Rationally Designed Dendritic Silica Nanoparticles for Oral Delivery of Exenatide

 , , , and
, , , and 
Abstract
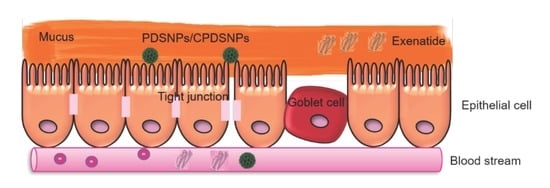
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of Dendritic Silica Nanoparticles
2.2. Synthesis of Phosphonate Modified DSNPs (PDSNPs)
2.3. Synthesis of Amino-Propyl Modified DSNPs (ADSNPs)
2.4. Synthesis of Succinic Anhydride Modified DSNPs (SDSNPs)
2.5. Physicochemical Characterization of DSNPs and Derivatives
2.6. Loading of Exenatide into Pristine and Modified DSNPs
2.7. Coating of PDSNPs with Chitosan
2.8. In Vitro Release of Exenatide from PDSNPs
2.9. In Vitro Permeability Assay of Exenatide- and Exenatide-Loaded PDSNPs Using a Caco-2 Monolayer Model
2.10. In Vitro Permeability Assay of Exenatide- and Exenatide-Loaded PDSNPs Using Caco-2/HT29-MTX/Raji-B Triple Co-Culture Model
2.11. Statistical Analysis
3. Results and Discussion
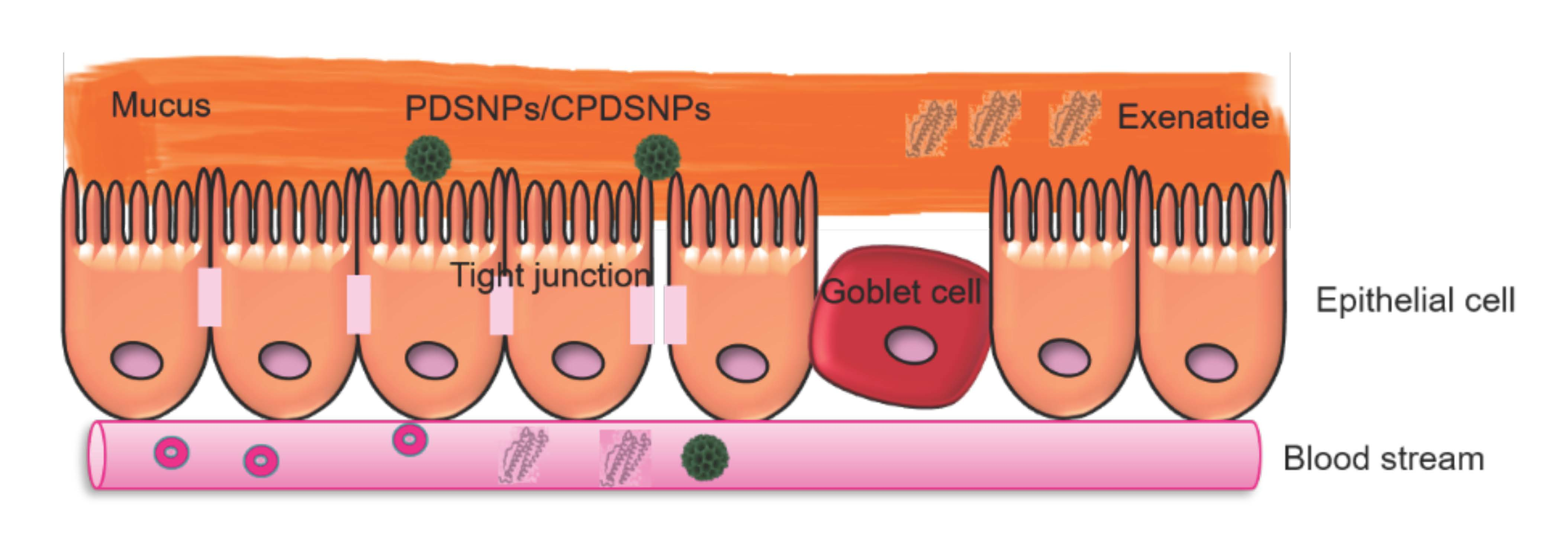
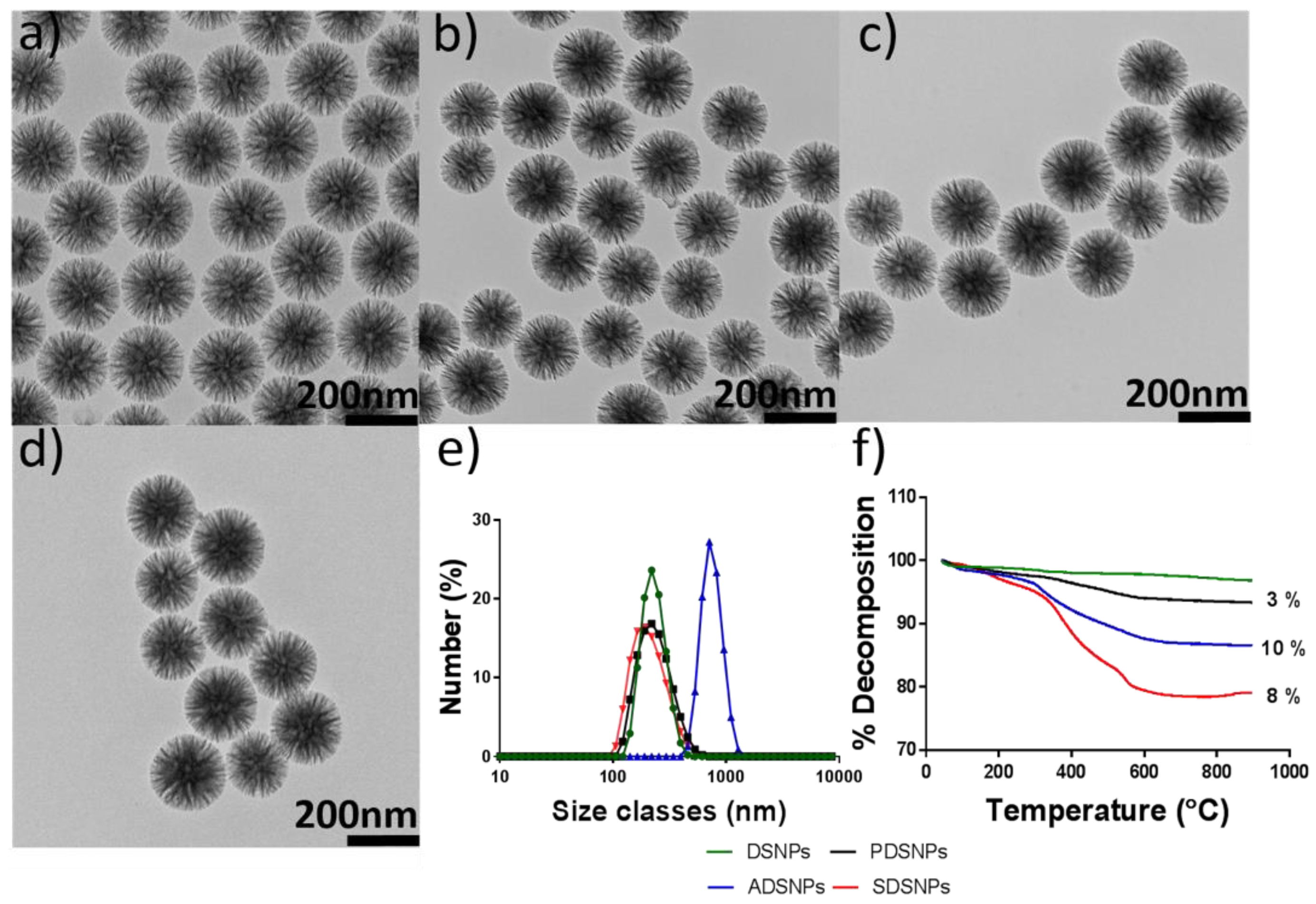
3.1. Preparation and Characterization of Pristine and Modified DSNPs
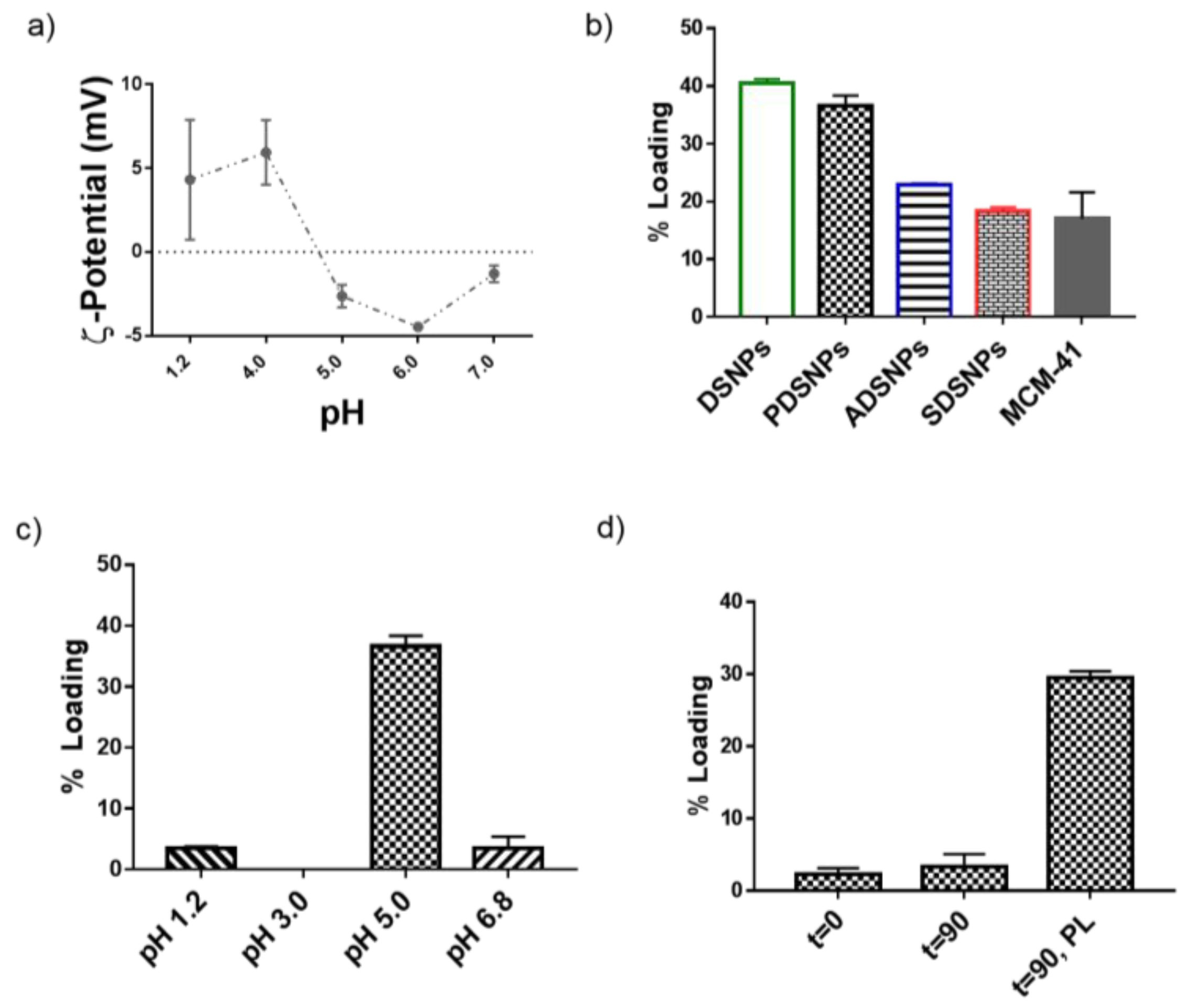
3.2. Loading of Exenatide into Pristine and Modified DSNPs
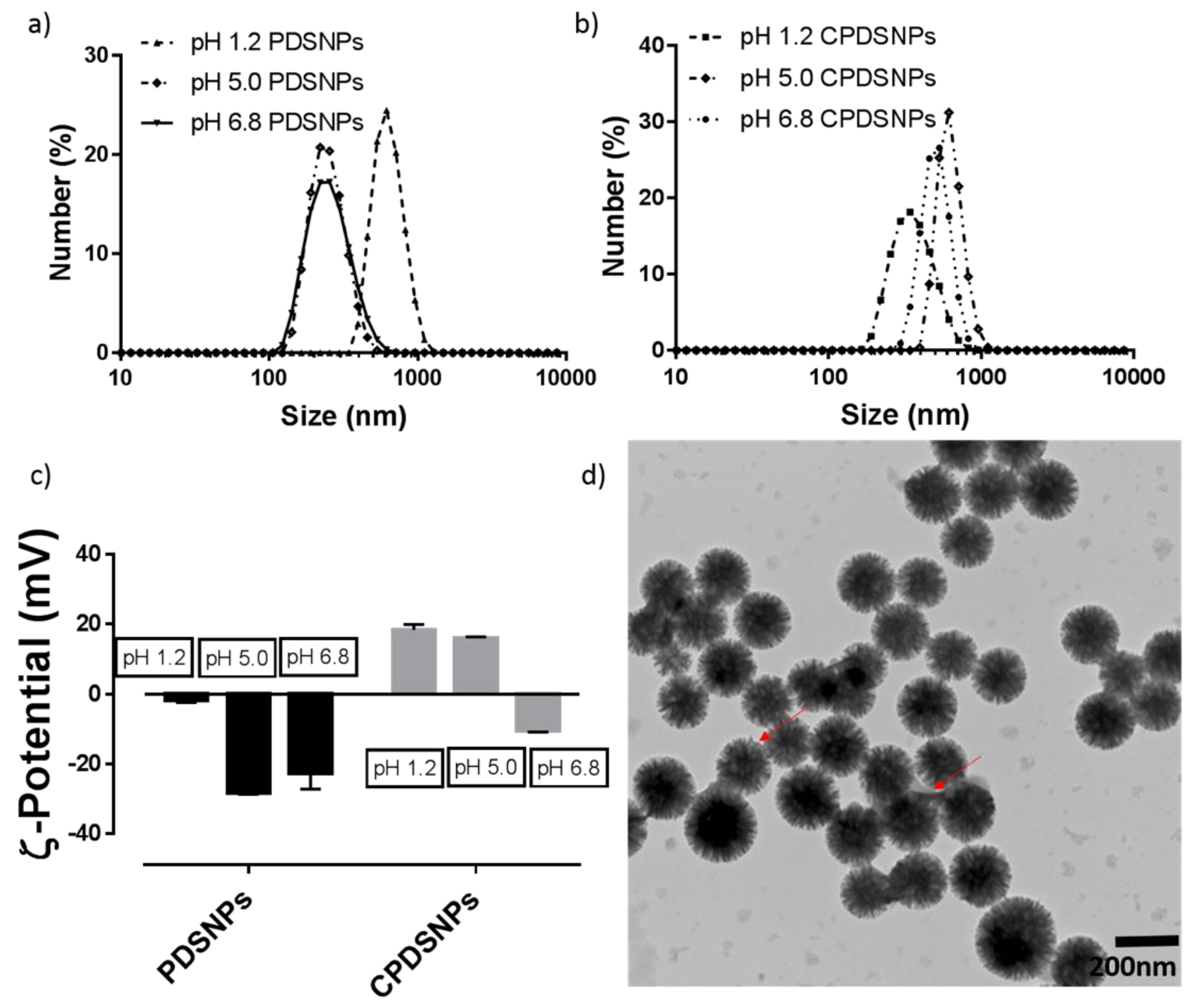
3.3. Particle Size and ζ-Potential Analysis of PDSNPs and CPDNSPs at Different pH
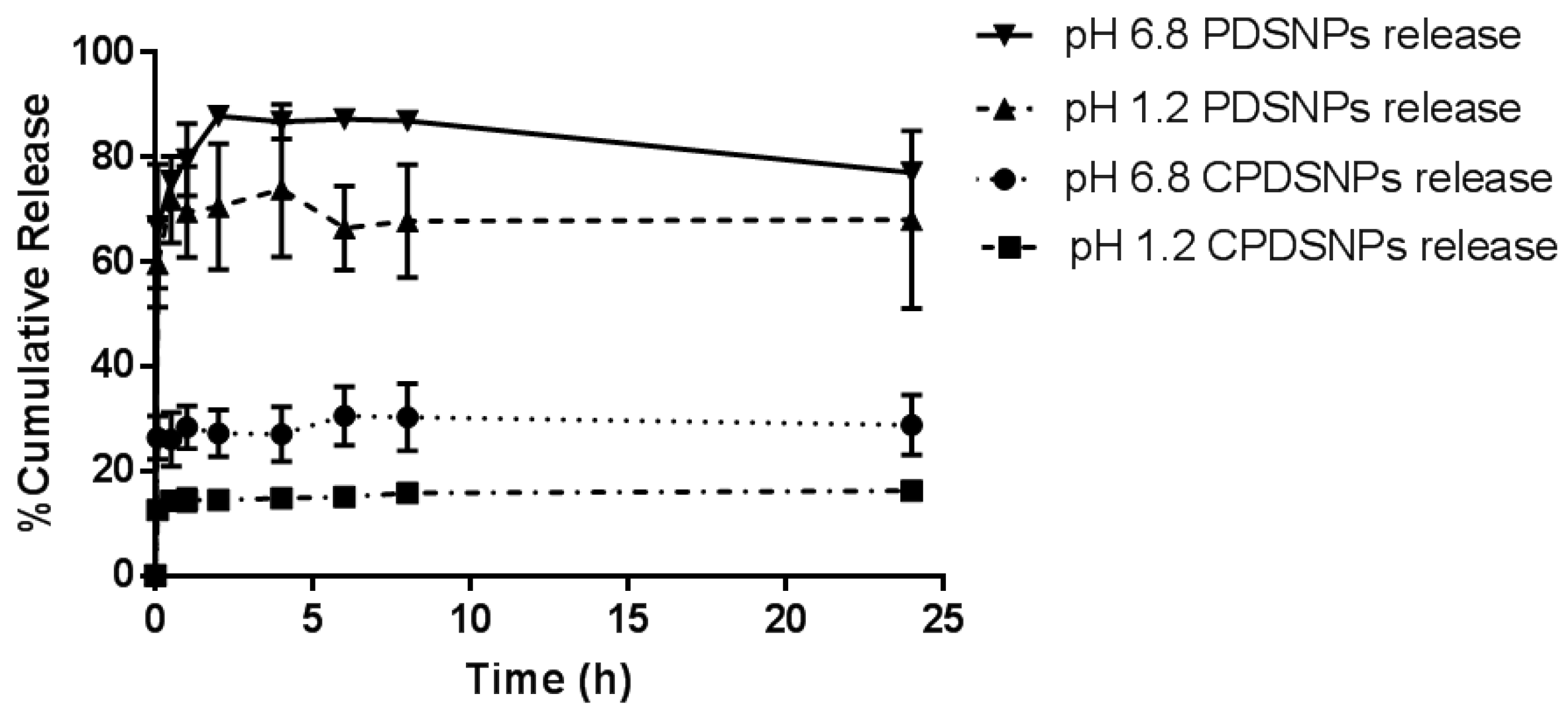
3.4. In Vitro Release Study of Exenatide from PDSNPs and CPDSNPs
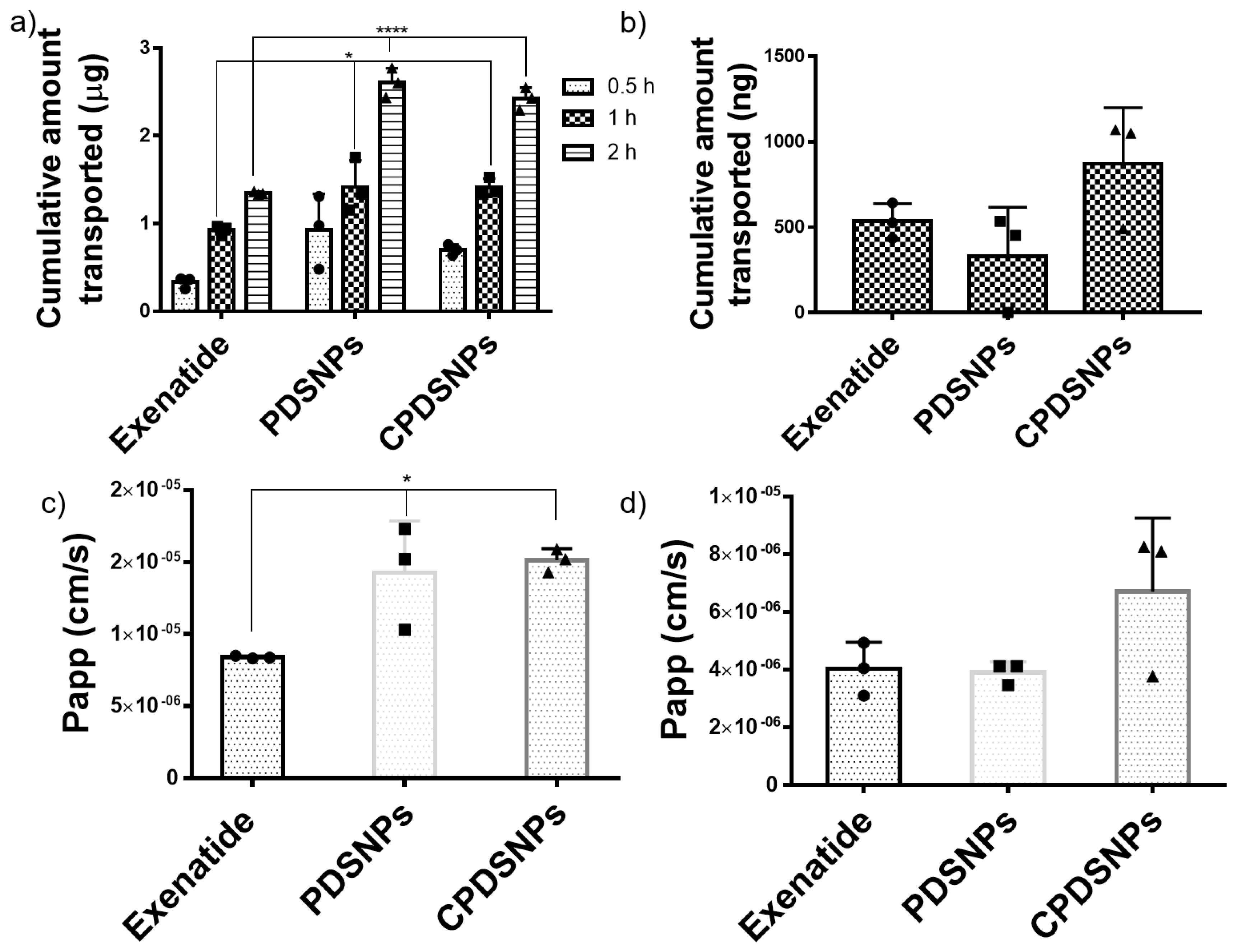
3.5. In Vitro Permeation of Exenatide-Loaded PDSNPs and Coated PDSNPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Tsomaia, N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Canepa, C.I.; Berini, J.C.; Lewicki, C.A.; Sosnik, M.; Biglione, A. Development of a Drug Delivery System Based on Chitosan Nanoparticles for Oral Administration of Interferon-Alpha. Biomacromolecules 2017, 18, 3302–3309. [Google Scholar] [CrossRef]
- Rehmani, S.; Dixon, J.E. Oral delivery of anti-diabetes therapeutics using cell penetrating and transcytosing peptide strategies. Peptides 2018, 100, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Meloni, A.R.; Lowe, M.B.D.C.; Parkes, D.G. GLP-1 receptor activated insulin secretion from pancreatic β-cells: Mechanism and glucose dependence. Diabetes Obes. Metab. 2013, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Wittbrodt, E. All-cause and diabetes-related healthcare costs among US adults with type 2 diabetes initiating exenatide once weekly or insulin glargine. Diabetes Obes. Metab. 2018, 20, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Hedrington, M.S.; Davis, S.N. Oral semaglutide for the treatment of type 2 diabetes. Expert Opin. Pharmacother. 2018, 20, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Araújo, F. The impact of nanoparticles on the mucosal translocation and transport of GLP-1 across the intestinal epithelium. Biomaterials 2014, 35, 9199–9207. [Google Scholar] [CrossRef]
- Menzel, C. In vivo evaluation of an oral self-emulsifying drug delivery system (SEDDS) for exenatide. J. Control. Release 2018, 277, 165–172. [Google Scholar] [CrossRef]
- Banerjee, A. Ionic liquids for oral insulin delivery. Proc. Natl. Acad. Sci. USA 2018, 115, 7296–7301. [Google Scholar] [CrossRef] [Green Version]
- Li, D. Influence of Particle Geometry on Gastrointestinal Transit and Absorption following Oral Administration. ACS Appl. Mater. Interfaces 2017, 9, 42492–42502. [Google Scholar] [CrossRef] [PubMed]
- Popat, A.S.J.; Zhang, J.; Yang, J.; Zhang, H.; Meka, A.; Yu, C. Programmable drug release using bioresponsive mesoporous silica nanoparticles for site-specific oral drug delivery. Chem. Commun. (Camb) 2014, 50, 4. [Google Scholar] [CrossRef] [PubMed]
- Florek, J.; Caillard, R.; Kleitz, F. Evaluation of mesoporous silica nanoparticles for oral drug delivery-current status and perspective of MSNs drug carriers. Nanoscale 2017, 9, 15252–15277. [Google Scholar] [CrossRef] [PubMed]
- Fu, C. The absorption, distribution, excretion and toxicity of mesoporous silica nanoparticles in mice following different exposure routes. Biomaterials 2013, 34, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Meka, A.K. Enhanced Solubility, Permeability and Anticancer Activity of Vorinostat Using Tailored Mesoporous Silica Nanoparticles. Pharmaceutics 2018, 10, 283. [Google Scholar] [CrossRef] [PubMed]
- Popat, J.L.; Lu, G.Q.; Qiao, S. A pH-responsive drug delivery system based on chitosan coated mesoporous silica nanoparticles. J. Mater. Chem. B 2012, 22. [Google Scholar] [CrossRef]
- Zhang, Q. Functionalized mesoporous silica nanoparticles with mucoadhesive and sustained drug release properties for potential bladder cancer therapy. Langmuir 2014, 30, 6151–6161. [Google Scholar] [CrossRef] [PubMed]
- Guha, A. Ph responsive cylindrical MSN for oral delivery of insulin-design, fabrication and evaluation. Drug Deliv. 2016, 23, 3552–3561. [Google Scholar] [CrossRef] [PubMed]
- Andreani, T. Preparation and characterization of PEG-coated silica nanoparticles for oral insulin delivery. Int. J. Pharm. 2014, 473, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Meka, A.K. A Vesicle Supra-Assembly Approach to Synthesize Amine-Functionalized Hollow Dendritic Mesoporous Silica Nanospheres for Protein Delivery. Small 2016, 12, 5169–5177. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Biphasic Synthesis of Large-Pore and Well-Dispersed Benzene Bridged Mesoporous Organosilica Nanoparticles for Intracellular Protein Delivery. Small 2015, 11, 2743–2749. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Evaluation of biomimetically synthesized mesoporous silica nanoparticles as drug carriers: Structure, wettability, degradation, biocompatibility and brain distribution. Mater. Sci. Eng. C 2019, 94, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Chen, C. Sustained-release study on Exenatide loaded into mesoporous silica nanoparticles: In vitro characterization and in vivo evaluation. Daru 2017, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Tu, J. Mesoporous Silica Nanoparticles with Large Pores for the Encapsulation and Release of Proteins. ACS Appl. Mater. Interfaces 2016, 8, 32211–32219. [Google Scholar] [CrossRef] [PubMed]
- Abbaraju, P.L. Asymmetric mesoporous silica nanoparticles as potent and safe immunoadjuvants provoke high immune responses. Chem. Commun. (Camb) 2018, 54, 2020–2023. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Small-sized and large-pore dendritic mesoporous silica nanoparticles enhance antimicrobial enzyme delivery. J. Mater. Chem. B 2016, 4, 2646–2653. [Google Scholar] [CrossRef]
- Hoang, T.T.T. Functionalized mesoporous silica nanoparticles and biomedical applications. Mater. Sci. Eng. C 2019, 99, 631–656. [Google Scholar] [CrossRef]
- Xu, C. Core-Cone Structured Monodispersed Mesoporous Silica Nanoparticles with Ultra-large Cavity for Protein Delivery. Small 2015, 11, 5949–5955. [Google Scholar] [CrossRef]
- Du, X. Developing functionalized dendrimer-like silica nanoparticles with hierarchical pores as advanced delivery nanocarriers. Adv. Mater. 2013, 25, 5981–5985. [Google Scholar] [CrossRef]
- Araujo, F.; Sarmento, B. Towards the characterization of an in vitro triple co-culture intestine cell model for permeability studies. Int. J. Pharm. 2013, 458, 128–134. [Google Scholar] [CrossRef]
- Shen, D. Biphase stratification approach to three-dimensional dendritic biodegradable mesoporous silica nanospheres. Nano Lett. 2014, 14, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.S.; Lee, J.K. Tunable synthesis of hierarchical mesoporous silica nanoparticles with radial wrinkle structure. Langmuir 2012, 28, 12341–12347. [Google Scholar] [CrossRef] [PubMed]
- Castangia, I. Faceted phospholipid vesicles tailored for the delivery of Santolina insularis essential oil to the skin. Colloids Surf. B Biointerfaces 2015, 132, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchoucha, M. Size-Controlled Functionalized Mesoporous Silica Nanoparticles for Tunable Drug Release and Enhanced Anti-Tumoral Activity. Chem. Mater. 2016, 28, 4243–4258. [Google Scholar] [CrossRef]
- Yuan, L.Y. Introduction of bifunctional groups into mesoporous silica for enhancing uptake of thorium(IV) from aqueous solution. ACS Appl. Mater. Interfaces 2014, 6, 4786–4796. [Google Scholar] [CrossRef]
- Wang, Y. Charge-Reversal APTES-Modified Mesoporous Silica Nanoparticles with High Drug Loading and Release Controllability. ACS Appl. Mater. Interfaces 2016, 8, 17166–17175. [Google Scholar] [CrossRef]
- Arantes, T.M. Synthesis and optimization of colloidal silica nanoparticles and their functionalization with methacrylic acid. Colloids Surf. A Physicochem. Eng. Asp. 2012, 415, 209–217. [Google Scholar] [CrossRef]
- Maciel, G.R. Siloxane-Based Solid Networks, from Silicas to Silicones, in Solid-State NMR Spectroscopy of Inorganic Materials; American Chemical Society: Washington, WA, USA, 1998; pp. 326–356. [Google Scholar]
- Chen, Y. Synthesis of Hollow Mesoporous Silica Nanoparticles by Silica-Etching Chemistry for Biomedical Applications; Chinese Academy of Sciences: Beijing, China, 2016; pp. 31–46. [Google Scholar]
- Perez, G. Synthesis and characterization of epoxy encapsulating silica microcapsules and amine functionalized silica nanoparticles for development of an innovative self-healing concrete. Mater. Chem. Phys. 2015, 165, 39–48. [Google Scholar] [CrossRef]
- An, Y. Preparation and self-assembly of carboxylic acid-functionalized silica. J. Colloid. Interface Sci. 2007, 311, 507–513. [Google Scholar] [CrossRef]
- Acres, R.G. Molecular Structure of 3-Aminopropyltriethoxysilane Layers Formed on Silanol-Terminated Silicon Surfaces. J. Phys. Chem. C 2012, 116, 6289–6297. [Google Scholar] [CrossRef]
- Hanafi-Bojd, M.Y. Surface functionalized mesoporous silica nanoparticles as an effective carrier for epirubicin delivery to cancer cells. Eur. J. Pharm. Biopharm. 2015, 89, 248–258. [Google Scholar] [CrossRef]
- Kwon, D. Extra-Large Pore Mesoporous Silica Nanoparticles for Directing in Vivo M2 Macrophage Polarization by Delivering IL-4. Nano Lett. 2017, 17, 2747–2756. [Google Scholar] [CrossRef]
- Shi, Y. Fc-modified exenatide-loaded nanoparticles for oral delivery to improve hypoglycemic effects in mice. Sci. Rep. 2018, 8, 726. [Google Scholar] [CrossRef]
- Kaasalainen, M. Electrostatic interaction on loading of therapeutic peptide GLP-1 into porous silicon nanoparticles. Langmuir 2015, 31, 1722–1729. [Google Scholar] [CrossRef]
- Maddala, S.P. Large pore raspberry textured phosphonate@silica nanoparticles for protein immobilization. J. Mater. Chem. B 2014, 2, 903–914. [Google Scholar] [CrossRef]
- Smith, A.; Perelman, M.; Hinchcliffe, M. Chitosan. Hum. Vaccines Immunother. 2013, 10, 797–807. [Google Scholar] [CrossRef]
- Fan, B. Ph-Responsive Thiolated Chitosan nanoparticles for oral low-molecular weight heparin delivery: In vitro and in vivo evaluation. Drug Deliv. 2016, 23, 238–247. [Google Scholar] [CrossRef]
- Fonte, P. Polymer-based nanoparticles for oral insulin delivery: Revisited approaches. Biotechnol. Adv. 2015, 33, 1342–1354. [Google Scholar] [CrossRef]
- Che, E. Development of phosphonate-terminated magnetic mesoporous silica nanoparticles for pH-controlled release of doxorubicin and improved tumor accumulation. Asian J. Pharm. Sci. 2014, 9, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W. Auricularia auricular polysaccharide-low molecular weight chitosan polyelectrolyte complex nanoparticles: Preparation and characterization. Asian J. Pharm. Sci. 2016, 11, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.H. Ph Effects on solubility, zeta potential, and correlation between antibacterial activity and molecular weight of chitosan. Carbohydr. Polym. 2015, 134, 74–81. [Google Scholar] [CrossRef]
- Dening, T.J.; Taylor, L.S. Supersaturation Potential of Ordered Mesoporous Silica Delivery Systems. Part 1: Dissolution Performance and Drug Membrane Transport Rates. Mol. Pharm. 2018, 15, 3489–3501. [Google Scholar] [CrossRef]
- Puddu, V.; Perry, C.C. Peptide Adsorption on Silica Nanoparticles: Evidence of Hydrophobic Interactions. ACS Nano 2012, 6, 6356–6363. [Google Scholar] [CrossRef]
- Costa, R.R.; Alatorre-Meda, M.; Mano, J.F. Drug nano-reservoirs synthesized using layer-by-layer technologies. Biotechnol. Adv. 2015, 33, 1310–1326. [Google Scholar] [CrossRef]
- Gupta, V. Delivery of Exenatide and Insulin Using Mucoadhesive Intestinal Devices. Ann. Biomed. Eng. 2016, 44, 1993–2007. [Google Scholar] [CrossRef]
- Press, B.G.D. Permeability for Intestinal Absorption Caco-2 Assay and Related Issues. Curr. Drug Metab. 2008, 9, 8. [Google Scholar] [CrossRef]
- Shrestha, N. Chitosan-modified porous silicon microparticles for enhanced permeability of insulin across intestinal cell monolayers. Biomaterials 2014, 35, 7172–7179. [Google Scholar] [CrossRef]
- Lundquist, P.; Artursson, P. Oral absorption of peptides and nanoparticles across the human intestine: Opportunities, limitations and studies in human tissues. Adv. Drug Deliv. Rev. 2016, 106, 256–276. [Google Scholar] [CrossRef]
- Antunes, F. Establishment of a triple co-culture in vitro cell models to study intestinal absorption of peptide drugs. Eur. J. Pharm. Biopharm. 2013, 83, 427–435. [Google Scholar] [CrossRef]
- Chen, C.H. Mutlifunctional nanoparticles prepared from arginine-modified chitosan and thiolated fucoidan for oral delivery of hydrophobic and hydrophilic drugs. Carbohydr. Polym. 2018, 193, 163–172. [Google Scholar] [CrossRef]
- Banerjee, A. Role of nanoparticle size, shape and surface chemistry in oral drug delivery. J. Control. Release 2016, 238, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Shan, W. Enhanced Oral Delivery of Protein Drugs Using Zwitterion-Functionalized Nanoparticles to Overcome both the Diffusion and Absorption Barriers. ACS Appl. Mater. Interfaces 2016, 8, 25444–25453. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dynamic Light Scattering | Nitrogen Sorption Analysis | ||||
|---|---|---|---|---|---|
| Particles | PDI (AU ± SD) | Particle Size (Mean Diameter nm ± SD) | ζ-Potential (mV) | BET Surface Area (m2/g) | Pore Volume (cm3/g) |
| DSNPs | 0.34 ± 0.05 | 226 ± 9 | −14.3 | 486 | 1.14 |
| PDSNPs | 0.26 ± 0.03 | 254 ± 7 | −30.5 | 305 | 0.88 |
| ADSNPs | 0.45 ± 0.04 | 742 ± 13 | 11.5 | 191 | 0.60 |
| SDSNPs | 0.15 ± 0.01 | 248 ± 23 | −23.2 | 83 | 0.39 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abeer, M.M.; Meka, A.K.; Pujara, N.; Kumeria, T.; Strounina, E.; Nunes, R.; Costa, A.; Sarmento, B.; Hasnain, S.Z.; Ross, B.P.; et al. Rationally Designed Dendritic Silica Nanoparticles for Oral Delivery of Exenatide. Pharmaceutics 2019, 11, 418. https://doi.org/10.3390/pharmaceutics11080418
Abeer MM, Meka AK, Pujara N, Kumeria T, Strounina E, Nunes R, Costa A, Sarmento B, Hasnain SZ, Ross BP, et al. Rationally Designed Dendritic Silica Nanoparticles for Oral Delivery of Exenatide. Pharmaceutics. 2019; 11(8):418. https://doi.org/10.3390/pharmaceutics11080418
Chicago/Turabian StyleAbeer, Muhammad Mustafa, Anand Kumar Meka, Naisarg Pujara, Tushar Kumeria, Ekaterina Strounina, Rute Nunes, Ana Costa, Bruno Sarmento, Sumaira Z. Hasnain, Benjamin P. Ross, and et al. 2019. "Rationally Designed Dendritic Silica Nanoparticles for Oral Delivery of Exenatide" Pharmaceutics 11, no. 8: 418. https://doi.org/10.3390/pharmaceutics11080418