Pharmaceutical Hydrates Analysis—Overview of Methods and Recent Advances


 ,
,  ,
,
Abstract
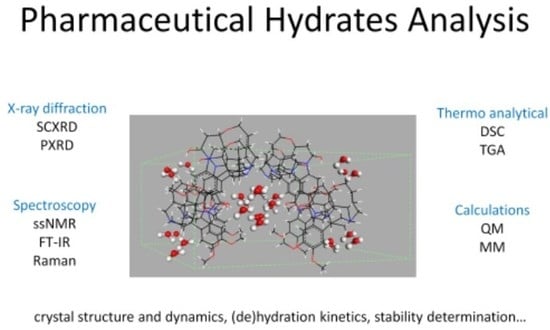
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods Applied for Analysis of Hydrates
2.1. Structure Determination
2.1.1. Structure Determination Techniques

2.1.2. Purely Computational Structure Determination Techniques
2.2. Kinetics of (De)Hydration Process
2.3. Stability Determination in the Industrial Production
3. Hydrates Structural Analysis, Selected Cases within the Last 10 Years
4. Mechanism of (De)Hydration, Selected Cases within the Last 10 Years
5. Hydrates’ Stability, Selected Cases within the Last 10 Years
6. Influence of Excipients on API Hydrates
7. Analysis of Commercial Solid Dosage Forms
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brog, J.-P.; Chanez, C.-L.; Crochet, A.; Fromm, K.M. Polymorphism, what it is and how to identify it: A systematic review. RSC Advances 2013, 3, 16905–16931. [Google Scholar] [CrossRef] [Green Version]
- Tieger, E.; Kiss, V.; Pokol, G.; Finta, Z.; Dušek, M.; Rohlíček, J.; Skořepová, E.; Brázda, P. Studies on the crystal structure and arrangement of water in sitagliptin l-tartrate hydrates. CrystEngComm 2016, 18, 3819–3831. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.E.; Gelbrich, T.; Kahlenberg, V.; Laus, G.; Weiser, J.; Griesser, U.J. Packing polymorphism of a conformationally flexible molecule (aprepitant). New J. Chem. 2008, 32, 1677–1685. [Google Scholar] [CrossRef]
- Seddon, K.R. Pseudopolymorph: A Polemic. Cryst. Growth Des. 2004, 4, 1087. [Google Scholar] [CrossRef]
- Bernstein, J. Another Comment on Pseudopolymorphism. Cryst. Growth Des. 2005, 5, 1661–1662. [Google Scholar] [CrossRef]
- Tian, F.; Qu, H.; Louhi-Kultanen, M.; Rantanen, J. Insight into Crystallization Mechanisms of Polymorphic Hydrate Systems. Chem Eng. Technol. 2010, 33, 833–838. [Google Scholar] [CrossRef]
- Fucke, K.; Steed, J.W. X-ray and Neutron Diffraction in the Study of Organic Crystalline Hydrates. Water 2010, 2, 333–350. [Google Scholar] [CrossRef] [Green Version]
- Authelin, J.-R. Thermodynamics of non-stoichiometric pharmaceutical hydrates. Int. J. Pharm. 2005, 303, 37–53. [Google Scholar] [CrossRef]
- Gossman, W.L.; Wilson, S.R.; Oldfield, E. Three hydrates of the bisphosphonate risedronate, consisting of one molecular and two ionic structures. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2003, 59, m33–m36. [Google Scholar] [CrossRef]
- Zimmermann, A.; Tian, F.; De Diego, H.L.; Frydenvang, K.; Bar-Shalom, D.; Elema, M.R.; Hovgaard, L. Structural Characterisation and Dehydration Behaviour of Siramesine Hydrochloride. J. Pharm. Sci. 2009, 98, 3596–3607. [Google Scholar] [CrossRef]
- Braun, D.E.; Griesser, U.J. Stoichiometric and Nonstoichiometric Hydrates of Brucine. Cryst. Growth Des. 2016, 16, 6111–6121. [Google Scholar] [CrossRef] [PubMed]
- Collin, S.; Moureau, F.; Quintero, F.M.; Vercauteren, D.P.; Evrard, G.; Durant, F. Stereoelectronic requirements of benzamide 5HT3 antagonists. Comparison with D2 antidopaminergic analogues. J. Chem. Soc. Perkin Trans. 1995, 2, 77. [Google Scholar] [CrossRef]
- Cambridge Crystallographic Data Centre. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 1 September 2020).
- Ma, X.; Müller, F.; Huang, S.; Lowinger, M.; Liu, X.; Schooler, R.; Williams, R.O., III. Influence of carbamazepine dihydrate on the preparation of amorphous solid dispersions by hot melt extrusion. Pharmaceutics 2020, 12, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [Green Version]
- Sathisaran, I.; Dalvi, S.V. Engineering Cocrystals of Poorly Water-Soluble Drugs to Enhance Dissolution in Aqueous Medium. Pharmaceutics 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Qu, H.; Zimmermann, A.; Munk, T.; Jørgensen, A.C.; Rantanen, J. Factors affecting crystallization of hydrates. J. Pharm. Pharmacol. 2010, 62, 1534–1546. [Google Scholar] [CrossRef] [PubMed]
- Scaramuzza, D.; Shneider Rauber, G.; Voinovich, D.; Hasa, D. Dehydration without Heating: Use of Polymer-Assisted Grinding for Understanding the Stability of Hydrates in the Presence of Polymeric Excipients. Cryst. Growth Des. 2018, 18, 5245–5253. [Google Scholar] [CrossRef]
- Brittain, G.H. Polymorphism and Solvatomorphism. J. Pharm. Sci. 2010, 99, 3648–3664. [Google Scholar] [CrossRef]
- Giron, D.A.; Mutz, M.; Goldbronn, C.; Pfeffer, S.; Piechon, P.; Schwab, P. Solid State Characterizations of Pharmaceutical Hydrates. J. Therm. Anal. Calorim. 2002, 68, 453–465. [Google Scholar] [CrossRef]
- Sood, J.; Sapra, B.; Bhandari, S.; Jindal, M.; Tiwary, A.K. Understanding pharmaceutical polymorphic transformations I: Influence of process variables and storage conditions. Ther. Del. 2014, 5, 1123–1142. [Google Scholar] [CrossRef]
- Allen, P.V.; Rahn, P.D.; Sarapu, A.C.; Vanderwielen, A.J. Physical characterization of erythromycin: Anhydrate, monohydrate, and dihydrate crystalline solids. J. Pharm. Sci. 1978, 67, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Bobrovs, L.; Seton, L.; Dempster, N. The reluctant polymorph: Investigation into the effect of self-association on the solvent mediated phase transformation and nucleation of theophylline. CrystEngComm 2015, 17, 5237–5251. [Google Scholar] [CrossRef] [Green Version]
- Law, D.; Henry, R.; Lou, X. Trihemihydrate, Anhydrate and Novel Hydrate Forms of Cefdinir. U.S. Patent Application No. 11/072,568, US20060025399A1, 1 September 2020. [Google Scholar]
- Brazil’s Patent Reform Innovation Towards National Competitiveness. p. 124. Available online: http://infojustice.org/wp-content/uploads/2013/09/Brazilian_Patent_Reform.pdf (accessed on 1 September 2020).
- Ho, C.M. Should All Drugs Be Patentable? A Comparative Perspective. Vand. J. Ent. Tech. L. 2014, 17, 295–348. [Google Scholar]
- Lactose Monohydrate Monographia in the US Pharmacopoeia. Available online: https://www.usp.org/harmonization-standards/pdg/excipients/lactose-monohydrate (accessed on 1 September 2020).
- Caffeine anhydrous, Caffeine Monohydrate Monographs in the International Pharmacopoeia 9th Edition. Available online: https://apps.who.int/phint/en/p/docf/ (accessed on 1 September 2020).
- Flicker, F.; Eberle, V.A.; Betz, G. Variability in commercial carbamazepine samples—Impact on drug release. Int. J. Pharm. 2011, 410, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Forster, A.; Gordon, K.; Schmierer, D.; Soper, N.; Wu, V.; Rades, T. Characterisation of two polymorphic forms of ranitidine-HCl. Internet J. Vib. Spectrosc. 1998, 2, 12. [Google Scholar]
- Burger, A.; Ramberger, R. On the polymorphism of pharmaceuticals and other molecular crystals. II. Microchim. Acta 1979, 72, 273–316. [Google Scholar] [CrossRef]
- Slobodin, B.V.; Samigullina, R.F. Thermoanalytical study of the polymorphism and melting behavior of Cu2V2O7. Inorg. Mat. 2010, 46, 196–200. [Google Scholar] [CrossRef]
- Han, J.; Suryanarayanan, R. A method for the rapid evaluation of the physical stability of pharmaceutical hydrates. Thermochim. Acta 1999, 329, 163–170. [Google Scholar] [CrossRef]
- Prendergast, R. Structure Determination of Small and Large Molecules Using Single Crystal X-ray Crystallography. Master’s Thesis, University of Manchester, Manchester, UK, 2010. [Google Scholar]
- Bukovec, P.; Meden, A.; Smrkolj, M.; Vrecer, F. Influence of Crystal Habit on the Dissolution of Simvastatin Single Crystals. Acta Chim. Slov. 2015, 62, 958–966. [Google Scholar] [CrossRef]
- Kirchmeyer, W.; Grassmann, O.; Wyttenbach, N.; Alsenz, J.; Kuentz, M. Miniaturized X-ray Powder Diffraction Assay (MixRay) for Quantitative Kinetic Analysis of Solvent-Mediated Phase Transformations in Pharmaceutics. J. Pharm. Biomed. Anal. 2016, 131, 195–201. [Google Scholar] [CrossRef] [PubMed]
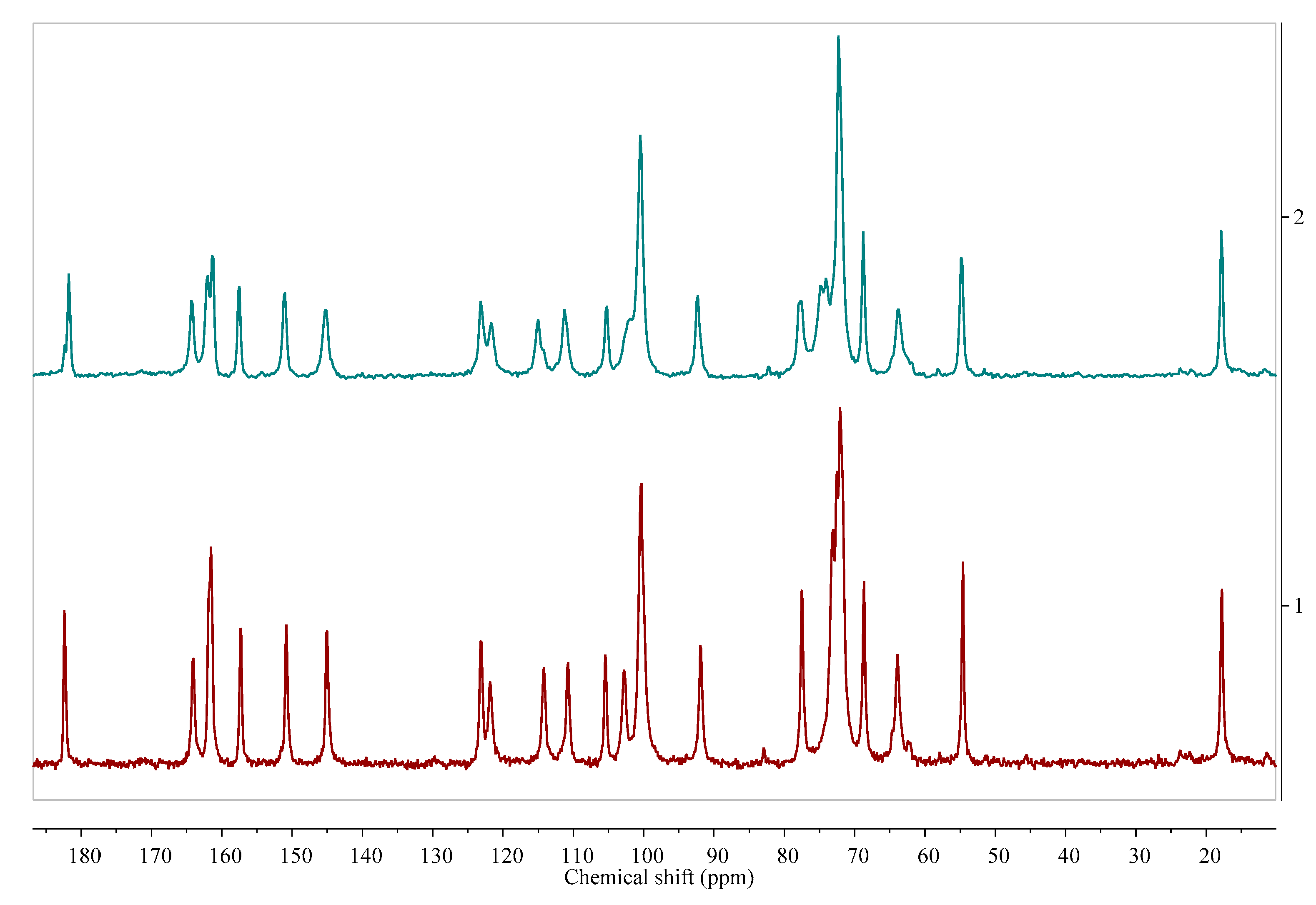
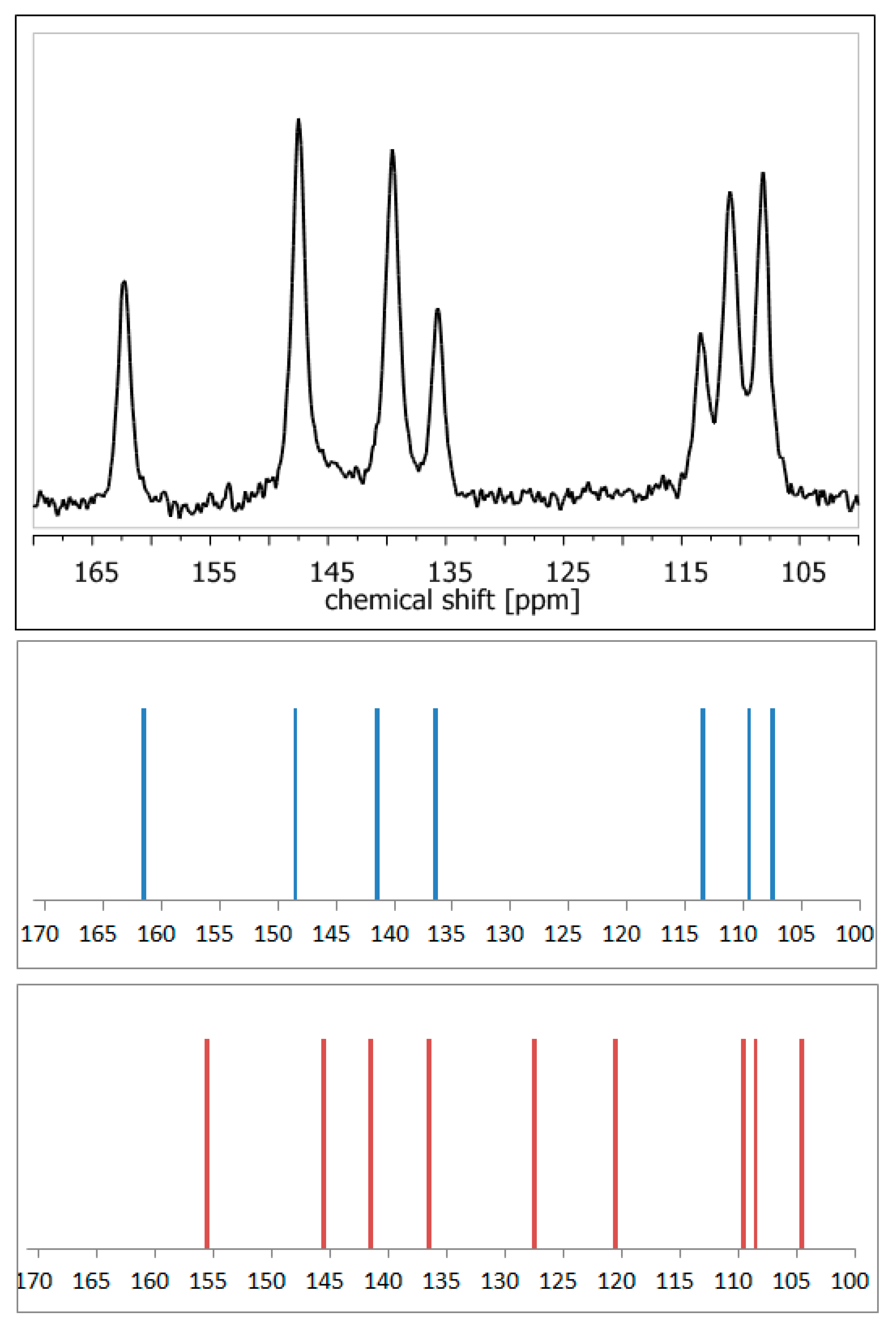
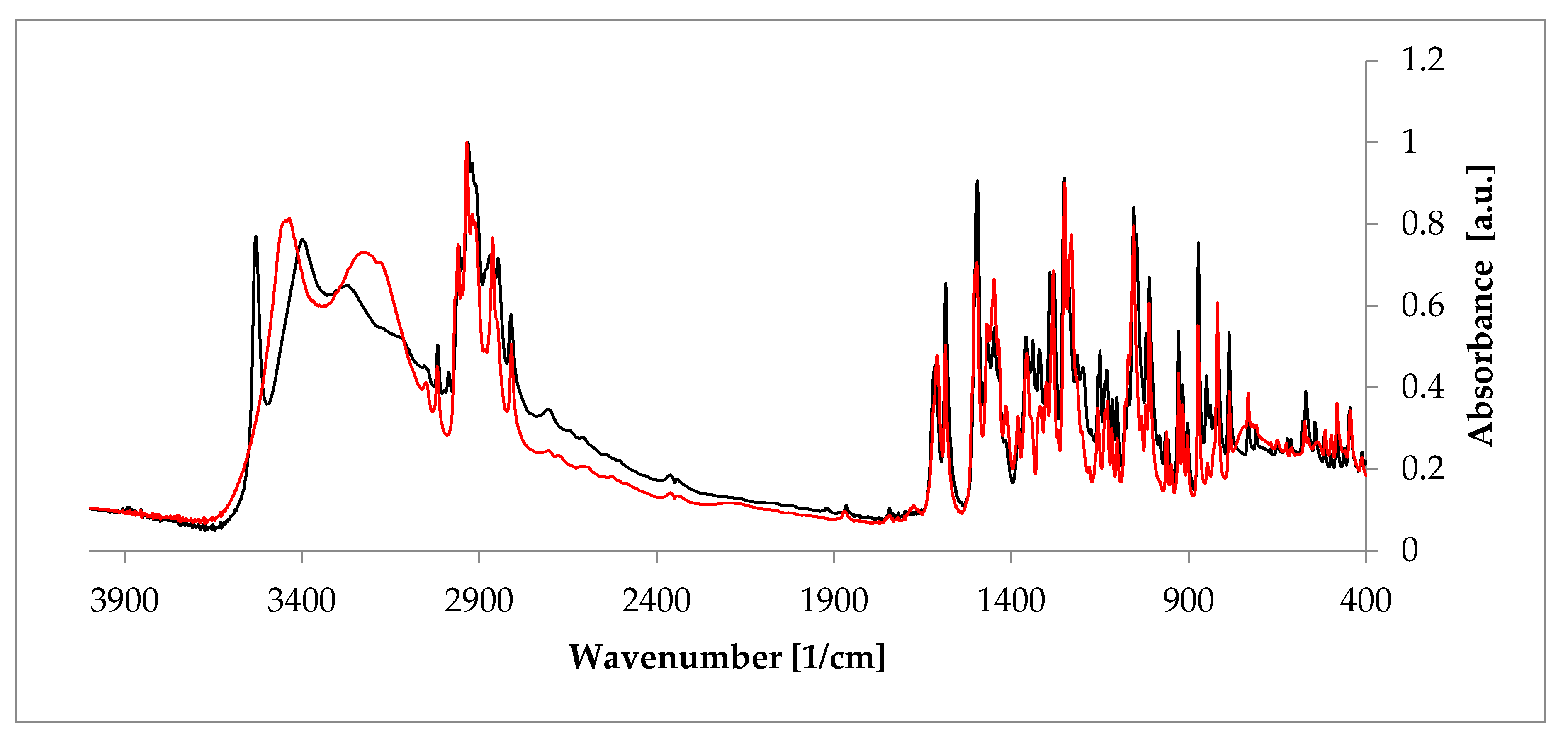
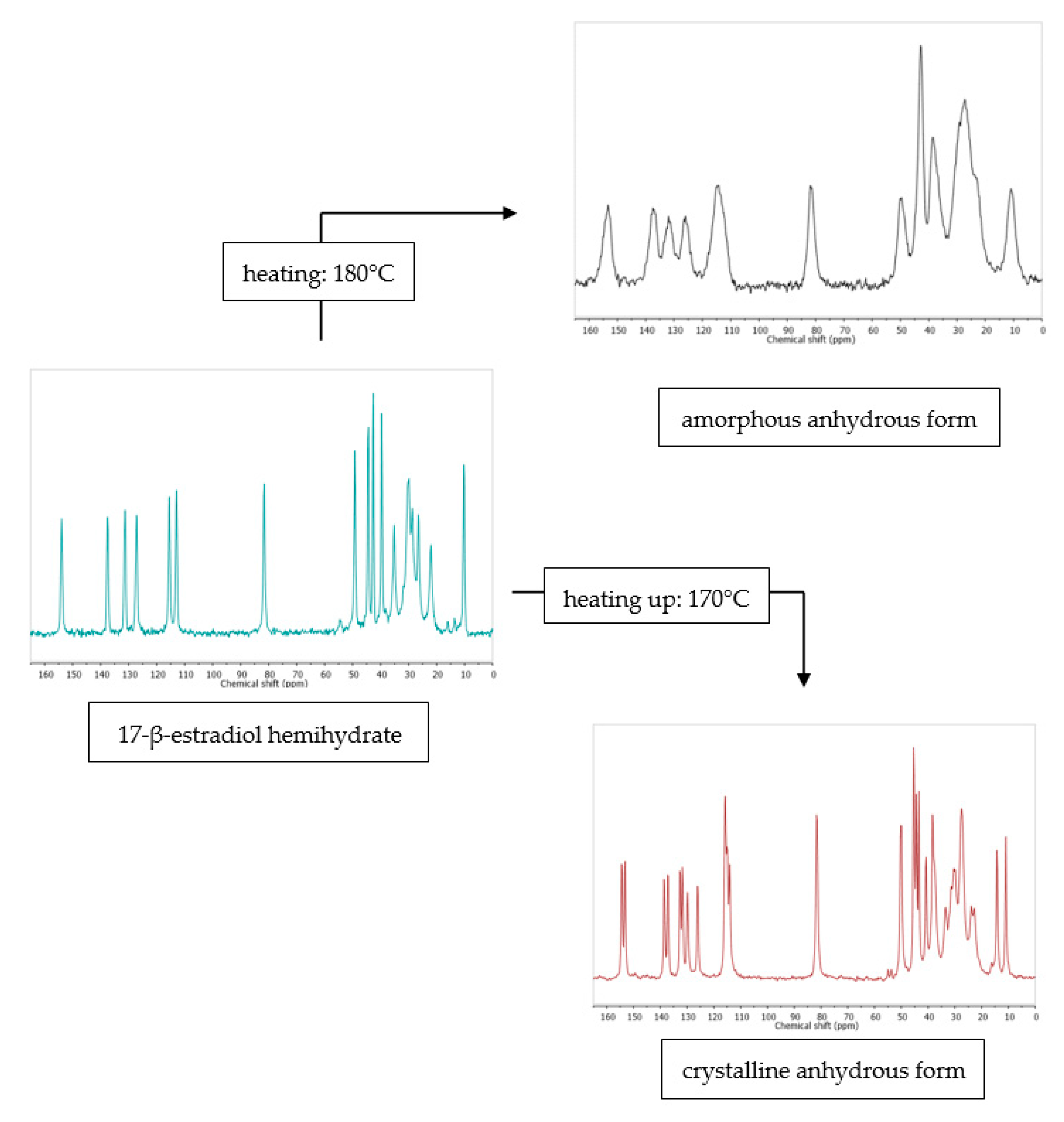
- Szeleszczuk, Ł.; Jurczak, E.; Zielińska-Pisklak, M.; Harwacki, J.; Pisklak, D.M. Comparison of the analytical methods (solid state NMR, FT-IR, PXRD) in the analysis of the solid drug forms with low concentration of an active ingredient—17-β-estradiol case. J. Pharm. Biomed. Anal. 2018, 149, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Nugrahani, I.; Slamet, I.; Mauludin, R.; Almira, M. Hydrate transformation study of fluoroquinolone antibiotics using Fourier transform infrared spectroscopy (FTIR). Int. J. Pharm. Pharm. Sci. 2015, 7, 246–252. [Google Scholar]
- Lu, J.; Wang, J.; Li, Z.; Rohani, S. Characterization and pseudopolymorphism of Lphenylalanine anhydrous and monohydrate forms. Afr. J. Pharm. Pharmacol. 2012, 6, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Seton, L.; Khamar, D.; Bradshaw, I.J.; Hutcheon, G.A. Solid State Forms of Theophylline: Presenting a New Anhydrous Polymorph. Cryst. Growth Des. 2010, 10, 3879–3886. [Google Scholar] [CrossRef]
- MS Reflex Plus Module within the Accelrys Materials Studio. Available online: https://www.3ds.com/fileadmin/PRODUCTS-SERVICES/BIOVIA/PDF/BIOVIA-material-studio-reflex-plus.pdf (accessed on 1 September 2020).
- Tishmack, P.A.; Bugay, D.A.; Byrn, S.R. Solid-state nuclear magnetic resonance spectroscopy—pharmaceutical application. J. Pharm. Sci. 2003, 92, 441–474. [Google Scholar] [CrossRef]
- Hirsh, D.A.; Wijesekara, A.V.; Carnahan, S.L.; Hung, I.; Lubach, J.W.; Nagapudi, K.; Rossini, A.J. Rapid Characterization of Formulated Pharmaceuticals Using Fast MAS 1H Solid-State NMR Spectroscopy. Mol. Pharm. 2019, 16, 3121–3132. [Google Scholar] [CrossRef] [Green Version]
- Chan-Huot, M.; Wimperis, S.; Gervais, C.; Bodenhausen, G.; Duma, L. Deuterium MAS NMR Studies of Dynamics on Multiple Timescales: Histidine and Oxalic Acid. Chem. Phys. Chem. 2014, 16, 204–215. [Google Scholar] [CrossRef]
- Szeleszczuk, Ł.; Pisklak, M.; Zielińska-Pisklak, M.; Wawer, I. Spectroscopic and structural studies of the diosmin monohydrate and anhydrous diosmin. Int. J. Pharm. 2017, 529, 193–199. [Google Scholar] [CrossRef]
- Gowda, V.; Laitinen, R.S.; Telkki, V.-V.; Larsson, A.C.; Antzutkin, O.N.; Lantto, P. DFT calculations in the assignment of solid-state NMR and crystal structure elucidation of a lanthanum(III) complex with dithiocarbamate and phenanthroline. Dalton Trans. 2016, 45, 19473–19484. [Google Scholar] [CrossRef] [Green Version]
- Costa, F.L.P.; Gomez, P.F.; Silva, K.A.; Liao, L.M. Conformational Analysis, Experimental and GIAO-DFT 13C NMR Chemical Shift Calculation on 2′-Hydroxy-3,4,5-trimethoxy-chalcone. J. Braz. Chem. Soc. 2017, 28, 2130–2135. [Google Scholar] [CrossRef]
- Charpentier, T. The PAW/GIPAW approach for computing NMR parameters: A new dimension added to NMR study of solids. Solid State NMR 2011, 40, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Iron, M.A. Evaluation of the Factors Impacting the Accuracy of 13C NMR Chemical Shift Predictions using Density Functional Theory—The Advantage of Long-Range Corrected Functionals. J. Chem. Theory Comput. 2017, 13, 5798–5819. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhang, J.; Peng, Q.; Glezakou, A.-V. A General Protocol for the Accurate Predictions of Molecular 13C/1H NMR Chemical Shifts via Machine Learning. ChemRxiv 2019. [Google Scholar]
- Czernek, J.; Brus, J. Exploring Accuracy Limits of Predictions of the 1H NMR Chemical Shielding Anisotropy in the Solid State. Molecules. 2019, 24, 1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryce, D.L. NMR crystallography: Structure and properties of materials from solid-state nuclear magnetic resonance observables. IUCrJ 2017, 4, 350–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeleszczuk, Ł.; Pisklak, D.M.; Zielińska-Pisklak, M.; Wawer, I. Effects of structural differences on the NMR chemical shifts in cinnamic acid derivatives: Comparison of GIAO and GIPAW calculations. Chem. Phys. Lett. 2016, 653, 35–41. [Google Scholar] [CrossRef]
- Colthup, N.L.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy, 3rd ed; Academic Press, Inc.: San Diego, CA, USA, 1990. [Google Scholar]
- Biswas, R.; Carpenter, W.; Voth, G.A.; Tokmakoff, A. Molecular modeling and assignment of IR spectra of the hydrated excess proton in isotopically dilute water. J. Chem. Phys. 2016, 145, 154504. [Google Scholar] [CrossRef]
- Hansen, P.E.; Spanget-Larsen, J. NMR and IR Investigations of Strong Intramolecular Hydrogen Bonds. Molecules 2017, 22, 552. [Google Scholar] [CrossRef] [Green Version]
- Van Eerdenbrugha, B.; Taylor, L.S. Application of mid-IR spectroscopy for the characterization of pharmaceutical systems. Int. J. Pharm. 2010, 417, 3–16. [Google Scholar] [CrossRef]
- Szakonyi, G.; Zelko, R. The effect of water on the solid state characteristics of pharmaceutical excipients: Molecular mechanisms, measurement techniques, and quality aspects of final dosage form. Int. J. Pharm. Investig. 2012, 2, 18–25. [Google Scholar] [PubMed] [Green Version]
- Oliver, K.V.; Marechal, A.; Rich, P.R. Effects of the Hydration State on the Mid-Infrared Spectra of Urea and Creatinine in Relation to Urine Analyses. Appl. Spectrosc. 2016, 70, 983–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv. Drug Deliver. Rev. 2017, 117, 25–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feth, M.P.; Nagel, N.; Baumgartner, B.; Bröckelmann, M.; Rigal, D.D.; Otto, B.; Spitzenberg, M.; Schulz, M.; Becker, B.D.; Fischer, F.; et al. Challenges in the development of hydrate phases as active pharmaceutical ingredients—An example. Eur. J. Pharm. Sci. 2011, 42, 116–129. [Google Scholar] [CrossRef]
- Jones, C.G.; Martynowycz, M.W.; Hattne, J.; Fulton, T.J.; Stoltz, B.J.; Rodriguez, J.A.; Nelson, H.M.; Gonen, T. The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent Sci. 2018, 4, 1587–1592. [Google Scholar] [CrossRef]
- Vasileiadis, M.; Pantelides, C.C.; Adjiman, C.S. Prediction of the crystal structures of axitinib, a polymorphic pharmaceutical molecule. Chem. Eng. Sci. 2015, 121, 60–76. [Google Scholar] [CrossRef] [Green Version]
- Baias, M.; Dumez, J.N.; Svensson, P.H.; Schantz, S.; Day, G.M.; Emsley, L. De Novo Determination of the Crystal Structure of a Large Drug Molecule by Crystal Structure Prediction-Based Powder NMR Crystallography. J. Am. Chem. Soc. 2013, 135, 17501–17507. [Google Scholar] [CrossRef] [Green Version]
- Woodley, S.M. Crystal structure prediction from first principles. Nat. Mater. 2008, 7, 937–946. [Google Scholar] [CrossRef]
- Hoja, J.; Ko, H.-Y.; Neumann, M.A.; Car, R.; DiStasio, R.A.; Tkatchenko, A. Reliable and practical computational description of molecular crystal polymorphs. Sci. Adv. 2019, 5, eaau3338. [Google Scholar] [CrossRef] [Green Version]
- Ratkova, E.L.; Abramov, Y.; Baskin, I.L.; Livingstone, D.J.; Fedorov, M.V.; Withnall, M.; Tetko, I.V. Background Information on Physical -Chemical Properties- Polymorphism. In Comprehensive Medicinal Chemistry III, 3rd ed.; Chackalamannil, S., Rotella, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017; pp. 396–400. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Abramov, Y.A. Current Computational Approaches to Support Pharmaceutical Solid Form Selection. Org. Process Res. Dev. 2013, 17, 472–485. [Google Scholar] [CrossRef]
- Day, G.M. Current approaches to predicting molecular organic crystal structures. Crystal. Rev. 2011, 17, 3–52. [Google Scholar] [CrossRef]
- Nyman, J.; Graeme, M.D. Modelling temperature-dependent properties of polymorphic organic molecular crystals. Phys. Chem. Chem. Phys. 2016, 18, 31132–31143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymorph Predictor BIOVIA Materials Studio. Available online: https://www.3ds.com/fileadmin/PRODUCTS-SERVICES/BIOVIA/PDF/BIOVIA-material-studio-polymorph-predictor.pdf (accessed on 1 September 2020).
- Braun, D.E.; Nartowski, K.P.; Khimyak, Y.Z.; Morris, K.R.; Byrn, S.R.; Griesser, U.J. Structural Properties, Order−Disorder Phenomena, and Phase Stability of Orotic Acid Crystal Forms. Mol. Pharm. 2016, 13, 1012–1029. [Google Scholar] [CrossRef] [Green Version]
- Dudek, M.K.; Paluch, P.; Pindelska, E. Crystal structures of two furazidin polymorphs revealed by a joint effort of crystal structure prediction and NMR crystallography. Acta Crystall. B 2020, 76, 322–335. [Google Scholar] [CrossRef]
- Dudek, M.K.; Paluch, P.; Śniechowska, J.; Nartowski, K.P.; Day, G.M.; Potrzebowski, M.J. Crystal structure determination of an elusive methanol solvate—Hydrate of catechin using crystal structure prediction and NMR crystallography. CrystEngComm 2020, 22, 4969–4981. [Google Scholar] [CrossRef]
- Braun, D.E.; Gelbrich, T.; Griesser, U.J. Experimental and computational approaches to produce and characterise isostructural solvates. CrystEngComm 2019, 21, 5533–5545. [Google Scholar] [CrossRef]
- May, J.C.; Grim, E.; Wheeler, R.M.; West, J. Determination of residual moisture in freeze-dried viral vaccines: Karl Fischer, gravimetric and thermogravimetric methodologies. J. Biol. Stand. 1982, 10, 249–259. [Google Scholar] [CrossRef]
- Bruttel, P.; Schlink, R. Water Determination by Karl Fischer Titration. In Pharmaceutical Sciences Encyclopedia; Gad, S.C., Ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA. [CrossRef]
- Kestens, V.; Conneely, P.; Bernreuther, A. Vaporisation coulometric Karl Fischer titration: A perfect tool for water content determination of difficult matrix reference materials. Food Chem. 2008, 106, 1454–1459. [Google Scholar] [CrossRef]
- U.S. Pharmacopoeia, Terpin Hydrate Monograph. Available online: http://www.pharmacopeia.cn/v29240/usp29nf24s0_m80920.html (accessed on 25 September 2020).
- Rezende., C.A.; San Gil, R.A.S.; Borré, L.B.; Pires, J.R.; Vaiss, V.S.; Resende, J.A.L.C.; Leitão, A.A.; De, A.; Katia, R.B.; Leal, K.Z. Combining Nuclear Magnetic Resonance Spectroscopy and Density Functional Theory Calculations to Characterize Carvedilol Polymorphs. J. Pharm. Sci. 2015, 105, 2648–2655. [Google Scholar] [CrossRef] [Green Version]
- Hédoux, A.; Paccou, L.; Derollez, P.; Guinet, Y. Dehydration mechanism of caffeine hydrate and structural description of driven metastable anhydrates analyzed by micro Raman spectroscopy. Int. J. Pharm. 2015, 486, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Basford, P.A.; Back, K.R.; Cram, M.; Docherty, R.; Davey, R.J.; Cruz-Cabeza, A.J. Impact of Crystal Structure and Molecular Conformation on the Hydration Kinetics of Fluconazole. Cryst. Growth 2019, 19, 7193–7205. [Google Scholar] [CrossRef]
- Braun, D.E.; Oberacher, H.; Arnhard, K.; Orlova, M.; Griesser, U.J. 4-Aminoquinaldine monohydrate polymorphism: Prediction and impurity aided discovery of a difficult to access stable form. CrystEngComm 2016, 22, 4053–4067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kons, A.; Rutkovska, L.; Bērziņš, A.; Bobrovs, R.; Actiņš, A. Three anhydrous forms and a dihydrate form of quifenadine hydrochloride: A structural study of the thermodynamic stability and dehydration mechanism. CrystEngComm 2015, 17, 3627–3635. [Google Scholar] [CrossRef]
- Braun, D.E.; Griesser, U.J. Why Do Hydrates (Solvates) Form in Small Neutral Organic Molecules? Exploring the Crystal Form Landscapes of the Alkaloids Brucine and Strychnine. Cryst. Growth 2016, 16, 6405–6418. [Google Scholar] [CrossRef] [Green Version]
- Leane, M.M.; Gamble, J.F.; Brown, J.; Hughes, H.; Crull, G.; Engstrom, J.; Gao, Q.; Bunker, M.; Rutherford, S.; Parker, A.; et al. Imaging Dehydration Kinetics of a Channel Hydrate Form of the HIV-1 Attachment Inhibitor Prodrug BMS-663068. J. Pharm. Sci. 2013, 102, 4375–4383. [Google Scholar] [CrossRef]
- Feth, M.P.; Jurascheck, J.; Spitzenberg, M.; Dillenz, J.; Bertele, G.; Stark, H. New technology for the investigation of water vapor sorption–induced crystallographic form transformations of chemical compounds: A water vapor sorption gravimetry–dispersive raman spectroscopy coupling. J. Pharm. Sci. 2011, 100, 1080–1092. [Google Scholar] [CrossRef]
- Goldstein, S.; Lebowitz, J.L.; Tumulka, R.; Zanghi, N. Gibbs and Boltzmann Entropy in Classical and Quantum Mechanics. In Statistical Mechanics and Scientific Explanation: Determinism, Indeterminism and Laws of Nature; Allori, V., Ed.; World Scientific: Singapore, 2020. [Google Scholar]
- Braun, E.; Gilmer, J.; Mayes, H.B.; Mobley, D.L.; Monroe, J.I.; Prasad, S.; Zuckerman, D.M. Best Practices for Foundations in Molecular Simulations. Living J. Comp. Mol. Sci. 2019, 1, 5957. [Google Scholar]
- Coates, R.A.; Armentrout, P.B. Binding energies of hydrated cobalt(II) by collision-induced dissociation and theoretical studies: Evidence for a new critical size. Phys. Chem. Chem. Phys. 2018, 20, 802–818. [Google Scholar] [CrossRef]
- Wen, S.; Beran, G.J.O. Crystal Polymorphism in Oxalyl Dihydrazide: Is Empirical DFT-D Accurate Enough? J. Chem. Theory Comput. 2012, 8, 2698–2705. [Google Scholar] [CrossRef]
- Rychkov, D.A.; Stare, J.; Boldyreva, E.V. Pressure-driven phase transition mechanisms revealed by quantum chemistry: L-serine polymorphs. Phys. Chem. Chem. Phys. 2017, 19, 6671–6676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thierfelder, C.; Hermann, A.; Schwerdtfeger, P.; Schmidt, W.G. Strongly bonded water monomers on the ice Ih basal plane: Density-functional calculations. Phys. Rev. B 2006, 74, 045422. [Google Scholar] [CrossRef] [Green Version]
- Skowronsky, L. Water Activity Applications in the Pharmaceutical Industry; Cundall, A., Fontana, A., Jr., Eds.; PDA/DHI: Houston, TX, USA, 2009; pp. 223–252. [Google Scholar]
- Skovronsky, L. Inhibition of Microbial Growth in Solid Dosages at ICH Stability Storage Conditions. Available online: https://www.europeanpharmaceuticalreview.com/article/8876/inhibition-of-microbial-growth-in-solid-dosages-at-ich-stability-storage-conditions/ (accessed on 1 September 2020).
- Touil, A.; Peczalski, R.; Zagrouba, F. Monitoring of theophylline dehydration in a vacuum contact dryer by near-infrared spectroscopy. Chem. Eng. Res. Des. 2013, 91, 1063–1070. [Google Scholar] [CrossRef]
- Chennuru, R.; Koya, R.T.; Kommavarapu, P.; Narasayya, S.V.; Muthudoss, P.; Vishweshwar, P.; Chandra, R.R.; Mahapatra, B.S. In Situ Metastable Form: A Route for the Generation of Hydrate and Anhydrous Forms of Ceritinib. Cryst. Growth Des. 2017, 17, 6341–6352. [Google Scholar] [CrossRef]
- Chadha, R.; Arora, P.; Saini, A.; Singh Jain, D. Solvated Crystalline Forms of Nevirapine: Thermoanalytical and Spectroscopic Studies. AAPS PharmSciTech 2010, 11, 1328–1339. [Google Scholar] [CrossRef]
- Jia, L.; Li, Z.; Gong, J. Two new polymorphs and one dihydrate of lenalidomide: Solid-state characterization study. Pharm. Dev. Technol. 2019, 24, 1175–1180. [Google Scholar] [CrossRef]
- Hsieh, W.; Cheng, W.; Chen, L.; Lin, S. Non-isothermal dehydration kinetic study of aspartame hemihydrate using DSC, TGA and DSC-FTIR microspectroscopy Characterization of polymorphic ampicillin forms. Asian J. Pharm. Sci. 2018, 13, 212–219. [Google Scholar] [CrossRef]
- Sorrenti, M.; Catenacci, L.; Cruickshank, D.L.; Caira, M.R. Lisinopril dihydrate: Single-crystal X-ray structure and physicochemical characterization of derived solid forms. J. Pharm. Sci. 2013, 102, 3596–3603. [Google Scholar] [CrossRef]
- Pajzderska, A.; Chudoba, D.M.; Mielcarek, J.; Wąsicki, J. Calorimetric, FTIR and 1H NMR measurements in combination with DFT calculations for monitoring solid-state changes of dynamics of sibutramine hydrochloride. J. Pharm. Sc. 2012, 101, 3799–3810. [Google Scholar] [CrossRef]
- Ferraboschi, P.; Sala, M.C.; Stradi, R.; Ragonesi, L.; Gagliardi, C.; Lanzarotti, P.; Ragg, E.M.; Mori, M.; Meneghetti, F. Full spectroscopic characterization of two crystal pseudopolymorphic forms of the antiandrogen cortexolone 17α-propionate for topic application. Steroids 2017, 128, 95–104. [Google Scholar] [CrossRef]
- Zhang, Q.; Lu, L.; Dai, W.; Mei, X. Polymorphism and isomorphism of Huperzine A solvates: Structure, properties and form transformation. CrystEngComm 2014, 16, 1919–1926. [Google Scholar] [CrossRef]
- Braun, D.E.; Gelbrich, T.; Wurst, K.; Griesser, U.J. Cputational and Experimental Characterization of Five Crystal Forms of Thymine: Packing Polymorphism, Polytypism/Disorder and Stoichiometric 0.8-Hydrate. Cryst. Growth Des. 2016, 16, 3480–3496. [Google Scholar] [CrossRef] [Green Version]
- Furuta, H.; Mori, S.; Yoshihashi, Y.; Yonemochi, E.; Uekusa, H.; Sugano, K.; Terada, K. Physicochemical and crystal structure analysis of pranlukast pseudo-polymorphs I: Anhydrates and hydrate. J. Pharm. Biomed. Anal. 2015, 107, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.K.; Thaimattam, R.; Ramanan, A. Multiple Crystal Forms of p-Aminosalicylic Acid: Salts, Salt Co-Crystal Hydrate, Co-Crystals, and Co-Crystal Polymorphs. Cryst. Growth Des. 2013, 13, 360–366. [Google Scholar] [CrossRef]
- Laxmi, P.; Varma, A.; Pai, A.; Sathyanarayana, M.B. Experimental Data of Fabricated Co-crystals of Doxorubicin HCL with Flavonoids. Indian J. Pharm. Educ. Res. 2019, 53, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Lange, L.; Sadowski, G. Polymorphs, hydrates, cocrystals, and cocrystal hydrates: Thermodynamic modeling of theophylline systems. Cryst. Growth Des. 2016, 16, 4439–4449. [Google Scholar] [CrossRef]
- Cherukuvada, S.; Bolla, G.; Sikligar, K.; Nangia, A. 4-Aminosalicylic acid adducts. Cryst. Growth Des. 2013, 13, 1551–1557. [Google Scholar] [CrossRef]
- Stepanovs, D.; Mishnev, A. Multicomponent pharmaceutical cocrystals: Furosemide and pentoxifylline. Acta Crystallogr. C Struct. Chem. 2012, 68, 488–491. [Google Scholar] [CrossRef]
- Báthori, N.B.; Lemmerer, A.; Venter, G.A.; Bourne, S.A.; Caira, M.R. Pharmaceutical Co-crystals with Isonicotinamide‒Vitamin B3, Clofibric Acid, and Diclofenac‒and Two Isonicotinamide Hydrates. Cryst. Growth Des. 2011, 11, 75–87. [Google Scholar] [CrossRef]
- Braun, D.E.; Vickers, M.; Griesser, U.J. Dapsone Form V: A Late Appearing Thermodynamic Polymorph of a Pharmaceutical. Mol. Pharm. 2019, 16, 3221–3236. [Google Scholar] [CrossRef]
- Pina, M.F.; Pinto, J.F.; Sousa, J.J.; Fábián, L.; Zhao, M.; Craig, D.Q. Identification and characterization of stoichiometric and nonstoichiometric hydrate forms of paroxetine HCl: Reversible changes in crystal dimensions as a function of water absorption. Mol. Pharm. 2012, 9, 3515–3525. [Google Scholar] [CrossRef] [PubMed]
- Lutker, K.M.; Quiñones, R.; Xu, J.; Ramamoorthy, A.; Matzger, A.J. Polymorphs and hydrates of acyclovir. J. Pharm. Sci. 2011, 100, 949–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsh, D.A.; Holmes, S.T.; Chakravarty, P.; Peach, A.A.; DiPasquale, A.G.; Nagapudi, K.; Schurko, R.W. In Situ Characterization of Waters of Hydration in a Variable-Hydrate Active Pharmaceutical Ingredient Using 35Cl Solid-State NMR and X-ray Diffraction. Cryst. Growth Des. 2019, 19, 7349–7362. [Google Scholar] [CrossRef]
- Nartowski, K.P.; Karabin, J.; Morritt, A.L.; Nowak, M.; Fábián, L.; Karolewicz, B.; Khimyak, Y.Z. Solvent driven phase transitions of acyclovir–the role of water and solvent polarity. CrystEngComm 2019, 21, 2180–2192. [Google Scholar] [CrossRef] [Green Version]
- Abraham, A.; Apperley, D.C.; Byard, S.J.; Ilott, A.J.; Robbins, A.J.; Zorin, V.; Hodgkinson, P. Characterising the role of water in sildenafil citrate by NMR crystallography. CrystEngComm 2016, 18, 1054–1063. [Google Scholar] [CrossRef] [Green Version]
- Kerr, H.E.; Mason, H.E.; Sparkes, H.A.; Hodgkinson, P. Testing the limits of NMR crystallography: The case of caffeine–citric acid hydrate. CrystEngComm 2016, 18, 6700–6707. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.E.; McMahon, J.A.; Bhardwaj, R.M.; Nyman, J.; Neumann, M.A.; van de Streek, J.; Reutzel-Edens, S.M. Inconvenient Truths about Solid Form Landscapes Revealed in the Polymorphs and Hydrates of Gandotinib. Cryst. Growth Des. 2019, 19, 2947–2962. [Google Scholar] [CrossRef]
- Kang, F.; Vogt, F.G.; Brum, J.; Forcino, R.; Copley, R.C.; Williams, G.; Carlton, R. Effect of particle size and morphology on the dehydration mechanism of a non-stoichiometric hydrate. Cryst. Growth Des. 2012, 12, 60–74. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, L.; Mei, X. Supramolecular structures and physicochemical properties of norfloxacin salts. Acta Crystallogr. B 2014, 70, 750–760. [Google Scholar] [CrossRef]
- Braun, D.E.; Koztecki, L.H.; McMahon, J.A.; Price, S.L.; Reutzel-Edens, S.M. Navigating the waters of unconventional crystalline hydrates. Mol. Pharm. 2012, 12, 3069–3088. [Google Scholar] [CrossRef]
- Braun, D.E.; Griesser, U.J. Supramolecular organization of nonstoichiometric drug hydrates: Dapsone. Front. Chem. 2018, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klitou, P.; Rosbottom, I.; Simone, E. Synthonic Modeling of Quercetin and Its Hydrates: Explaining Crystallization Behavior in Terms of Molecular Conformation and Crystal Packing. Cryst. Growth Des. 2019, 19, 4774–4783. [Google Scholar] [CrossRef]
- Wang, Y.; Du, S.; Wu, S.; Li, L.; Zhang, D.; Yu, B.; Zhou, L.; kiflegiorgis Bekele, H.; Gong, J. Thermodynamic and molecular investigation into the solubility, stability and self-assembly of gabapentin anhydrate and hydrate. J. Chem. Thermodyn. 2017, 113, 132–143. [Google Scholar] [CrossRef]
- Van de Streek, J.; Rantanen, J.; Bond, A.D. Structures of cefradine dihydrate and cefaclor dihydrate from DFT-D calculations. Acta Crystallogr. E 2013, 69, 1229–1233. [Google Scholar] [CrossRef]
- Grobelny, P.; Mukherjee, A.; Desiraju, G.R. Polymorphs and hydrates of Etoricoxib, a selective COX-2 inhibitor. CrystEngComm 2012, 14, 5785–5794. [Google Scholar] [CrossRef]
- Braun, D.E.; Gelbrich, T.; Kahlenberg, V.; Griesser, U.J. Insights into hydrate formation and stability of morphinanes from a combination of experimental and computational approaches. Mol. Pharm. 2014, 11, 3145–3163. [Google Scholar] [CrossRef]
- Aitipamula, S.; Chow, P.S.; Tan, R.B. Solvates and a monohydrate of N4-acetylsulfamerazine: Structural, thermochemical, and computational analysis. J. Mol. Struct. 2011, 1005, 134–140. [Google Scholar] [CrossRef]
- Petruševski, G.; Kajdžanoska, M.; Ugarkovic, S.; Micovski, I.; Bogoeva-Gaceva, G.; Jovanovski, G.; Makreski, P. Solvatomorphism of codeine phosphate sesquihydrate—Vibrational spectroscopy and thermoanalytical characterization. Vib. Spectrosc. 2012, 63, 460–468. [Google Scholar] [CrossRef]
- Bērziņš, A.; Actiņš, A. Dehydration of mildronate dihydrate: A study of structural transformations and kinetics. CrystEngComm 2014, 16, 3926–3934. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, H.; Xue, J.; Tang, W.; Fang, H.; Zhang, Q.; Li, Y.; Hong, Z. Vibrational spectroscopic study of polymorphism and polymorphic transformation of the anti-viral drug lamivudine. Spectrochim. Acta A 2015, 137, 1158–1163. [Google Scholar] [CrossRef]
- Sorrenti, M.; Catenacci, L.; Bruni, G.; Luppi, B.; Bigucci, F.; Bettinetti, G. Solid-state characterization of tacrine hydrochloride. J. Pharm. Biomed. Anal. 2012, 63, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Xue, J.; Wang, Q.; Du, Y. Investigation into structure and dehydration dynamic of gallic acid monohydrate: A Raman spectroscopic study. Spectrochim. Acta A 2018, 201, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.H.; Gordon, S.; Pajander, J.P.; Østergaard, J.; Rades, T.; Müllertz, A. Biorelevant characterisation of amorphous furosemide salt exhibits conversion to a furosemide hydrate during dissolution. Int. J. Pharm. 2013, 457, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Raijada, D.; Bond, A.D.; Larsen, F.H.; Cornett, C.; Qu, H.; Rantanen, J. Exploring the solid-form landscape of pharmaceutical hydrates: Transformation pathways of the sodium naproxen anhydrate-hydrate system. Pharm. Res. 2013, 30, 280–289. [Google Scholar] [CrossRef]
- Arora, K.K.; Thakral, S.; Suryanarayanan, R. Instability in theophylline and carbamazepine hydrate tablets: Cocrystal formation due to release of lattice water. Pharm. Res. 2013, 30, 1779–1789. [Google Scholar] [CrossRef]
- Pinto, M.A.L.; Ambrozini, B.; Ferreira, A.P.G.; Cavalheiro, É.T.G. Thermoanalytical studies of carbamazepine: Hydration/dehydration, thermal decomposition, and solid phase transitions. Brazilian J. Pharm. Sci. 2014, 50, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Gonzaga, E.V.; Viana, A.L.; Viana, O.M.; Doriguetto, A.C. Solid-state phase transition mechanism and physical–chemical study of the crystal forms of monosodium alendronate: Trihydrate versus anhydrate. Cryst. Growth Des. 2016, 16, 6891–6902. [Google Scholar] [CrossRef]
- Mizoguchi, R.; Uekusa, H. Elucidating the Dehydration Mechanism of Ondansetron Hydrochloride Dihydrate with a Crystal Structure. Cryst. Growth Des. 2018, 18, 6142–6149. [Google Scholar] [CrossRef]
- Dudek, M.K.; Jeziorna, A.; Potrzebowski, M.J. Computational and experimental study of reversible hydration/dehydration processes in molecular crystals of natural products—A case of catechin. CrystEngComm 2016, 18, 5267–5277. [Google Scholar] [CrossRef]
- Censi, R.; Martena, V.; Hoti, E.; Malaj, L.; Di Martino, P. Sodium ibuprofen dihydrate and anhydrous. J. Therm. Anal. Calorim. 2013, 111, 2009–2018. [Google Scholar] [CrossRef]
- ICH Topic Q 1 A (R2) Stability Testing of New Drug Substances and Products. Note for Guidance on Stability Testing. Stability Testing of New Drug Substances and Products. (CPMP/ICH/2736/99). Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances-products-step-5_en.pdf (accessed on 1 September 2020).
- Shah, H.S.; Chaturvedi, K.; Hamad, M.; Bates, S.; Hussain, A.; Morris, K. New insights on solid-state changes in the levothyroxine sodium pentahydrate during dehydration and its relationship to chemical instability. AAPS PharmSciTech 2019, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Sanphui, P.; Bolla, G.; Nangia, A. High solubility piperazine salts of the nonsteroidal anti-inflammatory drug (NSAID) meclofenamic acid. Cryst. Growth Des. 2012, 12, 2023–2036. [Google Scholar] [CrossRef]
- Allada, R.; Maruthapillai, A.; Devikala, S.; Pallepogu, R. Hydrated Moxonidine SaccharinateSalt: Synthesis, Characterization, Crystal structure determination and dissolution enhancement. Mater. Today 2019, 14, 618–629. [Google Scholar] [CrossRef]
- Bommaka, M.K.; Mannava, M.C.; Suresh, K.; Gunnam, A.; Nangia, A. Entacapone: Improving aqueous solubility, diffusion permeability, and cocrystal stability with theophylline. Cryst. Growth Des. 2018, 18, 6061–6069. [Google Scholar] [CrossRef]
- Inam, M.; Wu, J.; Shen, J.; Phan, C.U.; Tang, G.; Hu, X. Preparation and Characterization of Novel Pharmaceutical Co-Crystals: Ticagrelor with Nicotinamide. Crystals 2018, 8, 336. [Google Scholar] [CrossRef] [Green Version]
- Vangala, V.R.; Chow, P.S.; Tan, R.B. Co-crystals and co-crystal hydrates of the antibiotic nitrofurantoin: Structural studies and physicochemical properties. Cryst. Growth Des. 2012, 12, 5925–5938. [Google Scholar] [CrossRef]
- Reggane, M.; Wiest, J.; Saedtler, M.; Harlacher, C.; Gutmann, M.; Zottnick, S.H.; Piéchon, P.; Dix, I.; Müller-Buschbaum, K.; Holzgrabe, U.; et al. Bioinspired co-crystals of Imatinib providing enhanced kinetic solubility. Eur. J. Pharm. Biopharm. 2018, 128, 290–299. [Google Scholar] [CrossRef]
- Aitipamula, S.; Vangala, V.R.; Chow, P.S.; Tan, R.B. Cocrystal hydrate of an antifungal drug, griseofulvin, with promising physicochemical properties. Cryst. Growth Des. 2012, 12, 5858–5863. [Google Scholar] [CrossRef]
- Zhou, X.B.; Zhu, J.R.; Liu, J.Y.; Jin, Z.P.; Tang, F.Y.; Hu, X.R. Crystal structures and properties of two hydrated conglomerate forms of the heart-rate-lowering agent ivabradine hydrochloride. Acta Crystallogr. C 2019, 75, 545–553. [Google Scholar] [CrossRef]
- Grepioni, F.; Braga, D.; Chelazzi, L.; Shemchuk, O.; Maffei, P.; Sforzini, A.; Viscomi, G.C. Improving solubility and storage stability of rifaximin via solid-state solvation with Transcutol®. CrystEngComm 2019, 21, 5278–5283. [Google Scholar] [CrossRef]
- Warzecha, M.; Guo, R.M.; Bhardwaj, R.; Reutzel-Edens, S.M.; Price, S.L.; Lamprou, D.A.; Florence, A.J. Direct observation of templated two-step nucleation mechanism during olanzapine hydrate formation. Cryst. Growth Des. 2017, 17, 6382–6393. [Google Scholar] [CrossRef] [Green Version]
- Pinon, A.C.; Rossini, A.J.; Widdifield, C.M.; Gajan, D.; Emsley, L. Polymorphs of theophylline characterized by DNP enhanced solid-state NMR. Mol. Pharm. 2015, 12, 4146–4153. [Google Scholar] [CrossRef] [PubMed]
- Nugrahani, I.L.M.A.; Min, S.S. Hydrate transformation of sodium sulfacetamide and neomycin sulphate. Int. J. Pharm. Pharm. Sci. 2015, 7, 409–415. [Google Scholar]
- González-González, J.S.; Zúñiga-Lemus, O.; Hernández-Galindo, M.D. Hydrated Solid Forms of Theophylline and Caffeine Obtained by Mechanochemistry. IOSR J. Pharm. 2017, 7, 28–30. [Google Scholar] [CrossRef]
- Otsuka, M.; Kanai, Y.; Hattori, Y. Real-Time Monitoring of Changes of Adsorbed and Crystalline Water Contents in Tablet Formulation Powder Containing Theophylline Anhydrate at Various Temperatures During Agitated Granulation by Near-Infrared Spectroscopy. J. Pharm. Sci. 2014, 103, 2924–2936. [Google Scholar] [CrossRef]
- Chieng, N.; Rades, T.; Aaltonen, J. An overview of recent studies on the analysis of pharmaceutical polymorphs. J. Pharm. Biomed. Anal. 2011, 55, 618–644. [Google Scholar] [CrossRef]
- Fini, A.; Cavallari, C.; Bassini, G.; Ospitali, F.; Morigi, R. Diclofenac salts, part 7: Are the pharmaceutical salts with aliphatic amines stable? J. Pharm. Sci. 2012, 101, 3157–3168. [Google Scholar] [CrossRef]
- Arora, K.K.; Bhardwaj, S.P.; Mistry, P.; Suryanarayanan, R. Modulating the dehydration conditions of adefovir dipivoxil dihydrate to obtain different physical forms of anhydrate. J. Pharm. Sci. 2015, 104, 1056–1064. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurczak, E.; Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M.; Zielińska-Pisklak, M. Pharmaceutical Hydrates Analysis—Overview of Methods and Recent Advances. Pharmaceutics 2020, 12, 959. https://doi.org/10.3390/pharmaceutics12100959
Jurczak E, Mazurek AH, Szeleszczuk Ł, Pisklak DM, Zielińska-Pisklak M. Pharmaceutical Hydrates Analysis—Overview of Methods and Recent Advances. Pharmaceutics. 2020; 12(10):959. https://doi.org/10.3390/pharmaceutics12100959
Chicago/Turabian StyleJurczak, Ewa, Anna Helena Mazurek, Łukasz Szeleszczuk, Dariusz Maciej Pisklak, and Monika Zielińska-Pisklak. 2020. "Pharmaceutical Hydrates Analysis—Overview of Methods and Recent Advances" Pharmaceutics 12, no. 10: 959. https://doi.org/10.3390/pharmaceutics12100959