On-Chip Synthesis of Hyaluronic Acid-Based Nanoparticles for Selective Inhibition of CD44+ Human Mesenchymal Stem Cell Proliferation

 , , ,
, , ,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines
2.3. Microfluidic Device
2.4. HA/CS NPs on-Chip Preparation Method Set-up
2.5. Optimization of HA/CS NPs Preparation Method
2.6. Everolimus Loaded HA/CS NPs
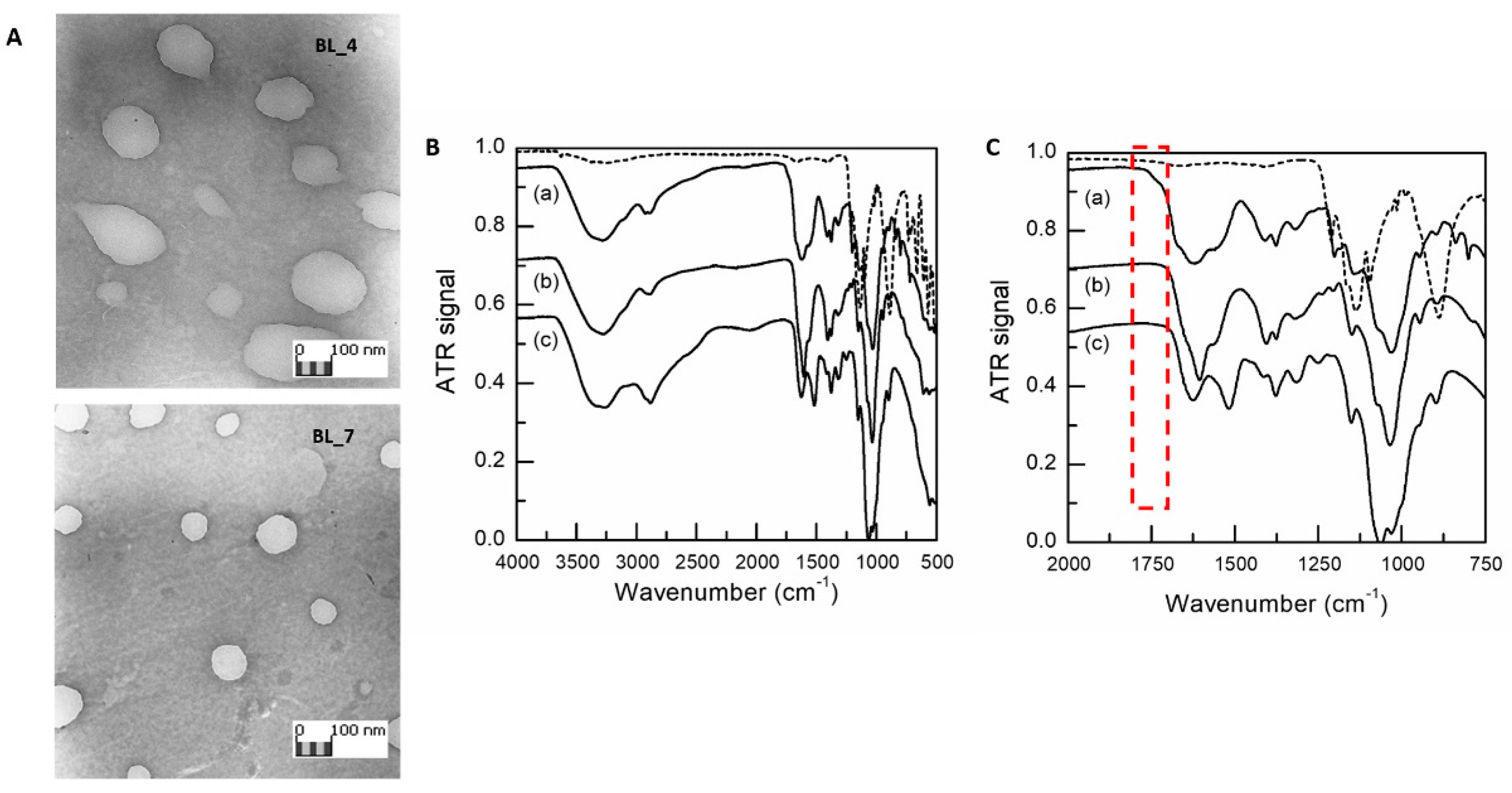
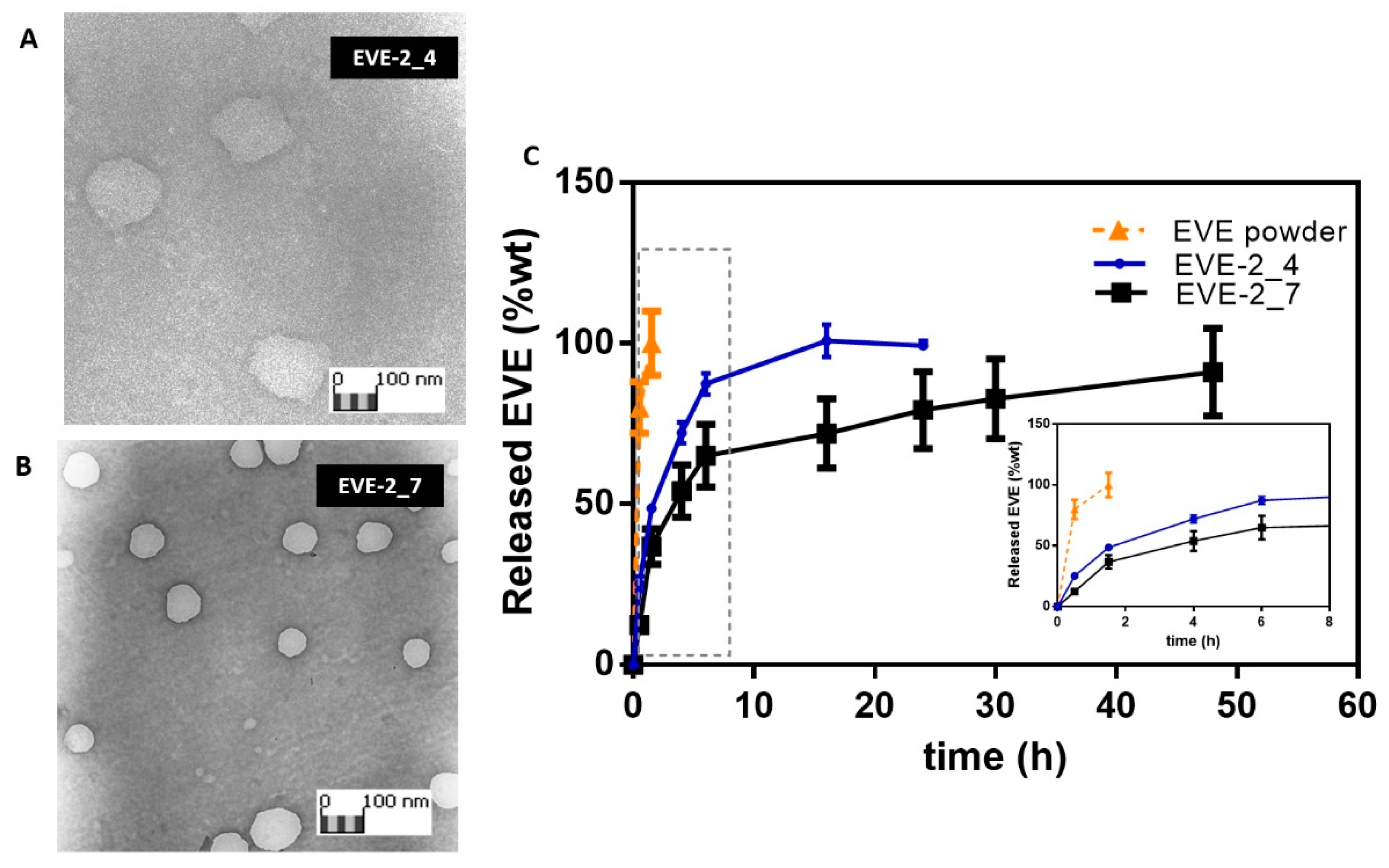
2.7. Characterization of NPs
2.8. Immunocytochemistry Assay
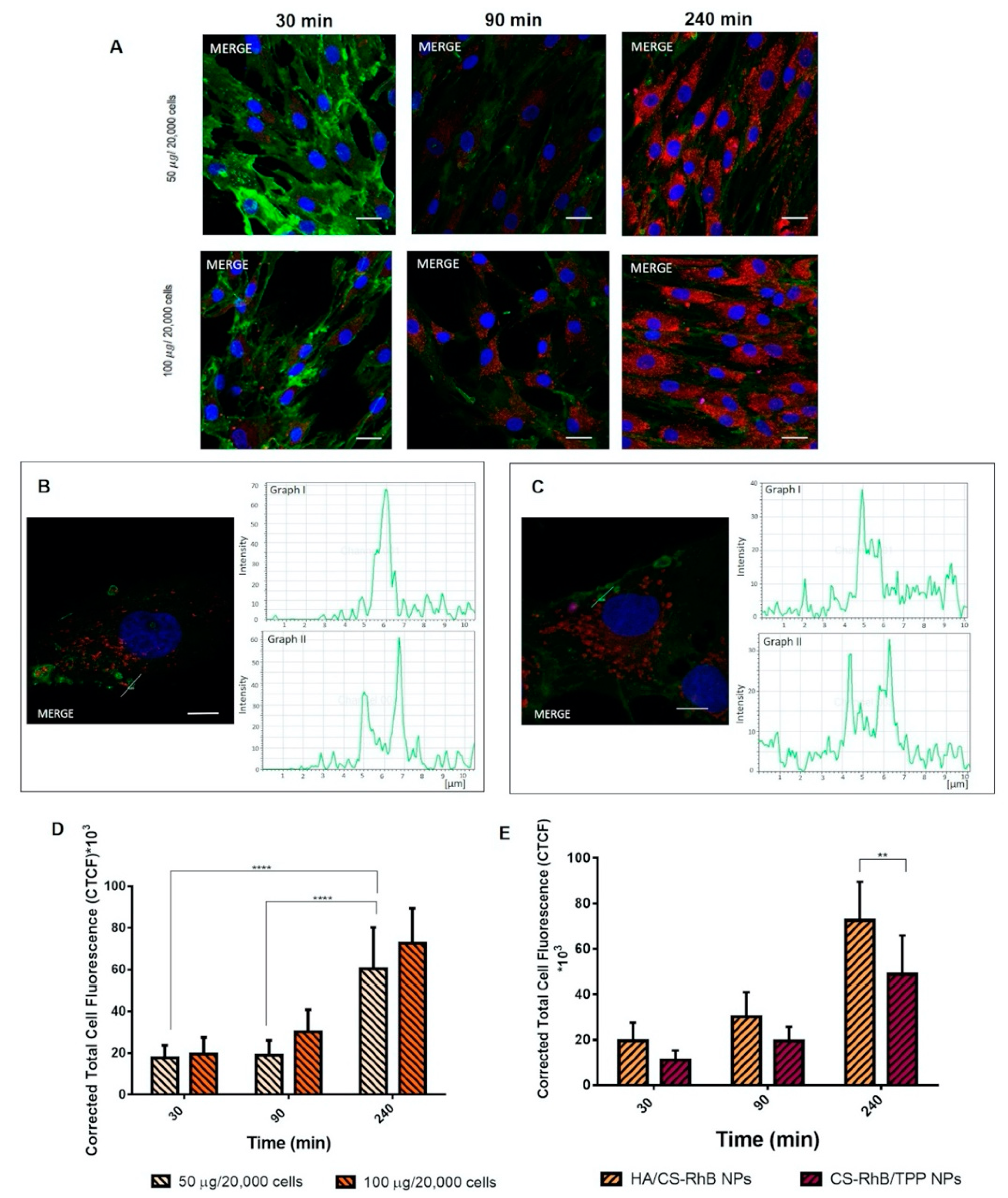
2.9. Cellular uptake of HA/CS NPs
2.9.1. Chitosan-Rhodamine B Conjugation
2.9.2. Placebo HA/CS-RhB NPs Preparation
2.9.3. Placebo CS-RhB/TPP NPs Preparation
2.10. Competitive Binding Experiment
2.11. In Vitro Cytotoxicity
2.12. Assessment of DNA Synthesis and Proliferative Activity by 5-Bromo-2′-Deoxyuridine Incorporation
2.13. Statistical Analysis
3. Results and Discussion
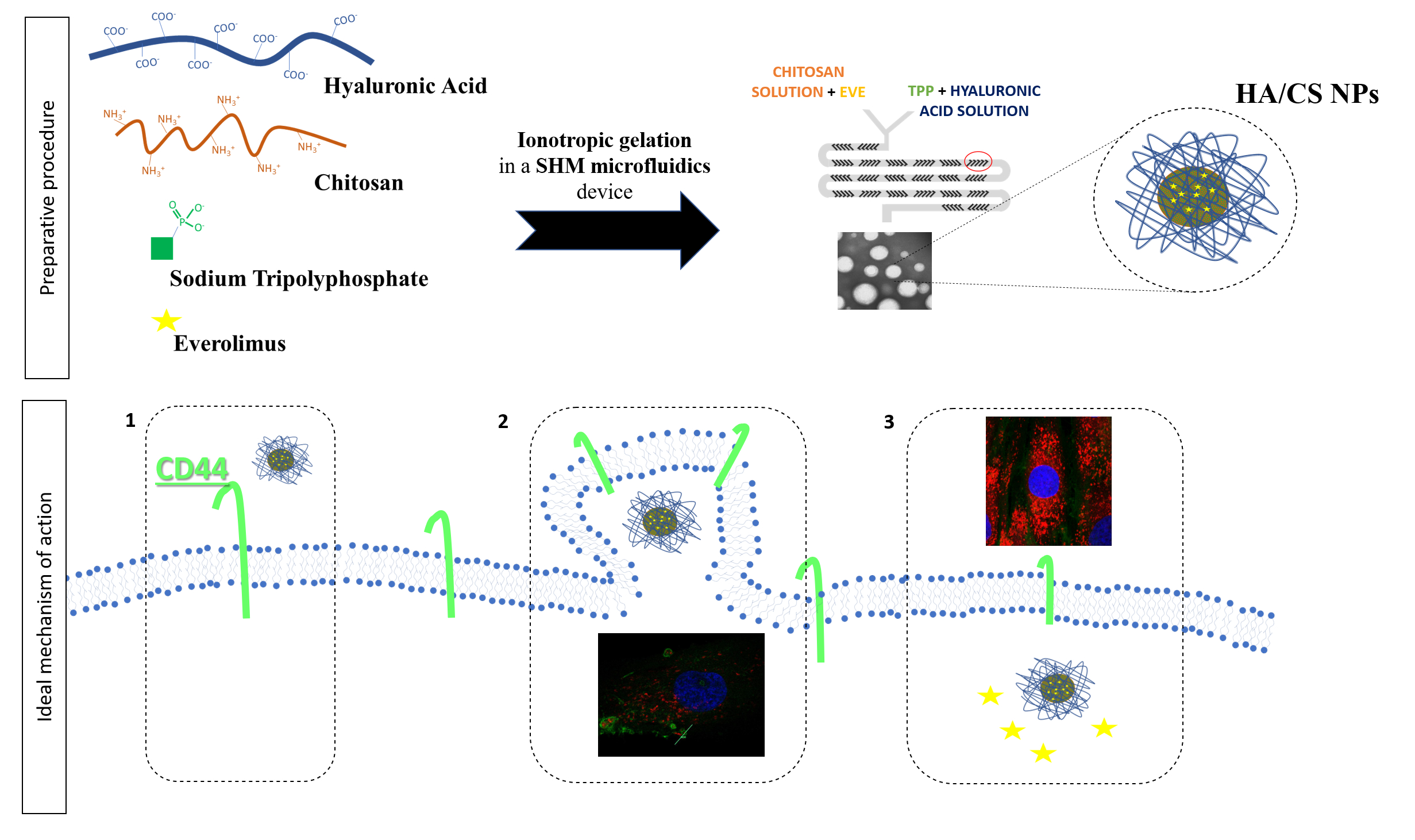
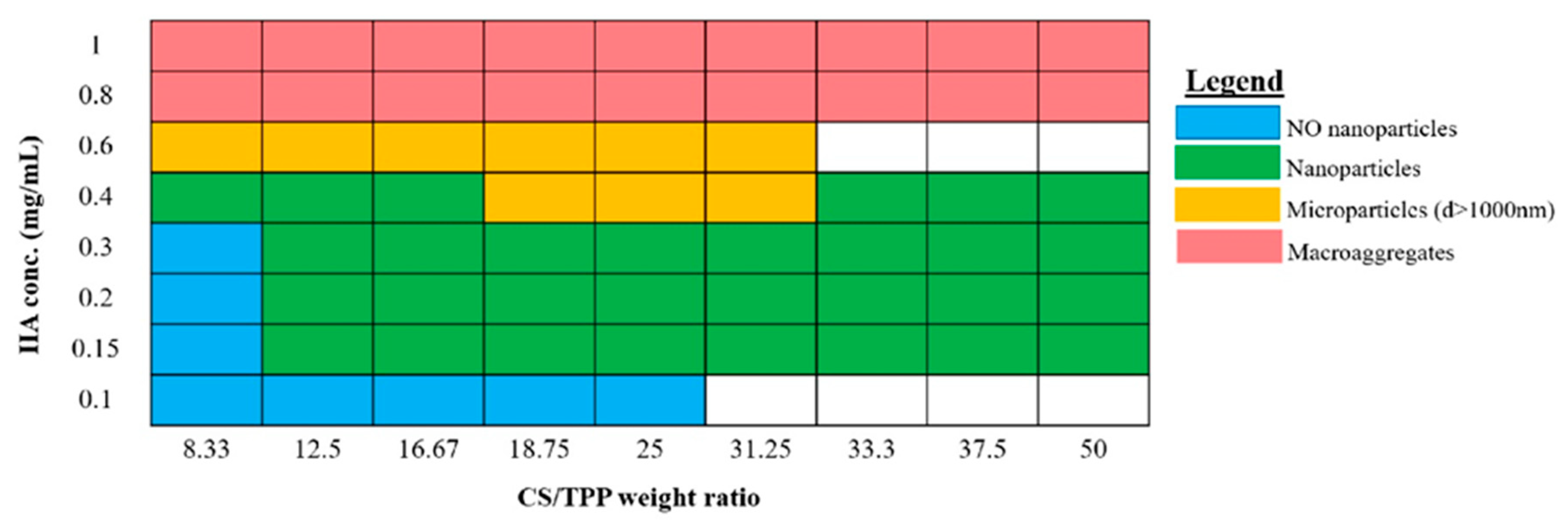
3.1. HA/CS NPs On-Chip Preparation Method Set Up
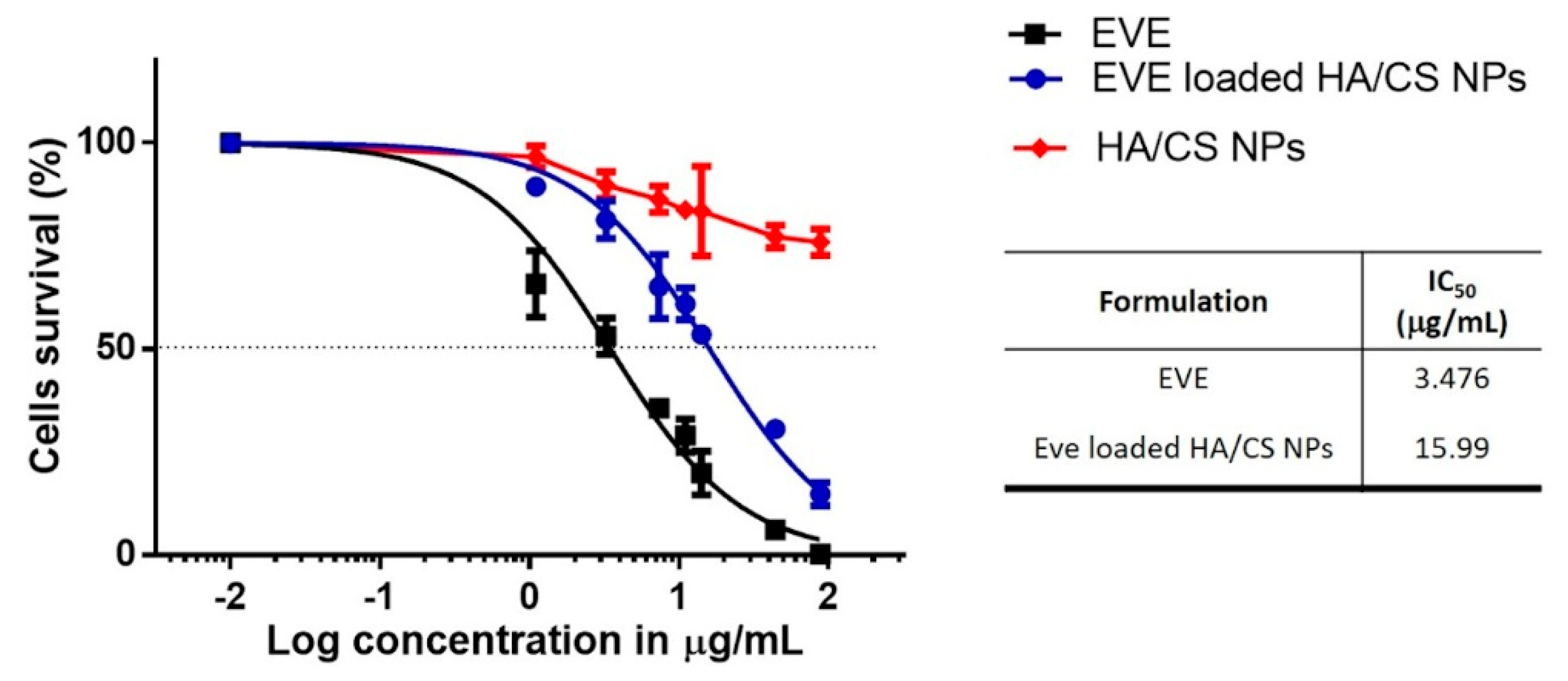
3.2. Everolimus Loading in HA/CS NPs
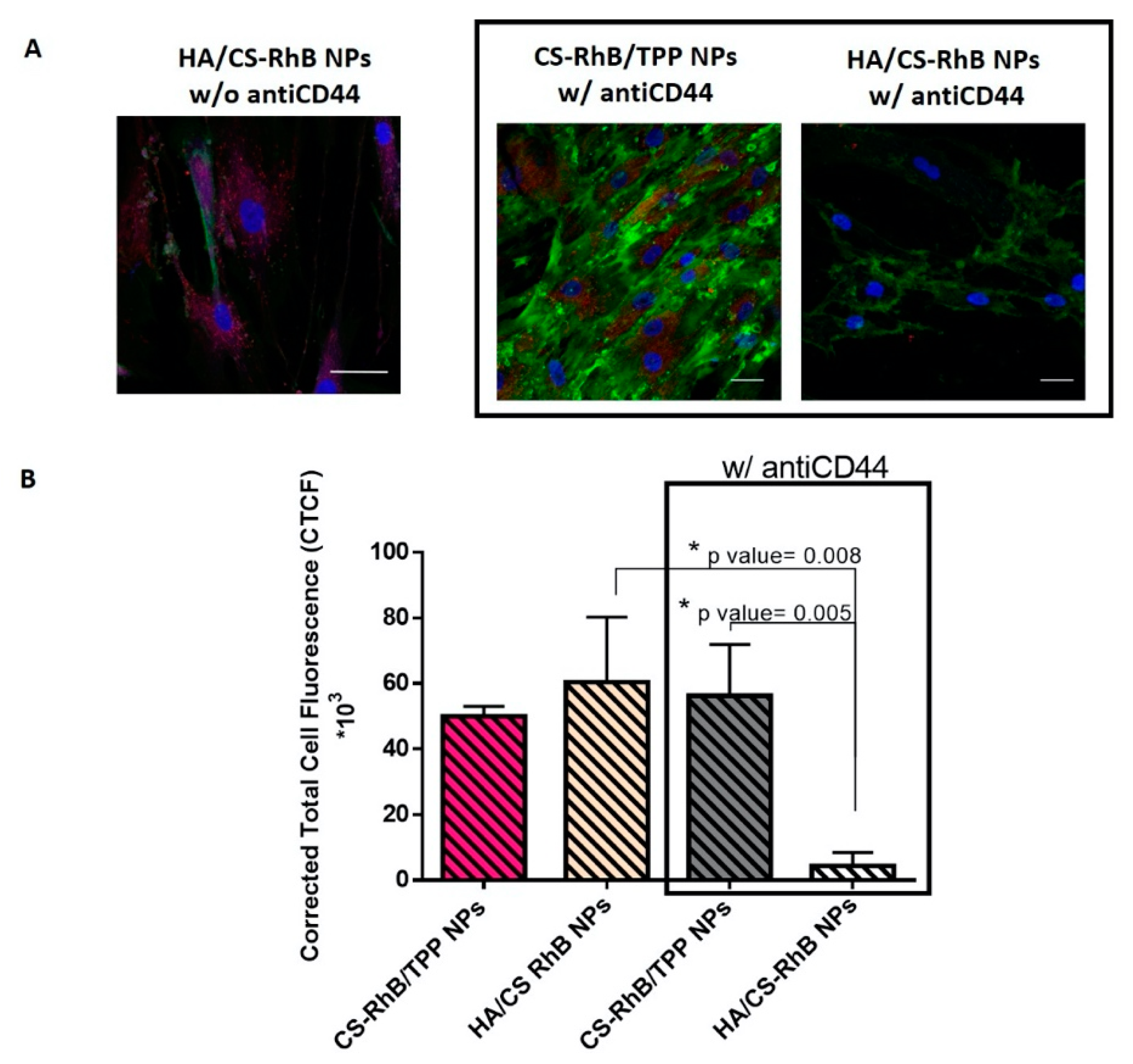
3.3. Cellular uptake of HA/CS NPs
3.4. Competitive Binding Experiment
3.5. In Vitro Cytotoxicity
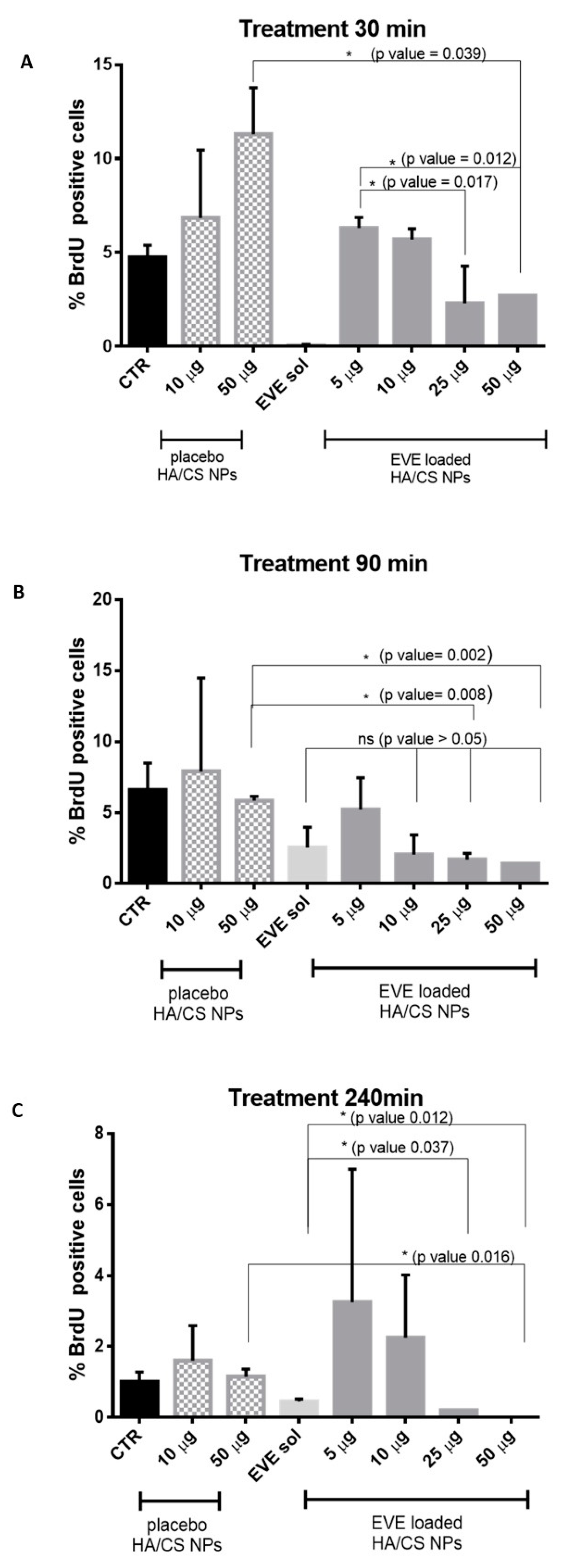
3.6. Assessment of DNA Synthesis and Proliferative Activity by 5-Bromo-2′-Deoxyuridine (BrdU) Incorporation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
HA/CS NPs On-Chip Preparation Method Set-Up: Design Space Definition

References
- Williams, K.; Motiani, K.; Giridhar, P.; Kasper, S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp. Biol. Med. 2013, 238, 324–338. [Google Scholar] [CrossRef]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.; Shiina, M.; Li, J.; Simpson, M.; Heldin, P. Hyaluronan-CD44 Interaction Promotes Oncogenic Signaling, microRNA Functions, Chemoresistance, and Radiation Resistance in Cancer Stem Cells Leading to Tumor Progression. Adv. Cancer Res. 2014, 123, 255–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, A.R.; Racine, R.R.; Hennig, M.J.; Lokeshwar, V.B. The Role of CD44 in Disease Pathophysiology and Targeted Treatment. Front. Immunol. 2015, 6, 182. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, K.; Minami, K.; Hayashi, N.; Yokoyama, Y.; Mori, S.; Yamamoto, H.; Koizumi, M. The CD44 standard isoform contributes to radioresistance of pancreatic cancer cells. J. Radiat. Res. 2017, 58, 816–826. [Google Scholar] [CrossRef] [Green Version]
- Senbanjo, L.; Chellaiah, M. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Alamgeer, M.; Watkins, D.; Banakh, I.; Kumar, B.; Gough, D.; Markman, B.; Ganju, V. A phase IIa study of HA-irinotecan, formulation of hyaluronic acid and irinotecan targeting CD44 in extensive-stage small cell lung cancer. Investig. New Drugs 2018, 36, 288–298. [Google Scholar] [CrossRef]
- Ossipov, D. Nanostructured hyaluronic acid-based materials for active delivery to cancer. Expert Opin. Drug Deliv. 2010, 7, 681–703. [Google Scholar] [CrossRef]
- Serafino, A.; Zonfrillo, M.; Andreola, F.; Psaila, R.; Mercuri, L.; Moroni, N.; Renier, D.; Campisi, M.; Secchieri, C.; Pierimarchi, P. CD44-Targeting for Antitumor Drug Delivery: A New SN-38-Hyaluronan Bioconjugate for Locoregional Treatment of Peritoneal Carcinomatosis. Curr. Cancer Drug Targets 2011, 11, 572–585. [Google Scholar] [CrossRef]
- Prestwich, G. Hyaluronic acid-based clinical biomaterials derived for cell and molecule delivery in regenerative medicine. J. Control. Release 2011, 155, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Knopf-Marques, H.; Pravda, M.; Wolfova, L.; Velebny, V.; Schaaf, P.; Vrana, N.; Lavalle, P. Hyaluronic Acid and Its Derivatives in Coating and Delivery Systems: Applications in Tissue Engineering, Regenerative Medicine and Immunomodulation. Adv. Healthc. Mater. 2016, 5, 2841–2855. [Google Scholar] [CrossRef] [PubMed]
- Cadete, A.; Alonso, M.J. Targeting cancer with hyaluronic acid-based nanocarriers: Recent advances and translational perspectives. Nanomedicine 2016, 11, 2341–2357. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Lim, Y.T. Degradation-Regulatable Architectured Implantable Macroporous Scaffold for the Spatiotemporal Modulation of Immunosuppressive Microenvironment and Enhanced Combination Cancer Immunotherapy. Adv. Funct. Mater. 2018, 28, 1804490. [Google Scholar] [CrossRef]
- Nasti, A.; Zaki, N.M.; de Leonardis, P.; Ungphaiboon, S.; Sansongsak, P.; Rimoli, M.G.; Tirelli, N. Chitosan/TPP and Chitosan/TPP-hyaluronic Acid Nanoparticles: Systematic Optimisation of the Preparative Process and Preliminary Biological Evaluation. Pharm. Res. 2009, 26, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Zaki, N.; Nasti, A.; Tirelli, N. Nanocarriers for Cytoplasmic Delivery: Cellular Uptake and Intracellular Fate of Chitosan and Hyaluronic Acid-Coated Chitosan Nanoparticles in a Phagocytic Cell Model. Macromol. Biosci. 2011, 11, 1747–1760. [Google Scholar] [CrossRef] [PubMed]
- Spadea, A.; de la Rosa, J.; Tirella, A.; Ashford, M.; Williams, K.; Stratford, I.; Tirelli, N.; Mehibel, M. Evaluating the Efficiency of Hyaluronic Acid for Tumor Targeting via CD44. Mol. Pharm. 2019, 16, 2481–2493. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Moon, M.; Kim, D.; Heo, S.; Jeong, Y. Hyaluronic Acid-Based Nanomaterials for Cancer Therapy. Polymers 2018, 10, 1133. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Hu, C.; Yang, Y.; Zhang, J.; Gao, H. Theranostic nanoparticleswithtumor-specific enzyme-triggeredsizereductionanddrugrelease to performphotothermaltherapyforbreast cancer treatment. Acta Pharm. Sin. B 2019, 9, 410–420. [Google Scholar] [CrossRef]
- Bellich, B.; D’Agostino, I.; Semeraro, S.; Gamini, A.; Cesaro, A. The Good, the Bad and the Ugly of Chitosans. Mar. Drugs 2016, 14, 99. [Google Scholar] [CrossRef] [Green Version]
- de la Fuente, M.; Seijo, B.; Alonso, M. Novel hyaluronan-based nanocarriers for transmucosal delivery of macromolecules. Macromol. Biosci. 2008, 8, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Cao, M.; Zhang, J.; Hu, K.; Yin, Z.; Zhou, Z.; Xiao, X.; Yang, Y.; Sheng, W.; Wu, Y.; et al. Hyaluronic acid-chitosan nanoparticles for co-delivery of MiR-34a and doxorubicin in therapy against triple negative breast cancer. Biomaterials 2014, 35, 4333–4344. [Google Scholar] [CrossRef] [PubMed]
- Oyarzun-Ampuero, F.A.; Brea, J.; Loza, M.I.; Torres, D.; Alonso, M.J. Chitosan-hyaluronic acid nanoparticles loaded with heparin for the treatment of asthma. Int. J. Pharm. 2009, 381, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Parajo, Y.; d’Angelo, I.; Welle, A.; Garcia-Fuentes, M.; Alonso, M. Hyaluronic acid/Chitosan nanoparticles as delivery vehicles for VEGF and PDGF-BB. Drug Deliv. 2010, 17, 596–604. [Google Scholar] [CrossRef]
- Naskar, S.; Sharma, S.; Kuotsu, K. Chitosan-based nanoparticles: An overview of biomedical applications and its preparation. J. Drug Deliv. Sci. Technol. 2019, 49, 66–81. [Google Scholar] [CrossRef]
- de la Fuente, M.; Seijo, B.; Alonso, M.J. Novel hyaluronic acid-chitosan nanoparticles for ocular gene therapy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2016–2024. [Google Scholar] [CrossRef] [Green Version]
- Almalik, A.; Alradwan, I.; Majrashi, M.; Alsaffar, B.; Algarni, A.; Alsuabeyl, M.; Alrabiah, H.; Tirelli, N.; Alhasan, A. Cellular responses of hyaluronic acid-coated chitosan nanoparticles. Toxicol. Res. 2018, 7, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, E.; Dorati, R.; Conti, B.; Modena, T.; Cova, E.; Meloni, F.; Genta, I. Hyaluronic Acid-Decorated Chitosan Nanoparticles for CD44-Targeted Delivery of Everolimus. Int. J. Mol. Sci. 2018, 19, 2310. [Google Scholar] [CrossRef] [Green Version]
- Gennari, A.; de la Rosa, J.M.; Hohn, E.; Pelliccia, M.; Lallana, E.; Donno, R.; Tirella, A.; Tirelli, N. The different ways to chitosan/hyaluronic acid nanoparticles: Template vs. direct complexation. Influence of particle preparation on morphology, cell uptake and silencing efficiency. Beilstein J. Nanotechnol. 2019, 10, 2594–2608. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.; de Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharmacol. 2018, 9, 790. [Google Scholar] [CrossRef]
- Karnik, R.; Gu, F.; Basto, P.; Cannizzaro, C.; Dean, L.; Kyei-Manu, W.; Langer, R.; Farokhzad, O.C. Microfluidic platform for controlled synthesis of polymeric nanoparticles. Nano Lett. 2008, 8, 2906–2912. [Google Scholar] [CrossRef]
- Serra, C.; Chang, Z. Microfluidic-assisted synthesis of polymer particles. Chem. Eng. Technol. 2008, 31, 1099–1115. [Google Scholar] [CrossRef]
- Valencia, P.; Farokhzad, O.; Karnik, R.; Langer, R. Microfluidic technologies for accelerating the clinical translation of nanoparticles. Nat. Nanotechnol. 2012, 7, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, E.; Dorati, R.; Pisani, S.; Conti, B.; Bergamini, G.; Modena, T.; Genta, I. The microfluidic technique and the manufacturing of polysaccharide nanoparticles. Pharmaceutics 2018, 10, 267. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Bevilacqua, P.; Netti, P.; Torino, E. A Microfluidic Platform to design crosslinked Hyaluronic Acid Nanoparticles (cHANPs) for enhanced MRI. Sci. Rep. 2016, 6, 37906. [Google Scholar] [CrossRef]
- Russo, M.; Bevilacqua, P.; Netti, P.A.; Torino, E. Commentary on A Microfluidic Platform to Design Crosslinked Hyaluronic Acid Nanoparticles (cHANPs) for Enhanced MRI. Mol. Imaging 2017, 16, 1536012117706237. [Google Scholar] [CrossRef] [Green Version]
- Souza, B.R.C.; Andrade, S.M.H. Production of hyaluronic acid (HA) nanoparticles by a continuous processinside microchannels: Effects of non-solvents, organic phase flow rate, and HA concentration. Chem. Eng. Sci. 2012, 84, 134–141. [Google Scholar] [CrossRef]
- Water, J.J.; Kim, Y.; Maltesen, M.J.; Franzyk, H.; Foged, C.; Nielsen, H.M. Hyaluronic Acid-Based Nanogels Produced by Microfluidics-Facilitated Self-Assembly Improves the Safety Profile of the Cationic Host Defense Peptide Novicidin. Pharm. Res. 2015, 32, 2727–2735. [Google Scholar] [CrossRef]
- Hasskarl, J. Everolimus. In Small Molecules in Oncology. Recent Results in Cancer Research, 3rd ed.; Martens, U.M., Ed.; Springer: Cham, Switzerland, 2018; Volume 201, pp. 101–123. [Google Scholar] [CrossRef]
- Chiesa, E.; Dorati, R.; Modena, T.; Conti, B.; Genta, I. Multivariate analysis for the optimization of microfluidics-assisted nanoprecipitation method intended for the loading of small hydrophilic drugs into PLGA nanoparticles. Int. J. Pharm. 2018, 536, 165–177. [Google Scholar] [CrossRef]
- Stroock, A.; Dertinger, S.; Ajdari, A.; Mezic, I.; Stone, H.; Whitesides, G. Chaotic mixer for microchannels. Science 2002, 295, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Iwase, Y.; Maitani, Y. Preparation and in Vivo Evaluation of Liposomal Everolimus for Lung Carcinoma and Thyroid Carcinoma. Biol. Pharm. Bull. 2012, 35, 975–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, F.; Omes, C.; Bassani, R.; Nappi, R.; Mazzini, G.; Cornaglia, A.; Casasco, A. In-vitro culture system for mesenchymal progenitor cells derived from waste human ovarian follicular fluid. Reprod. Biomed. Online 2014, 29, 457–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Yun, K. Fluorometric switch-on detection of heparin based on a system composed of rhodamine-labeled chitosan oligosaccharide lactate, and graphene oxide. Methods Appl. Fluoresc. 2018, 6, 035011. [Google Scholar] [CrossRef]
- dos Santos, T.; Varela, J.; Lynch, I.; Salvati, A.; Dawson, K. Effects of Transport Inhibitors on the Cellular Uptake of Carboxylated Polystyrene Nanoparticles in Different Cell Lines. PLoS ONE 2011, 6, e24438. [Google Scholar] [CrossRef] [Green Version]
- Amadori, M.; Soares-Filipe, J.F.; Riva, F.; Vitali, A.; Ruggeri, J.; Lacetera, N. Characterization of the blastogenic response to LPS of bovine peripheral blood mononuclear cells. PLoS ONE 2018, 13, e0204827. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Yan, W.; Xu, Z.S.; Ni, H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids Surf. B Biointerfaces 2012, 90, 21–27. [Google Scholar] [CrossRef]
- Richert, L.; Lavalle, P.; Payan, E.; Shu, X.; Prestwich, G.; Stoltz, J.; Schaaf, P.; Voegel, J.; Picart, C. Layer by layer buildup of polysaccharide films: Physical chemistry and cellular adhesion aspects. Langmuir 2004, 20, 448–458. [Google Scholar] [CrossRef]
- Bang, S.; Das, D.; Yu, J.; Noh, I. Evaluation of MC3T3 Cells Proliferation and Drug Release Study from Sodium Hyaluronate-1,4-butanediol Diglycidyl Ether Patterned Gel. Nanomaterials 2017, 7, 328. [Google Scholar] [CrossRef] [Green Version]
- Mattu, C.; Li, R.; Ciardelli, G. Chitosan nanoparticles as therapeutic protein nanocarriers: The effect of ph on particle formation and encapsulation efficiency. Polym. Compos. 2013, 34, 1538–1545. [Google Scholar] [CrossRef]
- Martins, A.; de Oliveira, D.; Pereira, A.; Rubira, A.; Muniz, E. Chitosan/TPP microparticles obtained by microemulsion method applied in controlled release of heparin. Int. J. Biol. Macromol. 2012, 51, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, Y.; Song, K.; Park, M. Preparation and properties of polyelectrolyte complex sponges composed of hyaluronic acid and chitosan and their biological behaviors. J. Appl. Polym. Sci. 2003, 90, 925–932. [Google Scholar] [CrossRef]
- Geven, M.; Francini, N.; Donno, R.; Tirelli, N. Microfluidic-Assisted Manufacturing of Materials for Nanomedicine. Mater. Matters™ 2019, 14, 83–89. [Google Scholar]
- Borro, B.C.; Bohr, A.; Bucciarelli, S.; Boetker, J.P.; Foged, C.; Rantanen, J.; Malmsten, M. Microfluidics-based self-assembly of peptide-loaded microgels: Effect of three dimensional (3D) printed micromixer design. J. Colloid Interface Sci. 2019, 538, 559–568. [Google Scholar] [CrossRef]
- Forbes, N.; Hussain, M.T.; Briuglia, M.L.; Edwards, D.P.; Horst, J.H.T.; Szita, N.; Perrie, Y. Rapid and scale-independent microfluidic manufacture of liposomes entrapping protein incorporating in-line purification and at-line size monitoring. Int. J. Pharm. 2019, 556, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Janes, K.; Fresneau, M.; Marazuela, A.; Fabra, A.; Alonso, M. Chitosan nanoparticles as delivery systems for doxorubicin. J. Control. Release 2001, 73, 255–267. [Google Scholar] [CrossRef]
- Meng, Q.; He, C.; Su, W.; Zhang, X.; Duan, C. A new rhodamine-chitosan fluorescent material for the selective detection of Hg2+ in living cells and efficient adsorption of Hg2+ in natural water. Sens. Actuators B Chem. 2012, 174, 312–317. [Google Scholar] [CrossRef]
- Xiao, G.; Gan, L.S. Receptor-mediated endocytosis and brain delivery of therapeutic biologics. Int. J. Cell Biol. 2013, 2013, 703545. [Google Scholar] [CrossRef] [Green Version]
- Ouasti, S.; Kingham, P.J.; Terenghi, G.; Tirelli, N. The CD44/integrins interplay and the significance of receptor binding and re-presentation in the uptake of RGD-functionalized hyaluronic acid. Biomaterials 2012, 33, 1120–1134. [Google Scholar] [CrossRef]
- Almalik, A.; Karimi, S.; Ouasti, S.; Donno, R.; Wandrey, C.; Day, P.J.; Tirelli, N. Hyaluronic acid (HA) presentation as a tool to modulate and control the receptor-mediated uptake of HA-coated nanoparticles. Biomaterials 2013, 34, 5369–5380. [Google Scholar] [CrossRef]
- Cai, S.; Alhowyan, A.A.B.; Yang, Q.; Forrest, W.C.M.; Shnayder, Y.; Forrest, M.L. Cellular Uptake and Internalization of Hyaluronan-based Doxorubicin and Cisplatin Conjugates. J. Drug Target. 2014, 22, 648–657. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | HA Conc (mg/mL) | HA:CS (w:w) | CS:TPP (w:w) | Mean Size ± SD (nm) | PDI ±SD | ζ Potential ± SD (mV) |
|---|---|---|---|---|---|---|
| BL_1 | 0.400 | 4:1 | 25:1 | 1025.45 ± 255.10 | 0.486 ± 0.045 | −24.88 ± 3.07 |
| BL_2 | 0.150 | 1.5:1 | 25:1 | 102.75 ± 3.23 | 0.297 ± 0.007 | +9.30 ± 1.89 |
| BL_3 | 0.150 | 1.5:1 | 50:1 | 159.85 ± 38.91 | 0.313 ± 0.039 | +17.34 ±4.13 |
| BL_4 | 0.400 | 8:1 | 12.5:1 | 584.00 ± 22.20 | 0.422 ± 0.027 | −24.02 ± 2.91 |
| BL_5 | 0.400 | 8:1 | 25:1 | 906.55± 103.21 | 0.393 ± 0.046 | −18.01 ± 8.31 |
| BL_6 | 0.150 | 3:1 | 12.5:1 | 239.20 ± 19.51 | 0.520 ± 0.059 | −21.79 ± 10.64 |
| BL_7 | 0.150 | 3:1 | 25:1 | 159.00 ± 7.28 | 0.306 ± 0.053 | −20.85 ± 3.16 |
| BL_8 | 0.275 | 3.6:1 | 25:1 | 758.45 ± 23.77 | 0.457 ± 0.055 | −23.89 ± 3.34 |
| Sample Code | Mean Size ± SD (nm) | PDI ± SD | ζ Potential ± SD (mV) | CS Amount ± SD (μg) | HA Amount ± SD (μg) |
|---|---|---|---|---|---|
| Et_CS_4 | 349.15±38.09 | 0.473 ± 0.058 | −25.80 ± 3.81 | 11.03 ± 3.23 | 238.21 ± 42.45 |
| Me_CS_4 | 406.80 ± 19.65 | 0.467 ± 0.012 | −31.72 ± 0.52 | 25.96 ± 17.92 | 189.42 ± 72.88 |
| Et_CS_7 | 117.50 ± 4.51 | 0.265 ± 0.024 | −22.52 ± 3.43 | 17.56 ± 3.96 | 220.76 ± 25.74 |
| Me_CS_7 | 120.03 ± 13.92 | 0.315 ± 0.025 | −20.18 ± 2.38 | 14.76 ± 6.46 | 222.69 ± 22.35 |
| Sample Code | CS/EVE Ratio | Mean Size ± SD (nm) | PDI ± SD | ζ Potential ± SD (mV) | DL ± SD (μg of EVE / 1 mg of NPs) | EE% ± SD |
|---|---|---|---|---|---|---|
| EVE-1_4 | 2:1 | 372.35 ± 71.50 | 0.440 ± 0.041 | −24.61 ± 7.26 | 33.09 ± 3.66 | 35.29 ± 3.90 |
| EVE-2_4 | 5:1 | 415.58 ± 84.56 | 0.490 ± 0.061 | −28.71 ± 2.78 | 48.91 ± 26.33 | 56.05 ± 11.91 |
| EVE-1_7 | 2:1 | 144.93 ± 13.31 | 0.286 ± 0.059 | −28.91 ± 6.18 | 78.81 ± 6.42 | 29.32 ± 10.64 |
| EVE-2_7 | 5:1 | 135.07 ± 45.81 | 0.356 ± 0.091 | −17.28 ± 2.30 | 88.12 ± 20.76 | 54.56 ± 7.45 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiesa, E.; Riva, F.; Dorati, R.; Greco, A.; Ricci, S.; Pisani, S.; Patrini, M.; Modena, T.; Conti, B.; Genta, I. On-Chip Synthesis of Hyaluronic Acid-Based Nanoparticles for Selective Inhibition of CD44+ Human Mesenchymal Stem Cell Proliferation. Pharmaceutics 2020, 12, 260. https://doi.org/10.3390/pharmaceutics12030260
Chiesa E, Riva F, Dorati R, Greco A, Ricci S, Pisani S, Patrini M, Modena T, Conti B, Genta I. On-Chip Synthesis of Hyaluronic Acid-Based Nanoparticles for Selective Inhibition of CD44+ Human Mesenchymal Stem Cell Proliferation. Pharmaceutics. 2020; 12(3):260. https://doi.org/10.3390/pharmaceutics12030260
Chicago/Turabian StyleChiesa, Enrica, Federica Riva, Rossella Dorati, Antonietta Greco, Stefania Ricci, Silvia Pisani, Maddalena Patrini, Tiziana Modena, Bice Conti, and Ida Genta. 2020. "On-Chip Synthesis of Hyaluronic Acid-Based Nanoparticles for Selective Inhibition of CD44+ Human Mesenchymal Stem Cell Proliferation" Pharmaceutics 12, no. 3: 260. https://doi.org/10.3390/pharmaceutics12030260