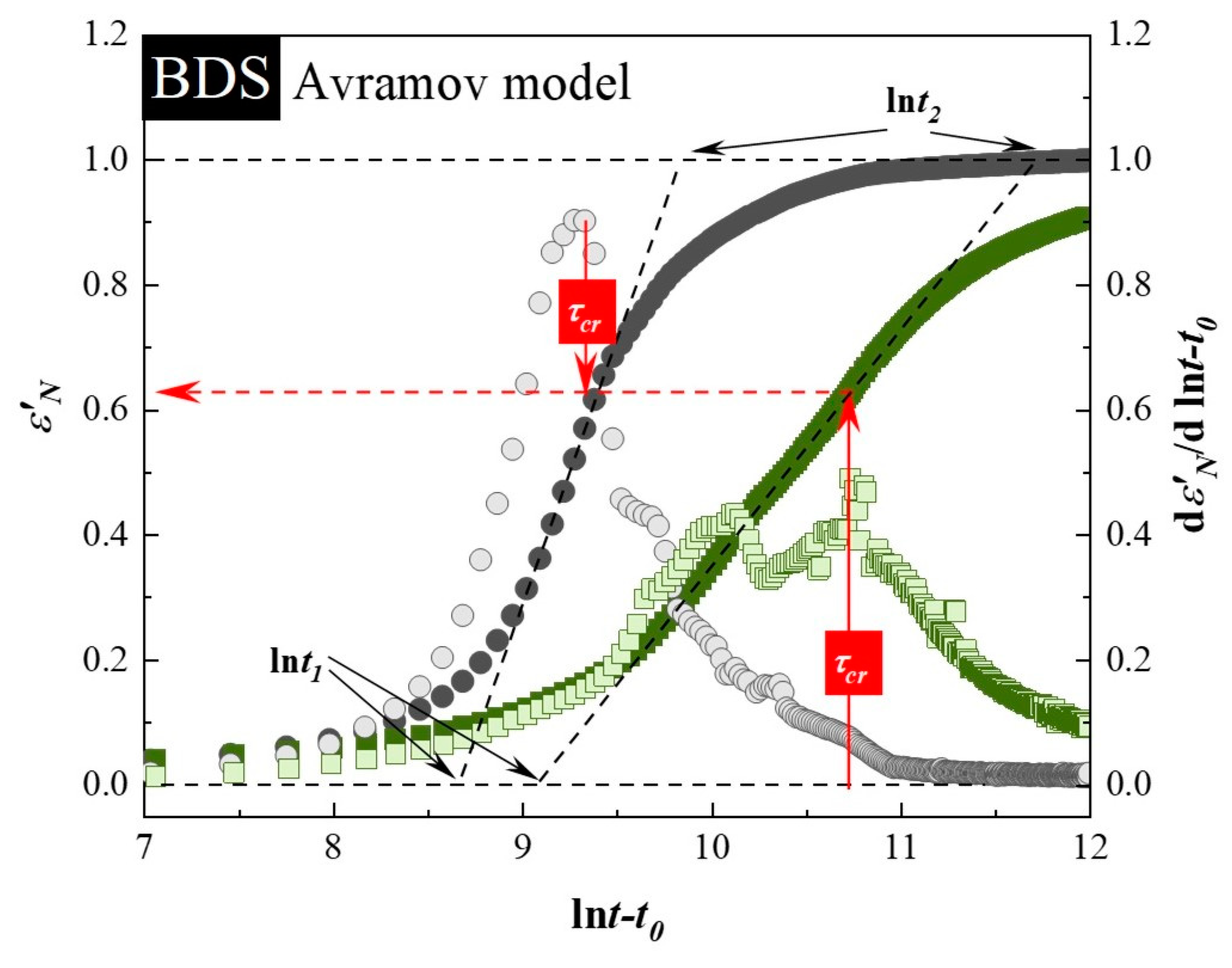
3.2. Isothermal Crystallization Studies Performed by BDS
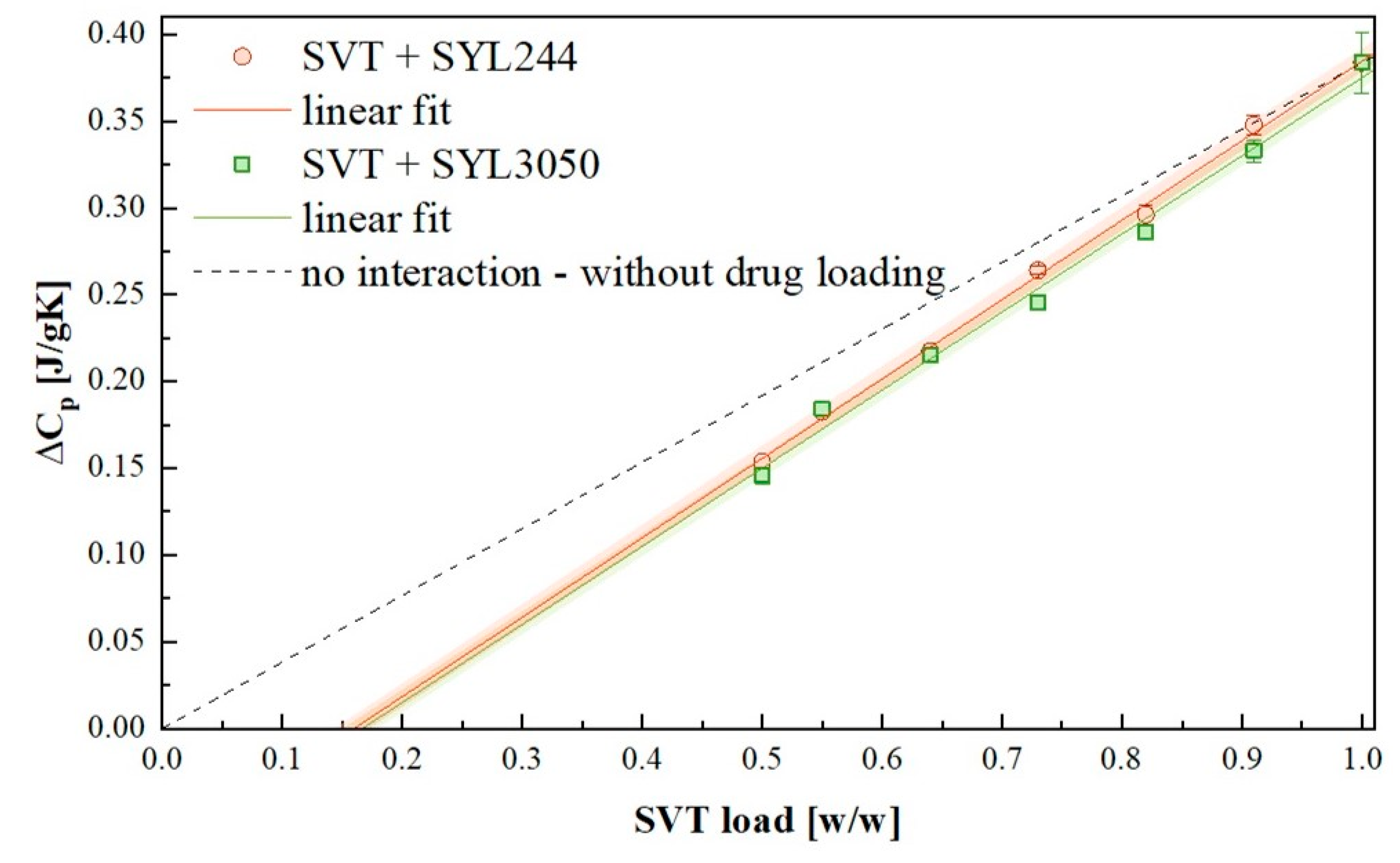
3.3. Loading Capacity of MSs for SVT
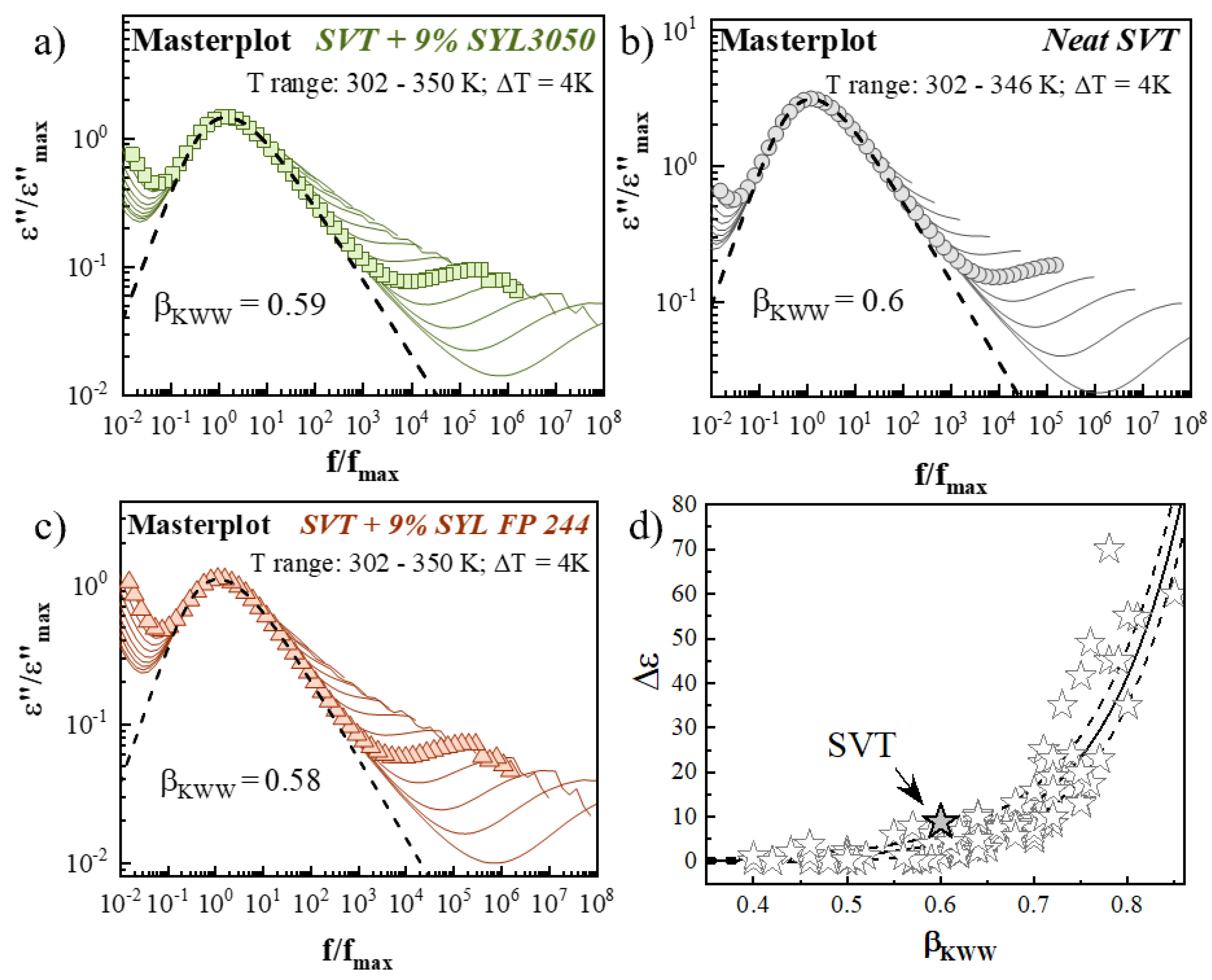
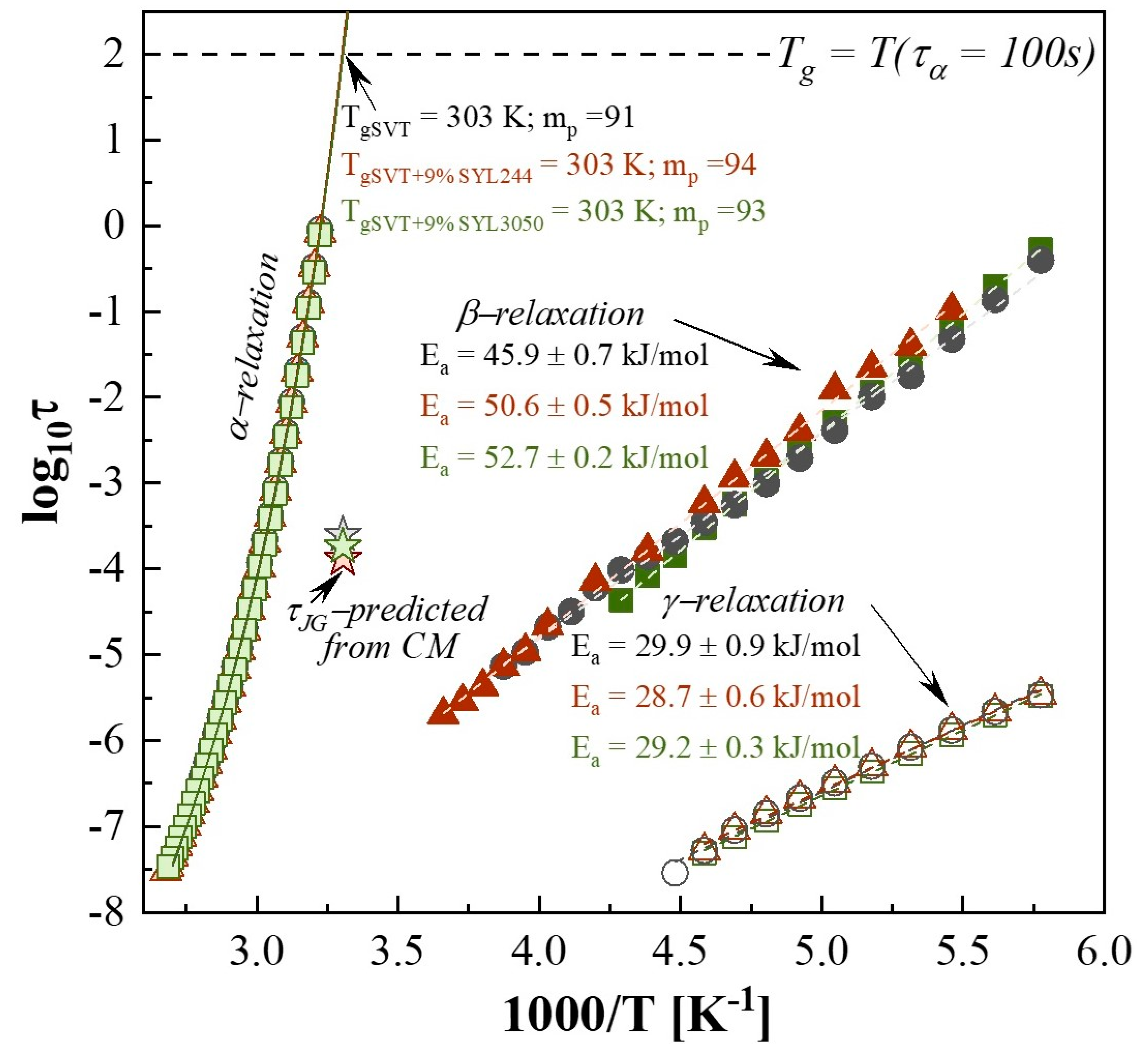
3.4. Effect of MS Materials on the Molecular Mobility of Supercooled SVT

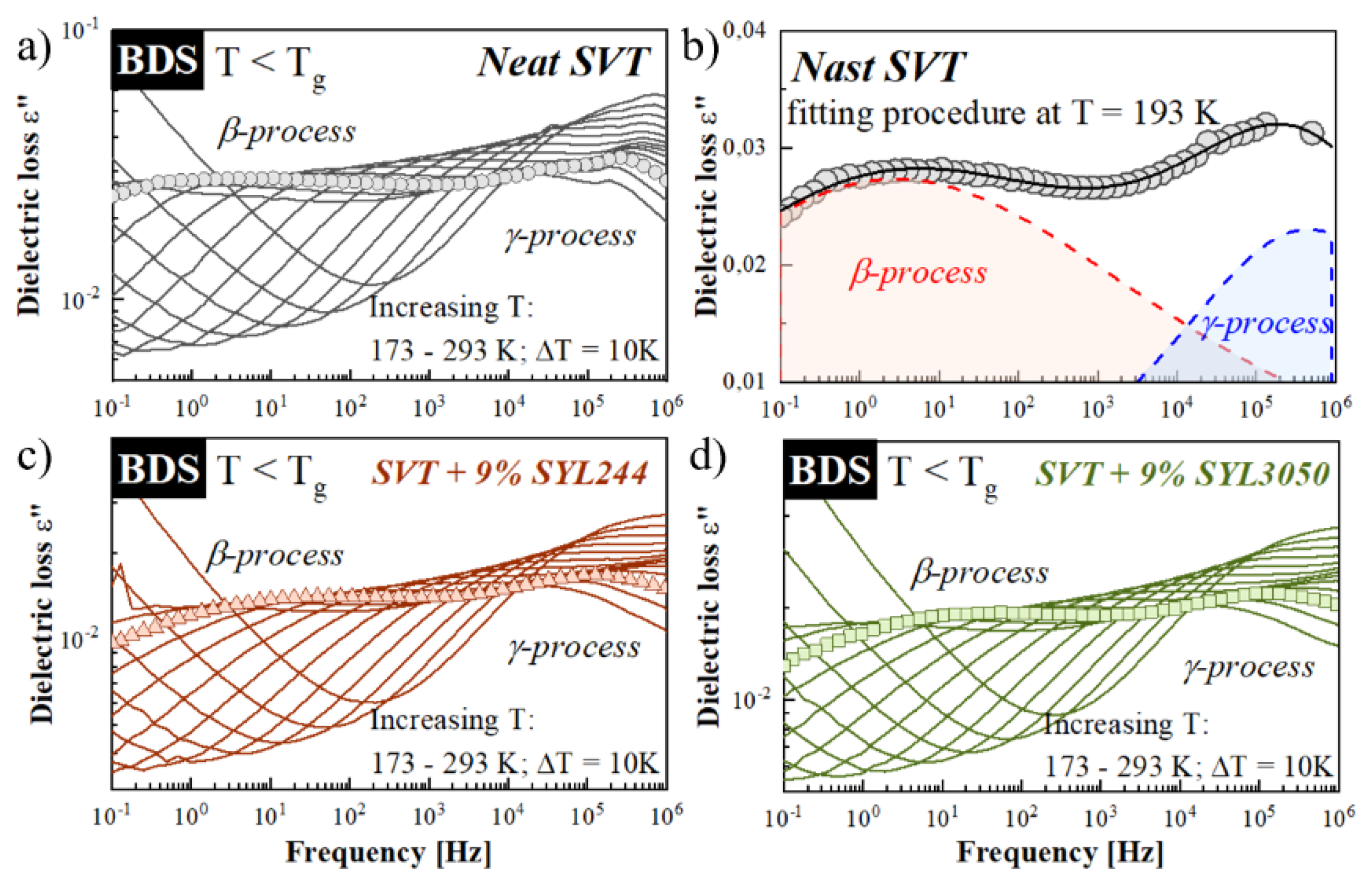
3.5. Effect of MS Materials on the Molecular Mobility of Glassy SVT
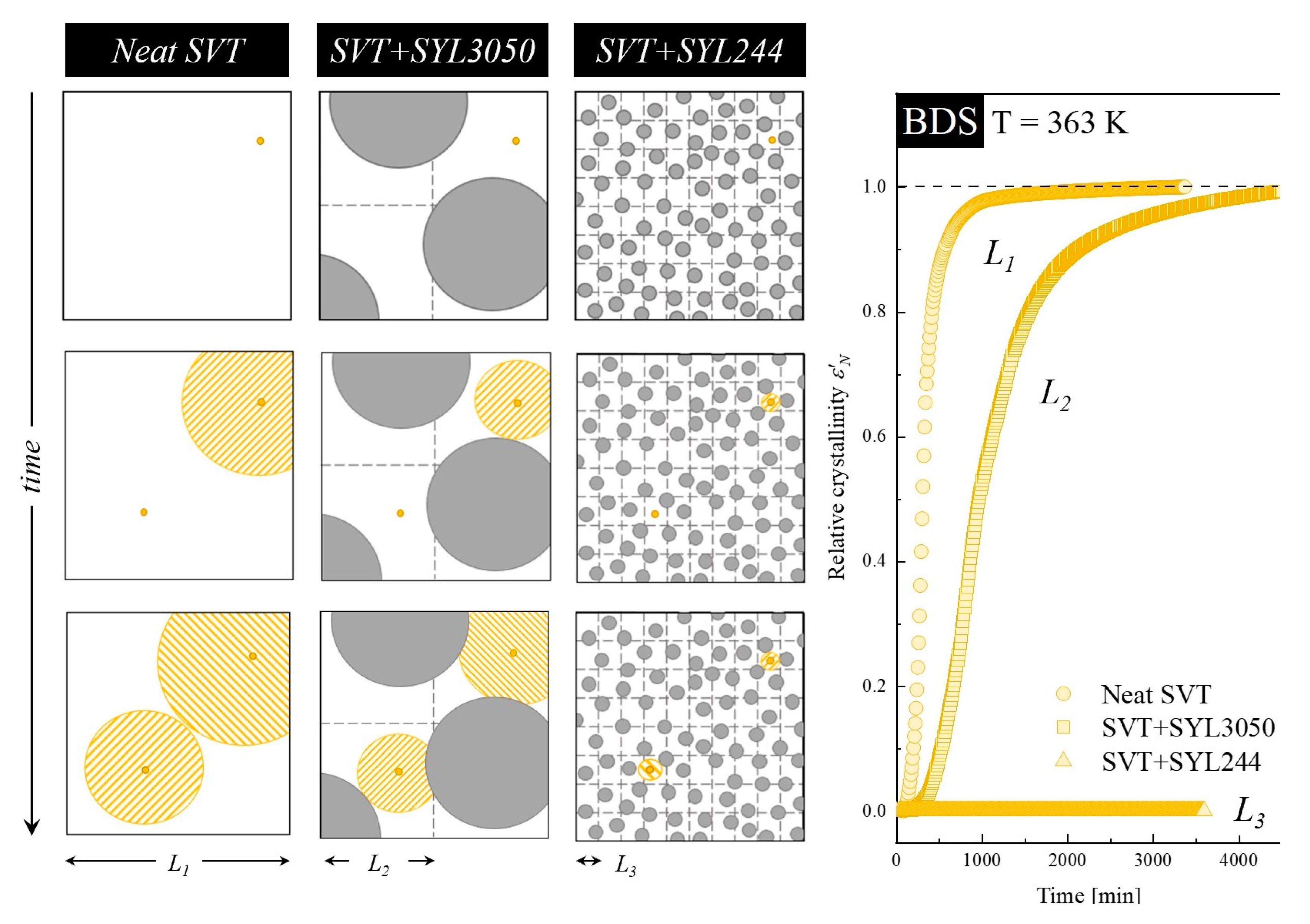
3.6. Mechanism of SVT Stabilization with MS Materials
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baird, J.A.; Taylor, L.S. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv. Drug Deliv. Rev. 2012, 64, 396–421. [Google Scholar] [CrossRef]
- Liu, X.; Fahr, A. Drug delivery strategies for poorly water-soluble drugs. Expert Opin. Drug Deliv. 2007, 4, 403–416. [Google Scholar]
- Williams, H.; Trevaskis, N.; Charman, S.; Shanker, R.; Charman, W.; Pouton, C.; Porter, C. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2016, 5, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, S.; Kushida, I.; Yamashita, T.; Hasebe, T.; Shirai, O.; Kano, K. Evaluation of drug supersaturation by thermodynamic and kinetic approaches for the prediction of oral absorbability in amorphous pharmaceuticals. J. Pharm. Sci. 2012, 101, 4220–4230. [Google Scholar] [CrossRef]
- Almeida E Sousa, L.; Reutzel-Edens, S.M.; Stephenson, G.A.; Taylor, L.S. Assessment of the amorphous “solubility” of a group of diverse drugs using new experimental and theoretical approaches. Mol. Pharm. 2015, 12, 484–495. [Google Scholar] [CrossRef]
- Bogner, R.H.; Murdande, S.B.; Pikal, M.J.; Shanker, R.M. Solubility advantage of amorphous pharmaceuticals: II. application of quantitative thermodynamic relationships for prediction of solubility enhancement in structurally diverse insoluble pharmaceuticals. Pharm. Res. 2010, 27, 2704–2714. [Google Scholar]
- Paradkar, A.R.; Chauhan, B.; Yamamura, S.; Pawar, A.P. Preparation and characterization of glassy celecoxib. Drug Dev. Ind. Pharm. 2003, 29, 739–744. [Google Scholar] [CrossRef]
- Chmiel, K.; Knapik-Kowalczuk, J.; Jurkiewicz, K.; Sawicki, W.; Jachowicz, R.; Paluch, M. A New Method to Identify Physically Stable Concentration of Amorphous Solid Dispersions (I): Case of Flutamide + Kollidon VA64. Mol. Pharm. 2017, 14, 3370–3380. [Google Scholar] [CrossRef]
- Kawakami, K. Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. Adv. Drug Deliv. Rev. 2012, 64, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Long-Term Physical Stability of PVP- and PVPVA-Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Szklarz, G.; Adrjanowicz, K.; Knapik-Kowalczuk, J.; Jurkiewicz, K.; Paluch, M. Crystallization of supercooled fenofibrate studied at ambient and elevated pressures. Phys. Chem. Chem. Phys. 2017, 19, 9879–9888. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, M.; Hancock, B.C.; Zografi, G. Crystallization of indomethacin from the amorphous state below and above its glass transition temperature. J. Pharm. Sci. 1994, 83, 1700–1705. [Google Scholar] [CrossRef]
- Kawakami, K.; Harada, T.; Miura, K.; Yoshihashi, Y.; Yonemochi, E.; Terada, K.; Moriyama, H. Relationship between crystallization tendencies during cooling from melt and isothermal storage: Toward a general understanding of physical stability of pharmaceutical glasses. Mol. Pharm. 2014, 11, 1835–1843. [Google Scholar] [CrossRef]
- Knapik, J.; Wojnarowska, Z.; Grzybowska, K.; Tajber, L.; Mesallati, H.; Paluch, K.J.; Paluch, M. Molecular Dynamics and Physical Stability of Amorphous Nimesulide Drug and Its Binary Drug-Polymer Systems. Mol. Pharm. 2016, 13, 1937–1946. [Google Scholar] [CrossRef] [Green Version]
- Knapik-Kowalczuk, J.; Tu, W.; Chmiel, K.; Rams-Baron, M.; Paluch, M. Co-Stabilization of Amorphous Pharmaceuticals—The Case of Nifedipine and Nimodipine. Mol. Pharm. 2018, 15, 2455–2465. [Google Scholar] [CrossRef]
- Kawakami, K. Ultraslow Cooling for the Stabilization of Pharmaceutical Glasses. J. Phys. Chem. B 2019, 123, 4996–5003. [Google Scholar] [CrossRef]
- Grzybowska, K.; Chmiel, K.; Knapik-Kowalczuk, J.; Grzybowski, A.; Jurkiewicz, K.; Paluch, M. Molecular factors governing the liquid and glassy states recrystallization of celecoxib in binary mixtures with excipients of different molecular weights. Mol. Pharm. 2017, 14, 1154–1168. [Google Scholar] [CrossRef]
- Knapik, J.; Wojnarowska, Z.; Grzybowska, K.; Jurkiewicz, K.; Tajber, L.; Paluch, M. Molecular dynamics and physical stability of coamorphous ezetimib and indapamide mixtures. Mol. Pharm. 2015, 12, 3610–3619. [Google Scholar] [CrossRef]
- Knapik-Kowalczuk, J.; Wojnarowska, Z.; Rams-Baron, M.; Jurkiewicz, K.; Cielecka-Piontek, J.; Ngai, K.L.; Paluch, M. Atorvastatin as a Promising Crystallization Inhibitor of Amorphous Probucol: Dielectric Studies at Ambient and Elevated Pressure. Mol. Pharm. 2017, 14, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Grzybowska, K.; Paluch, M.; Grzybowski, A.; Wojnarowska, Z.; Hawelek, L.; Kolodziejczyk, K.; Ngai, K.L. Molecular dynamics and physical stability of amorphous anti-inflammatory drug: Celecoxib. J. Phys. Chem. B 2010, 114, 12792–12801. [Google Scholar] [CrossRef] [PubMed]
- Priemel, P.A.; Laitinen, R.; Barthold, S.; Grohganz, H.; Lehto, V.P.; Rades, T.; Strachan, C.J. Inhibition of surface crystallisation of amorphous indomethacin particles in physical drug-polymer mixtures. Int. J. Pharm. 2013, 456, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Lehmkemper, K.; Kyeremateng, S.O.; Degenhardt, M.; Sadowski, G. Influence of Low-Molecular-Weight Excipients on the Phase Behavior of PVPVA64 Amorphous Solid Dispersions. Pharm. Res. 2018, 35, 25. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Moinuddin, S.M.; Cai, T. Advances in coamorphous drug delivery systems. Acta Pharm. Sin. B 2019, 9, 19–35. [Google Scholar] [CrossRef]
- Azaı, T.; Tourné-Péteilh, C.; Aussenac, F.; Baccile, N.; Coelho, C.; Devoisselle, J.-M.; Babonneau, F. Solid-State NMR Study of Ibuprofen Confined in MCM-41 Material. Chem. Mater. 2006, 18, 6382–6390. [Google Scholar] [CrossRef]
- Bahl, D.; Bogner, R.H. Amorphization of indomethacin by co-grinding with Neusilin US2: Amorphization kinetics, physical stability and mechanism. Pharm. Res. 2006, 23, 2317–2325. [Google Scholar] [CrossRef]
- Andersson, J.; Rosenholm, J.; Areva, S.; Lindén, M. Influences of material characteristics on ibuprofen drug loading and release profiles from ordered micro- and mesoporous silica matrices. Chem. Mater. 2004, 16, 4160–4167. [Google Scholar] [CrossRef]
- Bremmell, K.E.; Prestidge, C.A. Enhancing oral bioavailability of poorly soluble drugs with mesoporous silica based systems: Opportunities and challenges. Drug Dev. Ind. Pharm. 2019, 45, 349–358. [Google Scholar] [CrossRef]
- Bukara, K.; Schueller, L.; Rosier, J.; Martens, M.A.; Daems, T.; Verheyden, L.; Eelen, S.; Van Speybroeck, M.; Libanati, C.; Martens, J.A.; et al. Ordered mesoporous silica to enhance the bioavailability of poorly water-soluble drugs: Proof of concept in man. Eur. J. Pharm. Biopharm. 2016, 108, 220–225. [Google Scholar] [CrossRef]
- Cassano, D.; Mapanao, A.K.; Summa, M.; Vlamidis, Y.; Giannone, G.; Santi, M.; Guzzolino, E.; Pitto, L.; Poliseno, L.; Bertorelli, R.; et al. Biosafety and Biokinetics of Noble Metals: The Impact of Their Chemical Nature. ACS Appl. Bio Mater. 2019, 2, 4464–4470. [Google Scholar] [CrossRef]
- Croissant, J.G.; Fatieiev, Y.; Khashab, N.M. Degradability and Clearance of Silicon, Organosilica, Silsesquioxane, Silica Mixed Oxide, and Mesoporous Silica Nanoparticles. Adv. Mater. 2017, 29, 1604634. [Google Scholar] [CrossRef] [PubMed]
- Genina, N.; Hadi, B.; Löbmann, K. Hot Melt Extrusion as Solvent-Free Technique for a Continuous Manufacturing of Drug-Loaded Mesoporous Silica. J. Pharm. Sci. 2018, 107, 149–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rengarajan, G.T.; Enke, D.; Steinhart, M.; Beiner, M. Stabilization of the amorphous state of pharmaceuticals in nanopores. J. Mater. Chem. 2008, 18, 2537–2539. [Google Scholar] [CrossRef]
- Knapik, J.; Wojnarowska, Z.; Grzybowska, K.; Jurkiewicz, K.; Stankiewicz, A.; Paluch, M. Stabilization of the Amorphous Ezetimibe Drug by Confining Its Dimension. Mol. Pharm. 2016, 13, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, R.; Löbmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Riikonen, J.; Xu, W.; Lehto, V.P. Mesoporous systems for poorly soluble drugs—Recent trends. Int. J. Pharm. 2018, 536, 178–186. [Google Scholar] [CrossRef]
- Bavnhøj, C.G.; Knopp, M.M.; Madsen, C.M.; Löbmann, K. The role interplay between mesoporous silica pore volume and surface area and their effect on drug loading capacity. Int. J. Pharm. X 2019, 1, 100008. [Google Scholar] [CrossRef]
- Hempel, N.J.; Brede, K.; Olesen, N.E.; Genina, N.; Knopp, M.M.; Löbmann, K. A fast and reliable DSC-based method to determine the monomolecular loading capacity of drugs with good glass-forming ability in mesoporous silica. Int. J. Pharm. 2018, 544, 153–157. [Google Scholar] [CrossRef]
- Mellaerts, R.; Aerts, C.A.; Van Humbeeck, J.; Augustijns, P.; Van Den Mooter, G.; Martens, J.A. Enhanced release of itraconazole from ordered mesoporous SBA-15 silica materials. Chem. Commun. 2007, 1375–1377. [Google Scholar] [CrossRef]
- Antonino, R.S.C.M.Q.; Ruggiero, M.; Song, Z.; Nascimento, T.L.; Lima, E.M.; Bohr, A.; Knopp, M.M.; Löbmann, K. Impact of drug loading in mesoporous silica-amorphous formulations on the physical stability of drugs with high recrystallization tendency. Int. J. Pharm. X 2019, 1, 100026. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sailaja Chirravuri, S.V.; Shastri, N.R. Impact of surface area of silica particles on dissolution rate and oral bioavailability of poorly water soluble drugs: A case study with aceclofenac. Int. J. Pharm. 2014, 461, 459–468. [Google Scholar] [CrossRef]
- Yani, Y.; Chow, P.S.; Tan, R.B.H. Pore size effect on the stabilization of amorphous drug in a mesoporous material: Insights from molecular simulation. Microporous Mesoporous Mater. 2016, 221, 117–122. [Google Scholar] [CrossRef]
- Rao, M.; Mandage, Y.; Thanki, K.; Bhise, S. Dissolution improvement of simvastatin by surface solid dispersion technology. Dissolution Technol. 2010, 17, 27–34. [Google Scholar] [CrossRef]
- Vraníková, B.; Niederquell, A.; Ditzinger, F.; Šklubalová, Z.; Kuentz, M. Mechanistic aspects of drug loading in liquisolid systems with hydrophilic lipid-based mixtures. Int. J. Pharm. 2020, 578, 119099. [Google Scholar] [CrossRef]
- Rao, S.; Tan, A.; Boyd, B.J.; Prestidge, C.A. Synergistic role of self-emulsifying lipids and nanostructured porous silica particles in optimizing the oral delivery of lovastatin. Nanomedicine 2014, 9, 2745–2759. [Google Scholar] [CrossRef]
- Kremer, F.; Schonhals, A. Broadband Dielectric Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2003; ISBN 978-3-642-62809-2. [Google Scholar]
- Grzybowska, K.; Capaccioli, S.; Paluch, M. Recent developments in the experimental investigations of relaxations in pharmaceuticals by dielectric techniques at ambient and elevated pressure. Adv. Drug Deliv. Rev. 2016, 100, 158–182. [Google Scholar] [CrossRef]
- Descamps, M. Disordered Pharmaceutical Materials; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; ISBN 9783527652693. [Google Scholar]
- Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; Jamroz, W.; Paluch, M. Amorphous Drugs; Springer International Publishing: Cham, Switzerland, 2018; ISBN 9783319720012. [Google Scholar]
- Watson, E.S.; O’Neill, M.J.; Justin, J.; Brenner, N. A Differential Scanning Calorimeter for Quantitative Differential Thermal Analysis. Anal. Chem. 1964, 36, 1233–1238. [Google Scholar] [CrossRef]
- Höhne, G.W.H.; Hemminger, W.F.; Flammersheim, H.-J. Differential Scanning Calorimetry; Springer: Berlin/Heidelberg, Germany, 2003; ISBN 9783642055935. [Google Scholar]
- Kolodziejczyk, K.; Grzybowska, K.; Wojnarowska, Z.; Dulski, M.; Hawelek, L.; Paluch, M. Isothermal cold crystallization kinetics study of sildenafil. Cryst. Growth Des. 2014, 14, 3199–3209. [Google Scholar] [CrossRef]
- Avramov, I.; Avramova, K.; Rüssel, C. New method to analyze data on overall crystallization kinetics. J. Cryst. Growth 2005, 285, 394–399. [Google Scholar] [CrossRef]
- Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; Jamroz, W.; Paluch, M. Amorphous Drugs: Benefits and Challenges; Springer International Publishing: Cham, Switzerland, 2018; ISBN 9783319720029. [Google Scholar]
- Dantuluri, A.K.R.; Amin, A.; Puri, V.; Bansal, A.K. Role of r-Relaxation on Crystallization of Amorphous Celecoxib above T g Probed by Dielectric Spectroscopy. Mol. Pharm. 2011, 8, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Knapik-Kowalczuk, J.; Chmiel, K.; Paluch, M. Glass Transition Dynamics and Physical Stability of Amorphous Griseofulvin in Binary Mixtures with Low-T g Excipients. Mol. Pharm. 2019, 16, 3626–3635. [Google Scholar] [CrossRef] [PubMed]
- Hargis, M.J.; Grady, B.P. Effect of sample size on isothermal crystallization measurements performed in a differential scanning calorimeter: A method to determine avrami parameters without sample thickness effects. Thermochim. Acta 2006, 443, 147–158. [Google Scholar] [CrossRef]
- Kawakami, K. Surface effects on the crystallization of ritonavir glass. J. Pharm. Sci. 2015, 104, 276–279. [Google Scholar] [CrossRef]
- Wu, T.; Sun, Y.; Li, N.; De Villiers, M.M.; Yu, L. Inhibiting surface crystallization of amorphous indomethacin by nanocoating. Langmuir 2007, 23, 5148–5153. [Google Scholar] [CrossRef]
- Kawakami, K.; Harada, T.; Yoshihashi, Y.; Yonemochi, E.; Terada, K.; Moriyama, H. Correlation between Glass-Forming Ability and Fragility of Pharmaceutical Compounds. J. Phys. Chem. B 2015, 119, 4873–4880. [Google Scholar] [CrossRef]
- Havriliak, S.; Negami, S. A complex plane representation of dielectric and mechanical relaxation processes in some polymers. Polymer 1967, 8, 161–210. [Google Scholar] [CrossRef]
- Vogel, H. Das Temperaturabhangigkeitgesetz der Viskosität von Flüssigkeiten. J. Phys. Z. 1921, 22, 645–646. [Google Scholar]
- Fulcher, G.S. Analysis of Recent Measurements of the Viscosity of Glasses. J. Am. Ceram. Soc. 1925, 8, 339–355. [Google Scholar] [CrossRef]
- Tammann, G.; Hesse, W. Die Abhängigkeit der Viscosität von der Temperatur bie unterkühlten Flüssigkeiten. Z. Anorg. Allg. Chem. 1926, 156, 245–257. [Google Scholar] [CrossRef]
- Böhmer, R.; Ngai, K.L.; Angell, C.A.; Plazek, D.J. Nonexponential relaxations in strong and fragile glass formers. J. Chem. Phys. 1993, 99, 4201–4209. [Google Scholar] [CrossRef]
- Kawakami, K. Pharmaceutical Applications of Thermal Analysis. In Handbook of Thermal Analysis and Calorimetry; Elsevier: Amsterdam, The Netherlands, 2018; Volume 6, pp. 613–641. ISBN 9780444640628. [Google Scholar]
- Knapik, J.; Wojnarowska, Z.; Grzybowska, K.; Hawelek, L.; Sawicki, W.; Wlodarski, K.; Markowski, J.; Paluch, M. Physical stability of the amorphous anticholesterol agent (Ezetimibe): The role of molecular mobility. Mol. Pharm. 2014, 11, 4280–4290. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H. Relationship among glass-forming ability, fragility, and short-range bond ordering of liquids. J. Non. Cryst. Solids 2005, 351, 678–690. [Google Scholar] [CrossRef]
- Williams, G.; Watts, D.C. Non-symmetrical dielectric relaxation behaviour arising from a simple empirical decay function. Trans. Faraday Soc. 1970, 66, 80. [Google Scholar] [CrossRef]
- Shamblin, S.L.; Tang, X.; Chang, L.; Hancock, B.; Pikal, M.J. Characterization of the Time Scales of Molecular Motion in Pharmaceutically Important Glasses. J. Phys. Chem. B 1999, 103, 4113–4121. [Google Scholar] [CrossRef]
- Paluch, M.; Knapik, J.; Wojnarowska, Z.; Grzybowski, A.; Ngai, K.L. Universal Behavior of Dielectric Responses of Glass Formers: Role of Dipole-Dipole Interactions. Phys. Rev. Lett. 2016, 116, 025702. [Google Scholar] [CrossRef]
- Knapik-Kowalczuk, J.; Wojnarowska, Z.; Chmiel, K.; Rams-Baron, M.; Tajber, L.; Paluch, M. Can storage time improve the physical stability of amorphous pharmaceuticals with tautomerization ability exposed to compression? The case of chloramphenicol drug. Mol. Pharm. 2018, 15, 1928–1940. [Google Scholar] [CrossRef]
- Knapik-Kowalczuk, J.; Gündüz, M.G.; Chmiel, K.; Jurkiewicz, K.; Kurek, M.; Tajber, L.; Jachowicz, R.; Paluch, M. Molecular dynamics, viscoelastic properties and physical stability studies of a new amorphous dihydropyridine derivative with T-type calcium channel blocking activity. Eur. J. Pharm. Sci. 2020, 141, 105083. [Google Scholar] [CrossRef]
- Adrjanowicz, K.; Zakowiecki, D.; Kaminski, K.; Hawelek, L.; Grzybowska, K.; Tarnacka, M.; Paluch, M.; Cal, K. Molecular dynamics in supercooled liquid and glassy states of antibiotics: Azithromycin, clarithromycin and roxithromycin studied by dielectric spectroscopy. Advantages given by the amorphous state. Mol. Pharm. 2012, 9, 1748–1763. [Google Scholar] [CrossRef]
- Vogel, M.; Tschirwitz, C.; Schneider, G.; Koplin, C.; Medick, P.; Rössler, E. A2H NMR and dielelectric spectroscopy study of the slow β-process in organic glass formers. J. Non. Cryst. Solids 2002, 307–310, 326–335. [Google Scholar] [CrossRef]
- Grzybowska, K.; Paluch, M.; Wlodarczyk, P.; Grzybowski, A.; Kaminski, K.; Hawelek, L.; Zakowiecki, D.; Kasprzycka, A.; Jankowska-Sumara, I. Enhancement of amorphous celecoxib stability by mixing it with octaacetylmaltose: The molecular dynamics study. Mol. Pharm. 2012, 9, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, K.; Paluch, M.; Grzybowska, K.; Grzybowski, A.; Wojnarowska, Z.; Hawelek, L.; Ziolo, J.D. Relaxation dynamics and crystallization study of sildenafil in the liquid and glassy states. Mol. Pharm. 2013, 10, 2270–2282. [Google Scholar] [CrossRef] [PubMed]
- Descamps, M.; Willart, J.-F. Scaling laws and size effects for amorphous crystallization kinetics: Constraints imposed by nucleation and growth specificities. Int. J. Pharm. 2018, 542, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Delcourt, O.; Descamps, M.; Hilhorst, H.J. Size effect in a nucleation and growth transformation. Ferroelectrics 1991, 124, 109–114. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
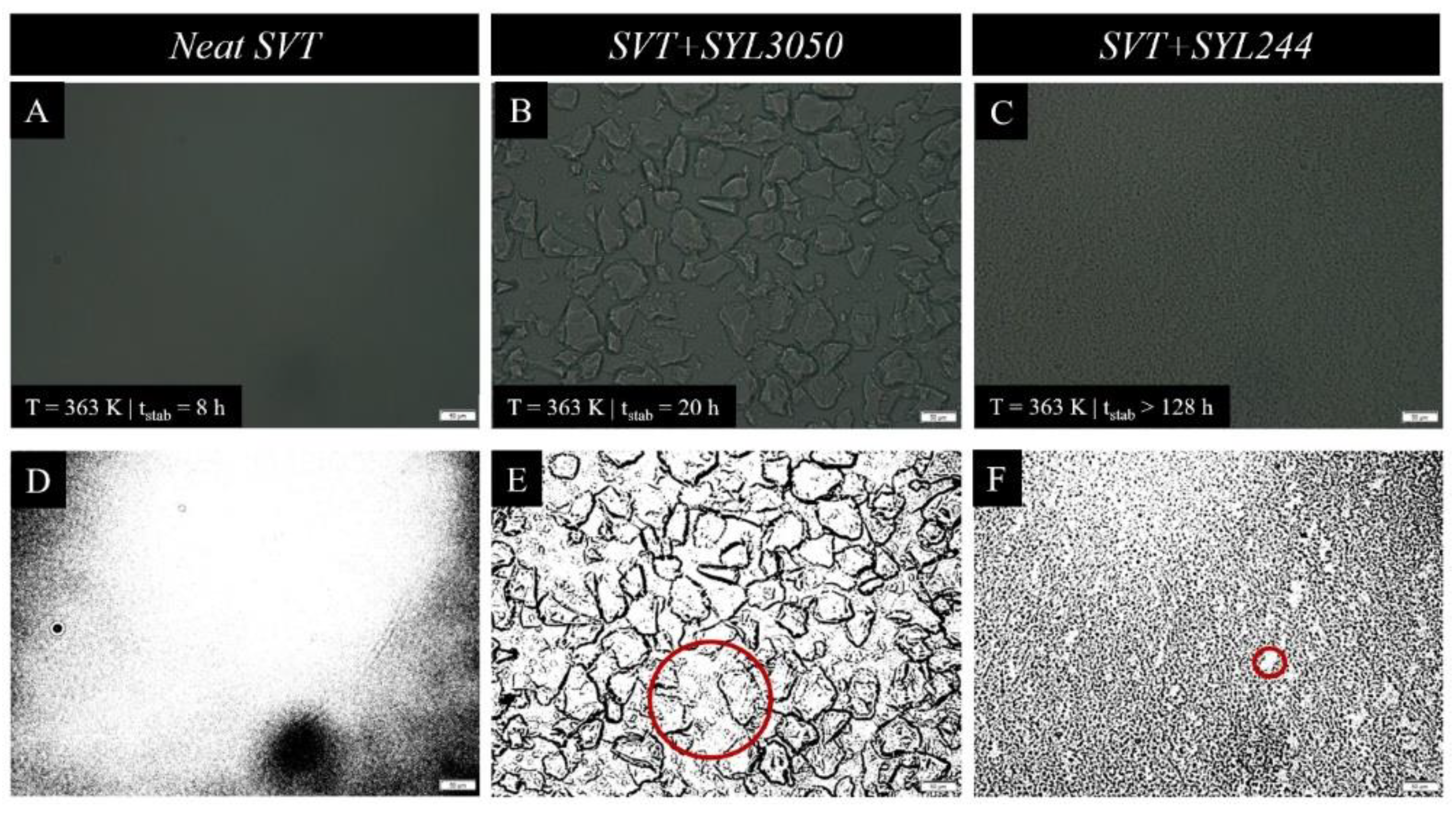{kind=link}
| MS Name: | BATCH/LOS: | Surface Area (m2/g) | Average Particle Size (μm) | Pore Diameter (nm) | Pore Volume (mL/g) |
|---|---|---|---|---|---|
| SYL244 | 1000320678 | 314 | 2.5–3.7 | 23 | 1.6 |
| SYL3050 | 1000298877 | 320 | 59 | 22.9 | 1.7 |
| Sample: | t0 (s) | τcr (min) | ln t1 | ln t2 | n (Equation (3)) | α(t)′ max | n (Equation (2)) |
|---|---|---|---|---|---|---|---|
| neat SVT | 8800 ± 100 | 55 ± 1 | 7.65 ± 0.02 | 8.361 ± 0.001 | 3.8 ± 0.1 | 1.57 ± 0.09 | 4.3 ± 0.2 |
| SVT + SYL3050 | 9150 ± 50 | 107 ± 3 | 7.79 ± 0.04 | 9.33 ± 0.08 | 1.8 ± 0.1 | 0.73 ± 0.06 | 2.0 ± 0.2 |
| Sample: | t0 (s) | τcr (min) | ln1 | ln2 | n (Equation (3)) | α(t)max’ | n (Equation (2)) |
|---|---|---|---|---|---|---|---|
| neat SVT | 13,600 ± 400 | 201 ± 12 | 8.71 ± 0.06 | 9.88 ± 0.02 | 2.34 ± 0.07 | 0.91 ± 0.01 | 2.48 ± 0.04 |
| SVT + SYL3050 | 29,750 ± 250 | 737 ± 28 | 9.02 ± 0.08 | 11.67 ± 0.08 | 1.026 ± 0.003 | 0.488 ± 0.001 | 1.326 ± 0.001 |
| Sample: | Tg (K) | log τ∞ | T0 (K) | BT0 | mp |
|---|---|---|---|---|---|
| SVT | 303 | −15.68 ± 0.13 | 244.01 ± 0.89 | 2386 ± 51 | 91 |
| SVT + SYL3050 | 303 | −15.23 ± 0.11 | 246.23 ± 0.77 | 2240 ± 42 | 93 |
| SVT + SYL244 | 303 | −15.18 ± 0.16 | 247.64 ±1.13 | 2183 ± 54 | 94 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knapik-Kowalczuk, J.; Kramarczyk, D.; Chmiel, K.; Romanova, J.; Kawakami, K.; Paluch, M. Importance of Mesoporous Silica Particle Size in the Stabilization of Amorphous Pharmaceuticals—The Case of Simvastatin. Pharmaceutics 2020, 12, 384. https://doi.org/10.3390/pharmaceutics12040384
Knapik-Kowalczuk J, Kramarczyk D, Chmiel K, Romanova J, Kawakami K, Paluch M. Importance of Mesoporous Silica Particle Size in the Stabilization of Amorphous Pharmaceuticals—The Case of Simvastatin. Pharmaceutics. 2020; 12(4):384. https://doi.org/10.3390/pharmaceutics12040384
Chicago/Turabian StyleKnapik-Kowalczuk, Justyna, Daniel Kramarczyk, Krzysztof Chmiel, Jana Romanova, Kohsaku Kawakami, and Marian Paluch. 2020. "Importance of Mesoporous Silica Particle Size in the Stabilization of Amorphous Pharmaceuticals—The Case of Simvastatin" Pharmaceutics 12, no. 4: 384. https://doi.org/10.3390/pharmaceutics12040384