Acid-Treated Water-Soluble Chitosan Suitable for Microneedle-Assisted Intracutaneous Drug Delivery

Abstract
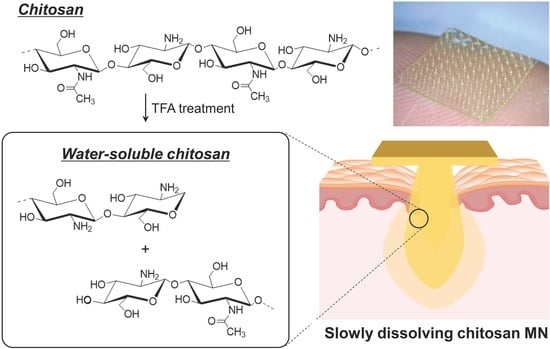
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of the WSCS by TFA Treatment
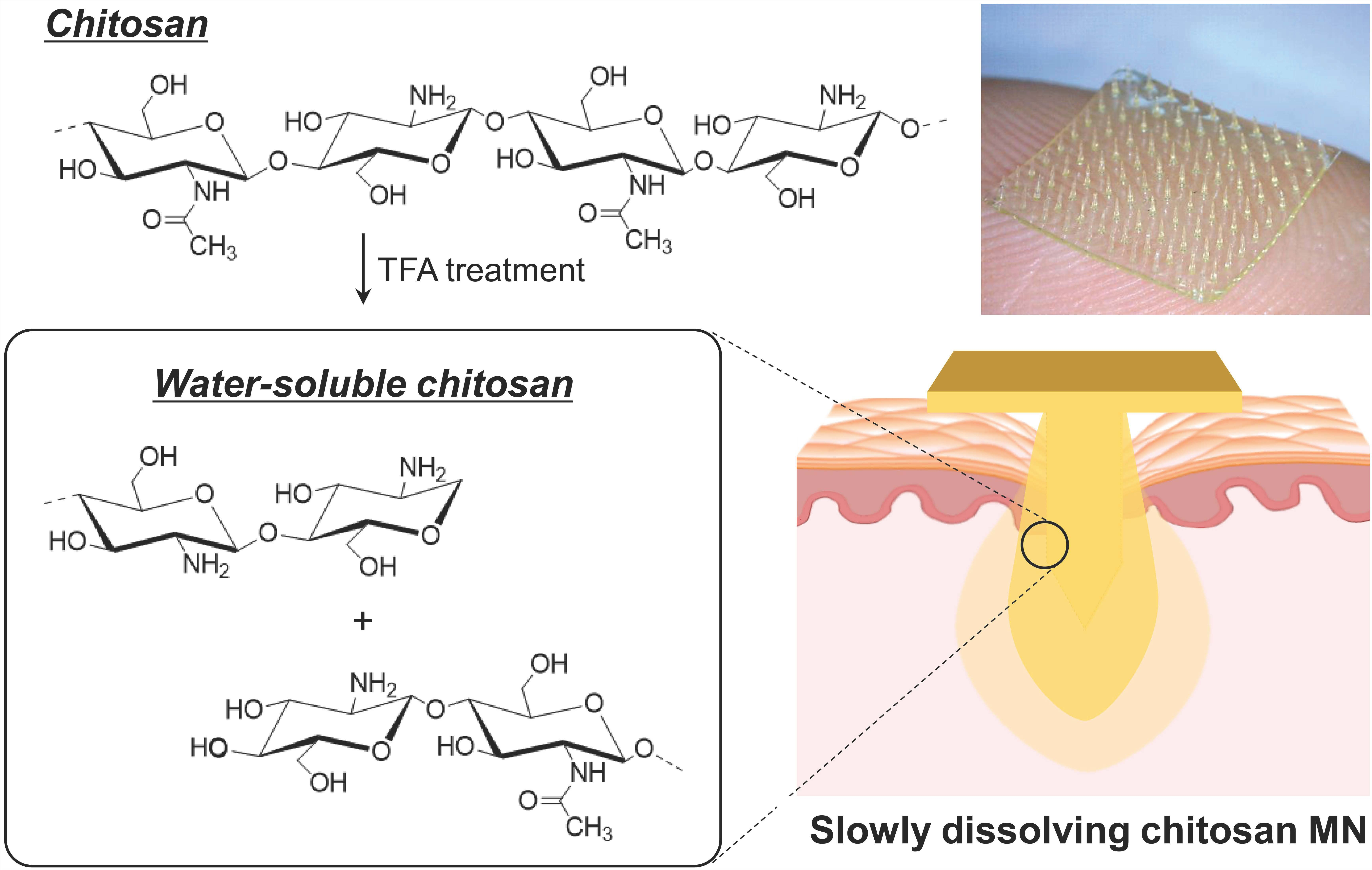
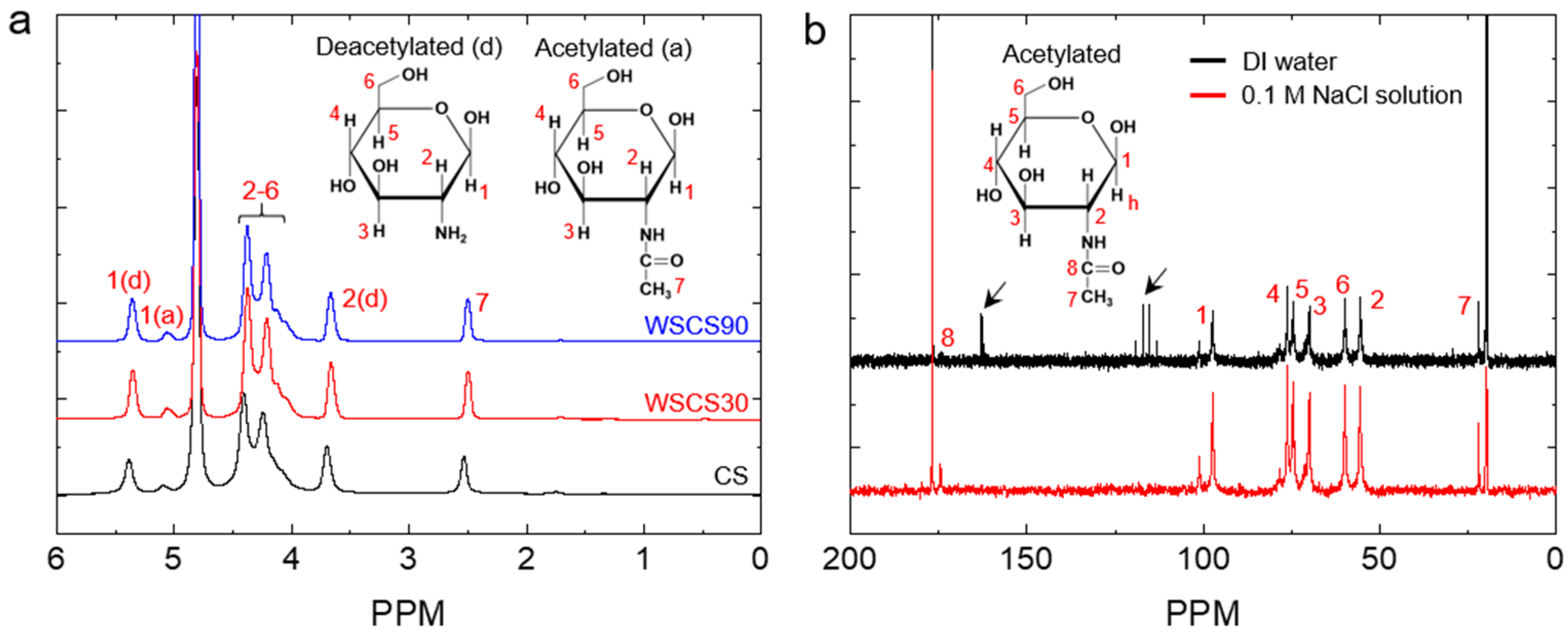
2.3. Characterization of the WSCS
2.4. Intrinsic Viscosity and Viscosity-Average MW
2.5. Solubility Test
2.6. Zeta Potential Test
2.7. Fabrication for the Array of WSCS MNs with Bullet-Shaped Geometry
2.8. Mechanical Tests of the WSCS MNs
2.9. In Vitro Dissolution Tests of the WSCS MNs
2.10. Ex Vivo Drug Release from the WSCS MN Patches
3. Results and Discussion
3.1. Preparation and Characterization of the WSCS
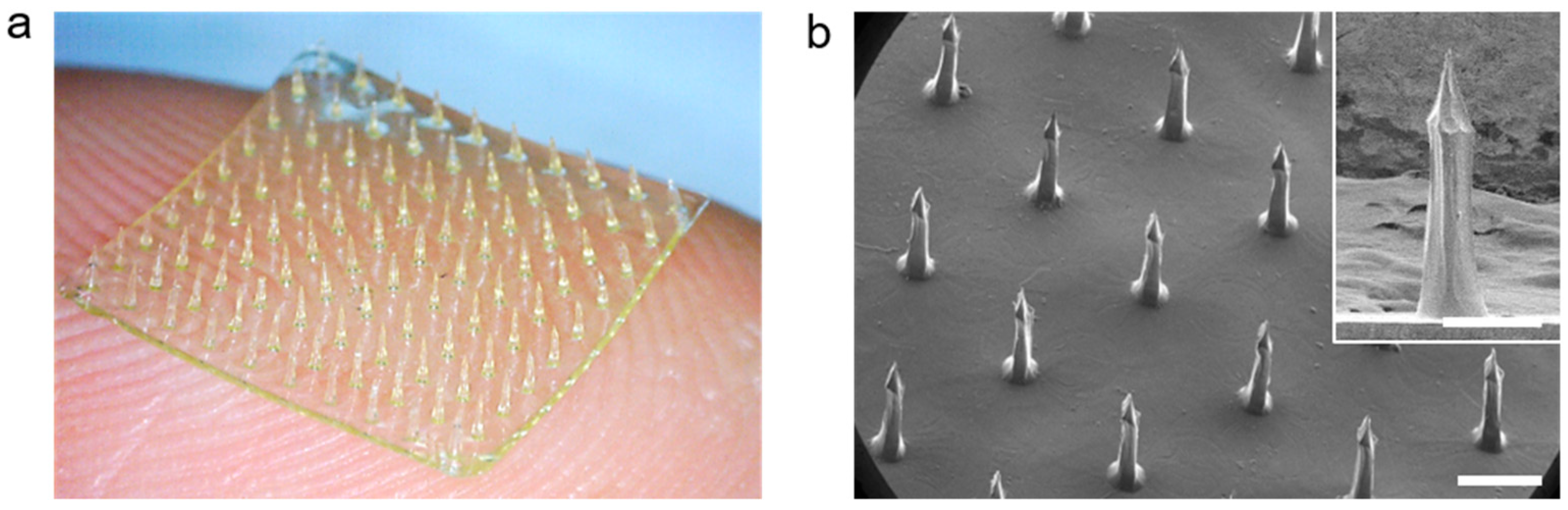
3.2. Preparation of the CS MN Array Using WSCS30 Solutions
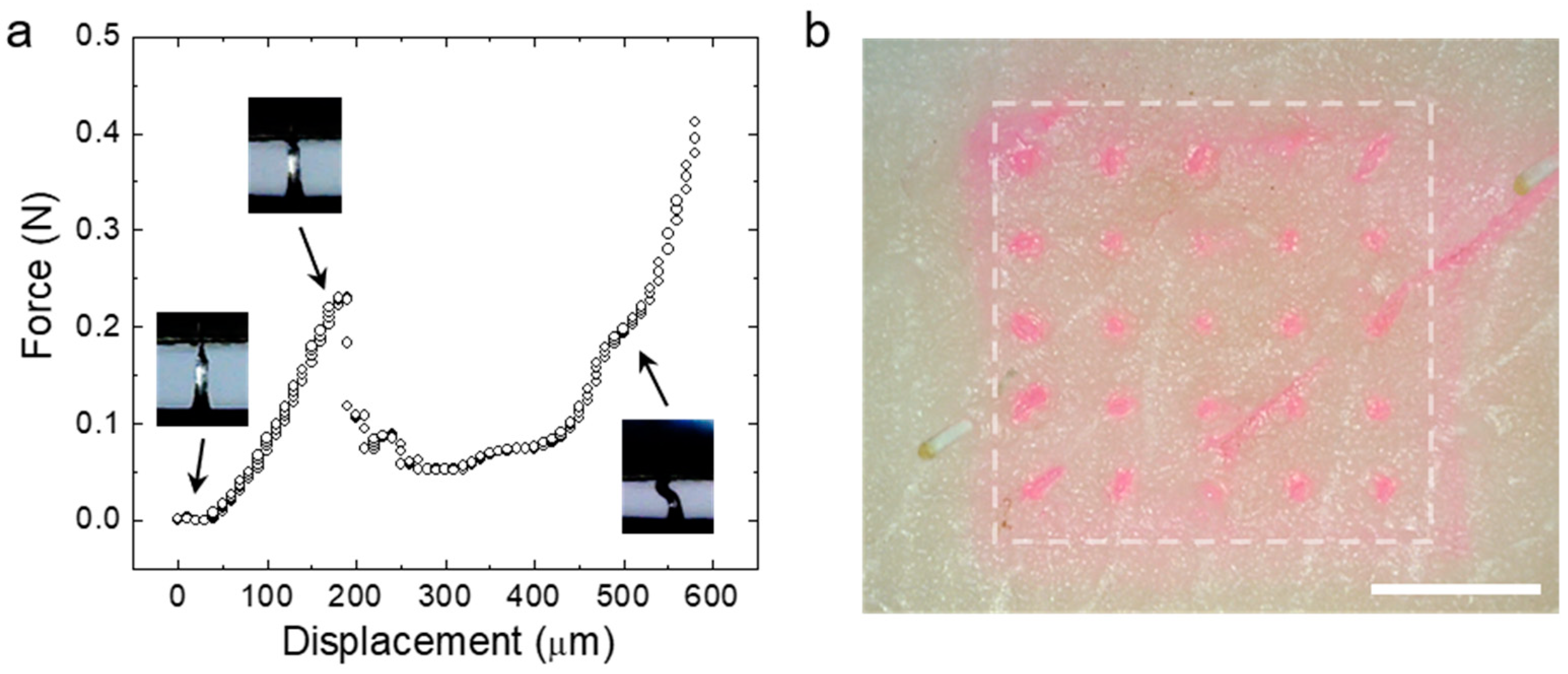
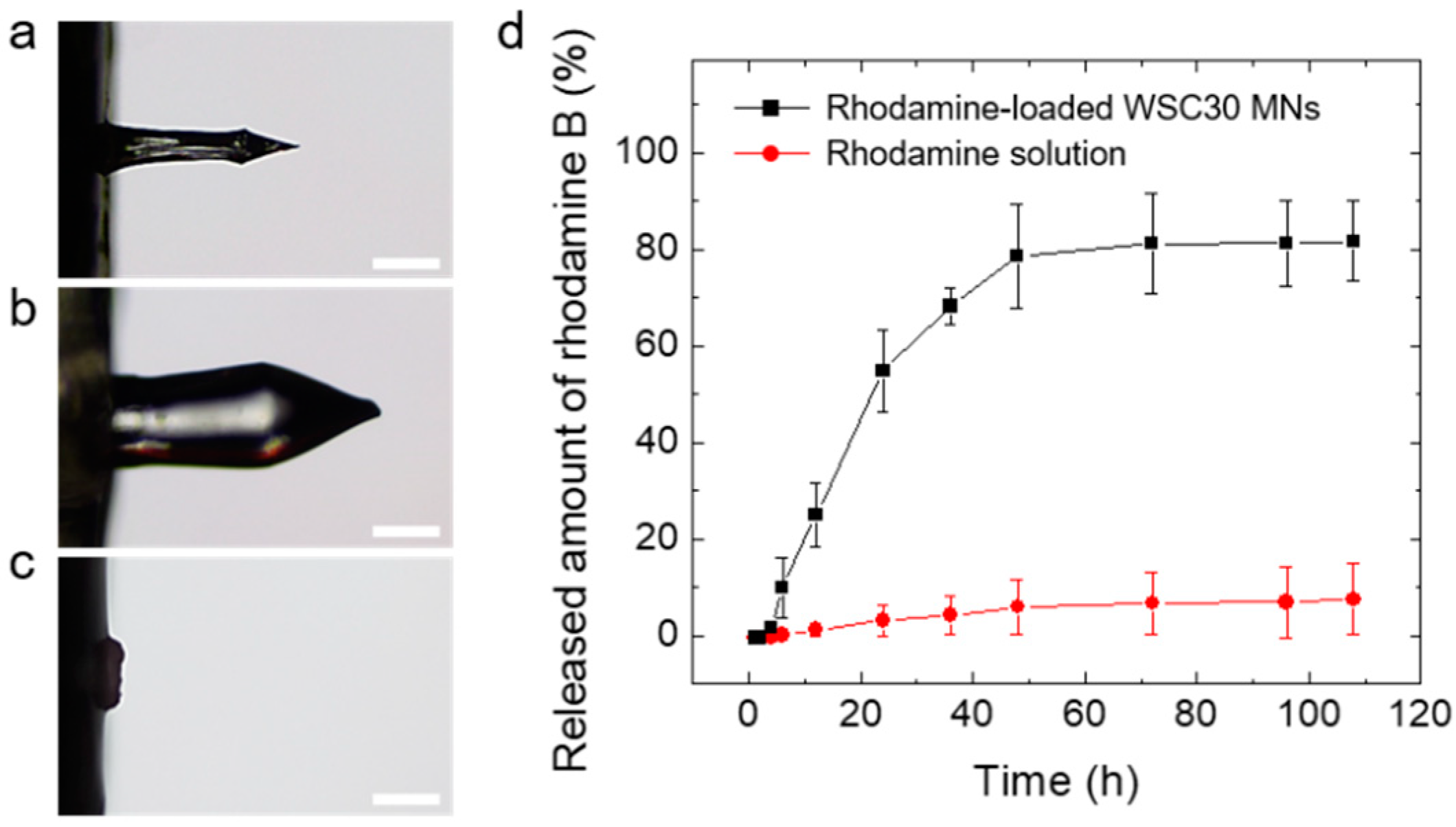
3.3. Fracture and Skin Insertion Tests Using the CS MNs
3.4. Water-Responsiveness of the CS MNs
3.5. In Vitro Drug Release Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Prausnitz, M.R.; Langer, R. Transdermal drug delivery. Nat. Biotechnol. 2008, 26, 1261. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.H. Transdermal drug delivery. In Drug Delivery; Springer: Berlin/Heidelberg, Germany, 2010; pp. 399–410. [Google Scholar]
- Alkilani, A.; McCrudden, M.T.; Donnelly, R. Transdermal drug delivery: Innovative pharmaceutical developments based on disruption of the barrier properties of the stratum corneum. Pharmaceutics 2015, 7, 438–470. [Google Scholar] [CrossRef]
- Schoellhammer, C.M.; Blankschtein, D.; Langer, R. Skin permeabilization for transdermal drug delivery: Recent advances and future prospects. Expert Opin. Drug Deliv. 2014, 11, 393–407. [Google Scholar] [CrossRef] [PubMed]
- N’Da, D. Prodrug strategies for enhancing the percutaneous absorption of drugs. Molecules 2014, 19, 20780–20807. [Google Scholar] [CrossRef]
- Todo, H. Transdermal permeation of drugs in various animal species. Pharmaceutics 2017, 9, 33. [Google Scholar] [CrossRef]
- Zhang, Z.; Michniak-Kohn, B.B. Tissue engineered human skin equivalents. Pharmaceutics 2012, 4, 26–41. [Google Scholar] [CrossRef]
- Tuan-Mahmood, T.-M.; McCrudden, M.T.; Torrisi, B.M.; McAlister, E.; Garland, M.J.; Singh, T.R.R.; Donnelly, R.F. Microneedles for intradermal and transdermal drug delivery. Eur. J. Pharm. Sci. 2013, 50, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.; DeLouise, L. Nanoparticle-enabled transdermal drug delivery systems for enhanced dose control and tissue targeting. Molecules 2016, 21, 1719. [Google Scholar] [CrossRef] [PubMed]
- Calatayud-Pascual, M.; Sebastian-Morelló, M.; Balaguer-Fernández, C.; Delgado-Charro, M.; López-Castellano, A.; Merino, V. Influence of Chemical Enhancers and Iontophoresis on the In Vitro Transdermal Permeation of Propranolol: Evaluation by Dermatopharmacokinetics. Pharmaceutics 2018, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Donalisio, M.; Leone, F.; Civra, A.; Spagnolo, R.; Ozer, O.; Lembo, D.; Cavalli, R. Acyclovir-Loaded Chitosan Nanospheres from Nano-Emulsion Templating for the Topical Treatment of Herpesviruses Infections. Pharmaceutics 2018, 10, 46. [Google Scholar] [CrossRef]
- Nguyen, H.; Banga, A. Electrically and Ultrasonically Enhanced Transdermal Delivery of Methotrexate. Pharmaceutics 2018, 10, 117. [Google Scholar] [CrossRef]
- Escobar-Chávez, J.J.; Rodríguez-Cruz, I.M.; Domínguez-Delgado, C.L.; Díaz-Torres, R.; Revilla-Vázquez, A.L.; Aléncaster, N.C. Nanocarrier systems for transdermal drug delivery. In Recent Advances in Novel Drug Carrier Systems; IntechOpen: London, UK, 2012; pp. 201–240. [Google Scholar]
- Nastiti, C.; Ponto, T.; Abd, E.; Grice, J.; Benson, H.; Roberts, M. Topical nano and microemulsions for skin delivery. Pharmaceutics 2017, 9, 37. [Google Scholar] [CrossRef]
- Abd, E.; Benson, H.; Roberts, M.; Grice, J. Minoxidil skin delivery from nanoemulsion formulations containing eucalyptol or oleic acid: Enhanced diffusivity and follicular targeting. Pharmaceutics 2018, 10, 19. [Google Scholar] [CrossRef]
- Yang, Y.; Kalluri, H.; Banga, A.K. Effects of chemical and physical enhancement techniques on transdermal delivery of cyanocobalamin (vitamin B12) in vitro. Pharmaceutics 2011, 3, 474–484. [Google Scholar] [CrossRef]
- Uchechi, O.; Ogbonna, J.D.; Attama, A.A. Nanoparticles for dermal and transdermal drug delivery. In Application of Nanotechnology in Drug Delivery; IntechOpen: London, UK, 2014; pp. 193–234. [Google Scholar]
- Goyal, R.; Macri, L.K.; Kaplan, H.M.; Kohn, J. Nanoparticles and nanofibers for topical drug delivery. J. Control. Release 2016, 240, 77–92. [Google Scholar] [CrossRef]
- Donnelly, R.F.; Singh, T.R.R.; Garland, M.J.; Migalska, K.; Majithiya, R.; McCrudden, C.M.; Kole, P.L.; Mahmood, T.M.T.; McCarthy, H.O.; Woolfson, A.D. Hydrogel-forming microneedle arrays for enhanced transdermal drug delivery. Adv. Funct. Mater. 2012, 22, 4879–4890. [Google Scholar] [CrossRef]
- Davidson, A.; Al-Qallaf, B.; Das, D.B. Transdermal drug delivery by coated microneedles: Geometry effects on effective skin thickness and drug permeability. Chem. Eng. Res. Des. 2008, 86, 1196–1206. [Google Scholar] [CrossRef]
- Cui, Y.; Mo, Y.; Zhang, Q.; Tian, W.; Xue, Y.; Bai, J.; Du, S. Microneedle-Assisted Percutaneous Delivery of Paeoniflorin-Loaded Ethosomes. Molecules 2018, 23, 3371. [Google Scholar] [CrossRef]
- Lee, K.; Park, S.H.; Lee, J.; Ryu, S.; Joo, C.; Ryu, W. Three-Step Thermal Drawing for Rapid Prototyping of Highly Customizable Microneedles for Vascular Tissue Insertion. Pharmaceutics 2019, 11, 100. [Google Scholar] [CrossRef]
- Chen, B.; Wei, J.; Tay, F.E.; Wong, Y.T.; Iliescu, C. Silicon microneedle array with biodegradable tips for transdermal drug delivery. Microsyst. Technol. 2008, 14, 1015–1019. [Google Scholar] [CrossRef]
- Mishra, R.; Maiti, T.K.; Bhattacharyya, T.K. Development of SU-8 hollow microneedles on a silicon substrate with microfluidic interconnects for transdermal drug delivery. J. Micromech. Microeng. 2018, 28, 105017. [Google Scholar] [CrossRef]
- Donnelly, R.F.; Singh, T.R.R.; Woolfson, A.D. Microneedle-based drug delivery systems: Microfabrication, drug delivery, and safety. Drug Deliv. 2010, 17, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Boks, M.A.; Unger, W.W.; Engels, S.; Ambrosini, M.; van Kooyk, Y.; Luttge, R. Controlled release of a model vaccine by nanoporous ceramic microneedle arrays. Int. J. Pharm. 2015, 491, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Feng, Y.; Zhang, L.; Chen, N.; Yuan, W.; Jin, T. A scalable fabrication process of polymer microneedles. Int. J. Pharm. 2012, 7, 1415. [Google Scholar]
- Jin, C.Y.; Han, M.H.; Lee, S.S.; Choi, Y.H. Mass producible and biocompatible microneedle patch and functional verification of its usefulness for transdermal drug delivery. Biomed. Microdevices 2009, 11, 1195. [Google Scholar] [CrossRef]
- Tsioris, K.; Raja, W.K.; Pritchard, E.M.; Panilaitis, B.; Kaplan, D.L.; Omenetto, F.G. Fabrication of silk microneedles for controlled-release drug delivery. Adv. Funct. Mater. 2012, 22, 330–335. [Google Scholar] [CrossRef]
- Wang, M.; Hu, L.; Xu, C. Recent advances in the design of polymeric microneedles for transdermal drug delivery and biosensing. Lab Chip 2017, 17, 1373–1387. [Google Scholar] [CrossRef] [PubMed]
- Indermun, S.; Luttge, R.; Choonara, Y.E.; Kumar, P.; Du Toit, L.C.; Modi, G.; Pillay, V. Current advances in the fabrication of microneedles for transdermal delivery. J. Control. Release 2014, 185, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Luangveera, W.; Jiruedee, S.; Mama, W.; Chiaranairungroj, M.; Pimpin, A.; Palaga, T.; Srituravanich, W. Fabrication and characterization of novel microneedles made of a polystyrene solution. J. Mech. Behav. Biomed. Mater. 2015, 50, 77–81. [Google Scholar] [CrossRef]
- Oh, J.-H.; Park, H.-H.; Do, K.-Y.; Han, M.; Hyun, D.-H.; Kim, C.-G.; Kim, C.-H.; Lee, S.S.; Hwang, S.-J.; Shin, S.-C. Influence of the delivery systems using a microneedle array on the permeation of a hydrophilic molecule, calcein. Eur. J. Pharm. Biopharm. 2008, 69, 1040–1045. [Google Scholar] [CrossRef]
- Li, W.; Terry, R.N.; Tang, J.; Feng, M.R.; Schwendeman, S.P.; Prausnitz, M.R. Rapidly separable microneedle patch for the sustained release of a contraceptive. Nat. Biomed. Eng. 2019, 3, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Li, W.; Zhou, X.; Yang, F.; Qian, Z. Microneedles-based transdermal drug delivery systems: A review. J. Biomed. Nanotechnol. 2017, 13, 1581–1597. [Google Scholar] [CrossRef]
- Naito, C.; Katsumi, H.; Suzuki, T.; Quan, Y.-s.; Kamiyama, F.; Sakane, T.; Yamamoto, A. Self-Dissolving Microneedle Arrays for Transdermal Absorption Enhancement of Human Parathyroid Hormone (1–34). Pharmaceutics 2018, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Migliore, A.; Granata, M. Intra-articular use of hyaluronic acid in the treatment of osteoarthritis. Clin. Interv. Aging 2008, 3, 365. [Google Scholar] [CrossRef] [PubMed]
- An, S.-M.; Seong, K.-Y.; Yim, S.-G.; Hwang, Y.J.; Bae, S.H.; Yang, S.Y.; An, B.-S. Intracutaneous delivery of gelatins induces lipolysis and suppresses lipogenesis of adipocytes. Acta Biomater. 2018, 67, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Larraneta, E.; Lutton, R.E.; Brady, A.J.; Vicente-Pérez, E.M.; Woolfson, A.D.; Thakur, R.R.S.; Donnelly, R.F. Microwave-Assisted Preparation of Hydrogel-Forming Microneedle Arrays for Transdermal Drug Delivery Applications. Macromol. Mater. Eng. 2015, 300, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-C.; Huang, S.-F.; Lai, K.-Y.; Ling, M.-H. Fully embeddable chitosan microneedles as a sustained release depot for intradermal vaccination. Biomaterials 2013, 34, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Pillai, C.; Paul, W.; Sharma, C.P. Chitin and chitosan polymers: Chemistry, solubility and fiber formation. Prog. Polym. Sci. 2009, 34, 641–678. [Google Scholar] [CrossRef]
- Bellich, B.; D’Agostino, I.; Semeraro, S.; Gamini, A.; Cesàro, A. “The Good, the Bad and the Ugly” of Chitosans. Mar. Drugs 2016, 14, 99. [Google Scholar] [CrossRef]
- Baldrick, P. The safety of chitosan as a pharmaceutical excipient. Regul. Toxicol. Pharmacol. 2010, 56, 290–299. [Google Scholar] [CrossRef]
- Vårum, K.; Ottøy, M.; Smidsrød, O. Acid hydrolysis of chitosans. Carbohydr. Polym. 2001, 46, 89–98. [Google Scholar] [CrossRef]
- Tian, M.; Tan, H.; Li, H.; You, C. Molecular weight dependence of structure and properties of chitosan oligomers. RSC Adv. 2015, 5, 69445–69452. [Google Scholar] [CrossRef]
- Chang, S.-H.; Lin, H.-T.V.; Wu, G.-J.; Tsai, G.J. pH Effects on solubility, zeta potential, and correlation between antibacterial activity and molecular weight of chitosan. Carbohydr. Polym. 2015, 134, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, J.C.; Van Cutsem, P. Preparation of chitooligosaccharides with degree of polymerization higher than 6 by acid or enzymatic degradation of chitosan. Biochem. Eng. J. 2005, 25, 165–172. [Google Scholar] [CrossRef]
- Qin, C.; Du, Y.; Xiao, L. Effect of hydrogen peroxide treatment on the molecular weight and structure of chitosan. Polym. Degrad. Stab. 2002, 76, 211–218. [Google Scholar] [CrossRef]
- Hasegawa, M.; Isogai, A.; Onabe, F.; Usuda, M. Dissolving states of cellulose and chitosan in trifluoroacetic acid. J. Appl. Polym. Sci. 1992, 45, 1857–1863. [Google Scholar] [CrossRef]
- Lin, H.; Wang, H.; Xue, C.; Ye, M. Preparation of chitosan oligomers by immobilized papain. Enzyme Microb. Technol. 2002, 31, 588–592. [Google Scholar] [CrossRef]
- Kasaai, M.R.; Arul, J.; Charlet, G. Fragmentation of chitosan by acids. Sci. World J. 2013, 2013, 508540. [Google Scholar] [CrossRef]
- Yan, X.; Evenocheck, H.M. Chitosan analysis using acid hydrolysis and HPLC/UV. Carbohydr. Polym. 2012, 87, 1774–1778. [Google Scholar] [CrossRef]
- Kasaai, M.R. Determination of the degree of N-acetylation for chitin and chitosan by various NMR spectroscopy techniques: A review. Carbohydr. Polym. 2010, 79, 801–810. [Google Scholar] [CrossRef]
- Singh, A.; Benjakul, S.; Prodpran, T. Ultrasound-Assisted Extraction of Chitosan from Squid Pen: Molecular Characterization and Fat Binding Capacity. J. Food Sci. 2019, 84, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Kasaai, M.R. Calculation of Mark-Houwink-Sakurada (MHS) equation viscometric constants for chitosan in any solvent–temperature system using experimental reported viscometric constants data. Carbohydr. Polym. 2007, 68, 477–488. [Google Scholar] [CrossRef]
- Seong, K.-Y.; Seo, M.-S.; Hwang, D.Y.; O’Cearbhaill, E.D.; Sreenan, S.; Karp, J.M.; Yang, S.Y. A self-adherent, bullet-shaped microneedle patch for controlled transdermal delivery of insulin. J. Control. Release 2017, 265, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Gaussier, H.; Morency, H.; Lavoie, M.C.; Subirade, M. Replacement of trifluoroacetic acid with HCl in the hydrophobic purification steps of pediocin PA-1: A structural effect. Appl. Environ. Microbiol. 2002, 68, 4803–4808. [Google Scholar] [CrossRef] [PubMed]
- Maderuelo, C.; Zarzuelo, A.; Lanao, J.M. Critical factors in the release of drugs from sustained release hydrophilic matrices. J.Control. Release 2011, 154, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Learoyd, T.P.; Burrows, J.L.; French, E.; Seville, P.C. Chitosan-based spray-dried respirable powders for sustained delivery of terbutaline sulfate. Eur. J. Pharm. Biopharm. 2008, 68, 224–234. [Google Scholar] [CrossRef]
- Choi, C.K.; Lee, K.J.; Youn, Y.N.; Jang, E.H.; Kim, W.; Min, B.-K.; Ryu, W. Spatially discrete thermal drawing of biodegradable microneedles for vascular drug delivery. Eur. J. Pharm. Biopharm. 2013, 83, 224–233. [Google Scholar] [CrossRef]
- Hennink, W.E.; van Nostrum, C.F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 223–236. [Google Scholar] [CrossRef]
- Ahmadi, F.; Oveisi, Z.; Samani, S.M.; Amoozgar, Z. Chitosan based hydrogels: Characteristics and pharmaceutical applications. Res. Pharm. Sci. 2015, 10, 1. [Google Scholar]
- Larraneta, E.; Lutton, R.E.; Woolfson, A.D.; Donnelly, R.F. Microneedle arrays as transdermal and intradermal drug delivery systems: Materials science, manufacture and commercial development. Mater. Sci. Eng. R Rep. 2016, 104, 1–32. [Google Scholar] [CrossRef]
- Yang, S.Y.; O’Cearbhaill, E.D.; Sisk, G.C.; Park, K.M.; Cho, W.K.; Villiger, M.; Bouma, B.E.; Pomahac, B.; Karp, J.M. A bio-inspired swellable microneedle adhesive for mechanical interlocking with tissue. Nat. Commun. 2013, 4, 1702. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.G.; Larrañeta, E.; Donnelly, R.F.; McGoldrick, N.; Migalska, K.; McCrudden, M.T.; Irwin, N.J.; Donnelly, L.; McCoy, C.P. Hydrogel-forming microneedle arrays made from light-responsive materials for on-demand transdermal drug delivery. Mol. Pharm. 2016, 13, 907–914. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | DDA (%) |
|---|---|
| CS | 78 |
| WSCS30 | 80 |
| WSCS90 | 81 |
| Samples | Intrinsic Viscosity (dL/g) | Mv (kDa)a |
|---|---|---|
| CS | 5.21 | 51.52 |
| WSCS30 | 1.92 | 14.51 |
| WSCS90 | 0.96 | 6.05 |
| Samples | Zeta Potential (mV) | |||
|---|---|---|---|---|
| pH 5.0 | pH 6.0 | pH 7.0 | pH 8.0 | |
| CS | 22.70 ± 1.13 | 14.33 ± 0.65 | 4.52 ± 0.15 | −1.12 ± 0.27 |
| WSCS30 | 29.95 ± 0.49 | 20.8 ± 0.61 | 6.89 ± 0.04 | −0.21 ± 0.01 |
| WSCS90 | 27.30 ± 1.87 | 16.43 ± 0.55 | 5.36 ± 0.34 | −0.24 ± 0.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekharan, A.; Hwang, Y.J.; Seong, K.-Y.; Park, S.; Kim, S.; Yang, S.Y. Acid-Treated Water-Soluble Chitosan Suitable for Microneedle-Assisted Intracutaneous Drug Delivery. Pharmaceutics 2019, 11, 209. https://doi.org/10.3390/pharmaceutics11050209
Chandrasekharan A, Hwang YJ, Seong K-Y, Park S, Kim S, Yang SY. Acid-Treated Water-Soluble Chitosan Suitable for Microneedle-Assisted Intracutaneous Drug Delivery. Pharmaceutics. 2019; 11(5):209. https://doi.org/10.3390/pharmaceutics11050209
Chicago/Turabian StyleChandrasekharan, Ajeesh, Young Jun Hwang, Keum-Yong Seong, Samdae Park, Sodam Kim, and Seung Yun Yang. 2019. "Acid-Treated Water-Soluble Chitosan Suitable for Microneedle-Assisted Intracutaneous Drug Delivery" Pharmaceutics 11, no. 5: 209. https://doi.org/10.3390/pharmaceutics11050209