Micelle-Forming Block Copolymers Tailored for Inhibition of P-gp-Mediated Multidrug Resistance: Structure to Activity Relationship

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis of Monomers and Initiator
2.3. Synthesis of Thiazolidine-2-Thione Functional CTA 2-Cyano-5-oxo-5-(2-thioxo-1,3-thiazolidin-3-yl)pentan-2-yl Ethyl Carbonotrithioate (CTA-TT)
2.4. Synthesis of Hydrophilic Polymer Blocks A1 and A2 Based on PHPMA Copolymers
2.5. Purification of Hydrophobic Polymer Block B Based on PPO before the Reaction
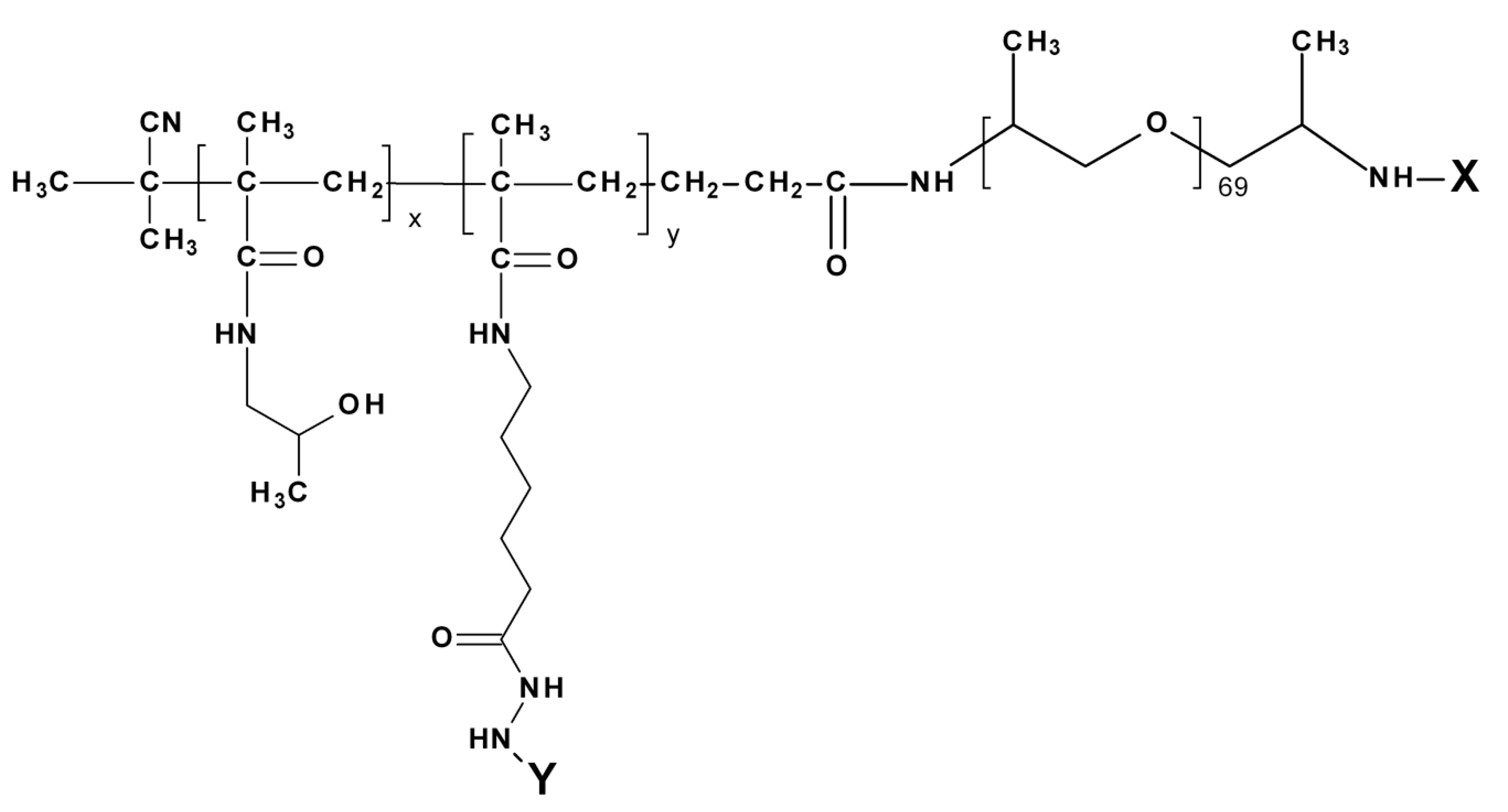
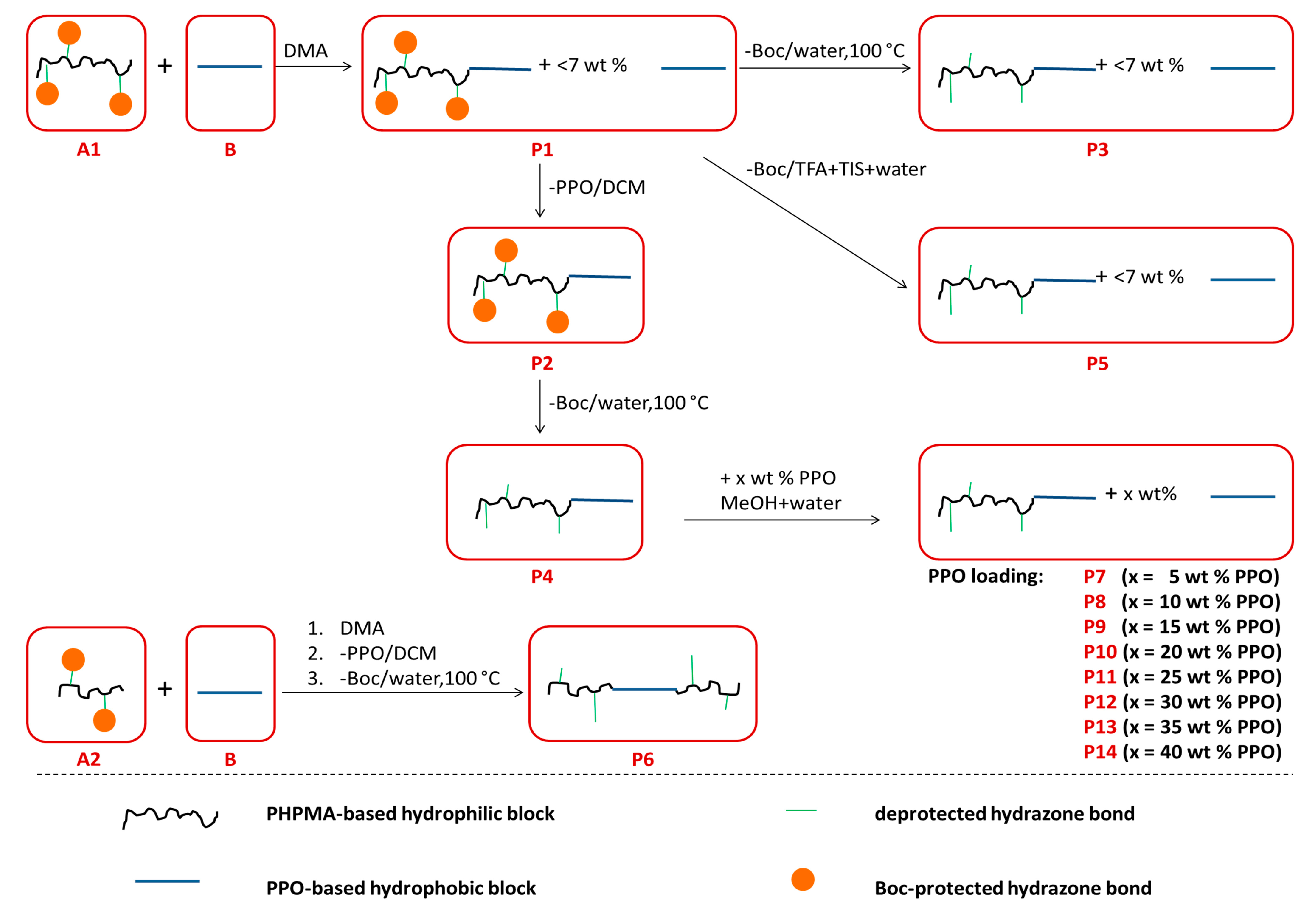
2.6. Synthesis of Polymer Carriers: Diblock and Triblock Micellar Copolymers
2.6.1. Boc-Protected Diblock Copolymer PHPMA-b-PPO P1 and P2
2.6.2. Diblock Copolymers P3 and P4, PHPMA-b-PPO, with Deprotected Hydrazide Groups in Distilled Water at Elevated Temperature and Pressure
2.6.3. Diblock Copolymer P5, PHPMA-b-PPO, with a Deprotected Hydrazide Group by the Mixture of TFA/TIS/Water
2.6.4. Triblock Copolymer P6, PHPMA-b-PPO-b-PHPMA
2.7. Loading of Free PPO into the Diblock Polymer Micelles
2.8. Time-Dependent Micellar Stability
2.9. Critical Micellar Concentration
2.10. Physico-Chemical Characterisation
2.11. Cell Lines
2.12. Calcein Efflux Assay
3. Results and Discussion
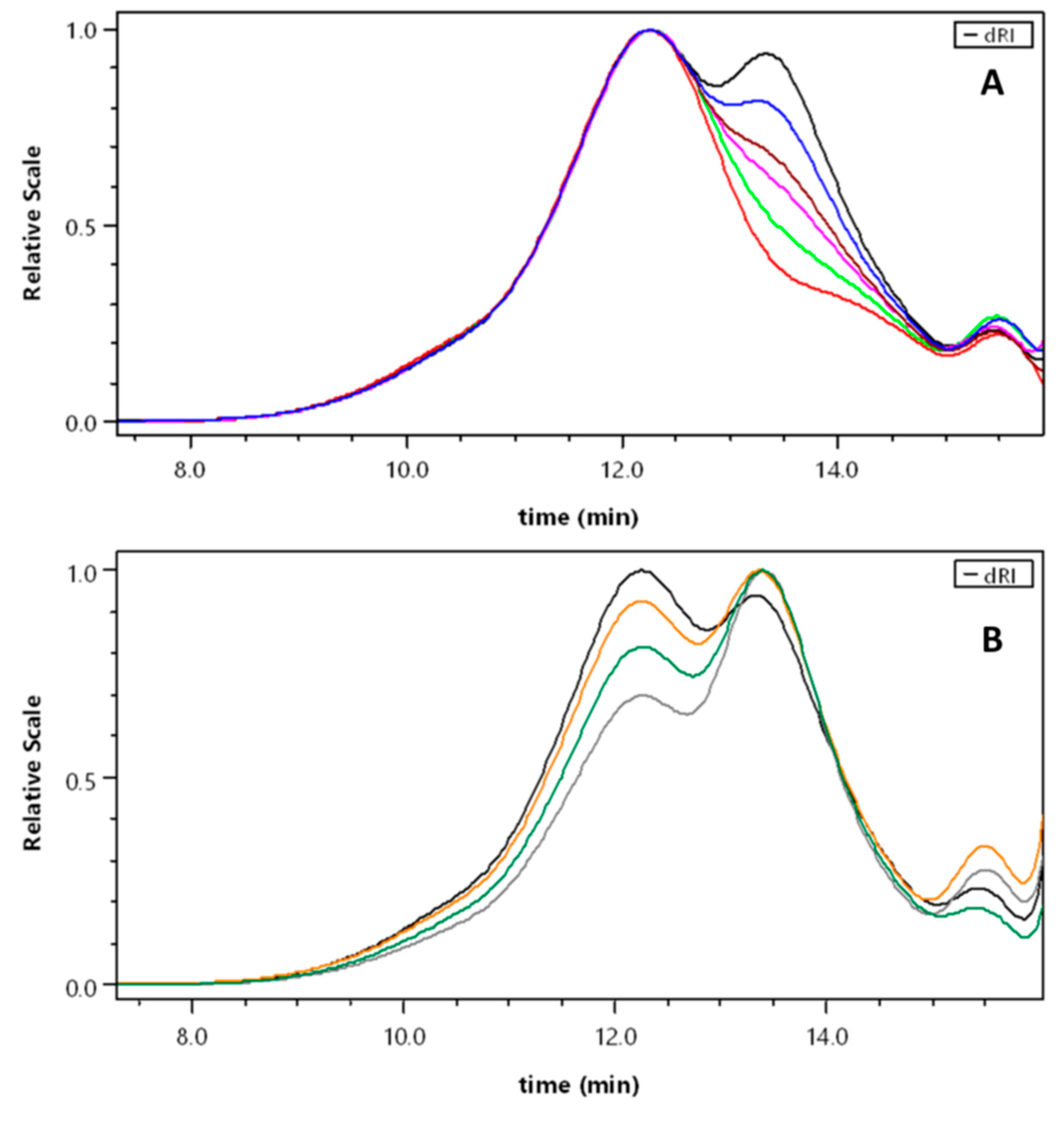
3.1. Synthesis of Hydrophilic Blocks A1, A2 and Unloaded Polymers P1–P6
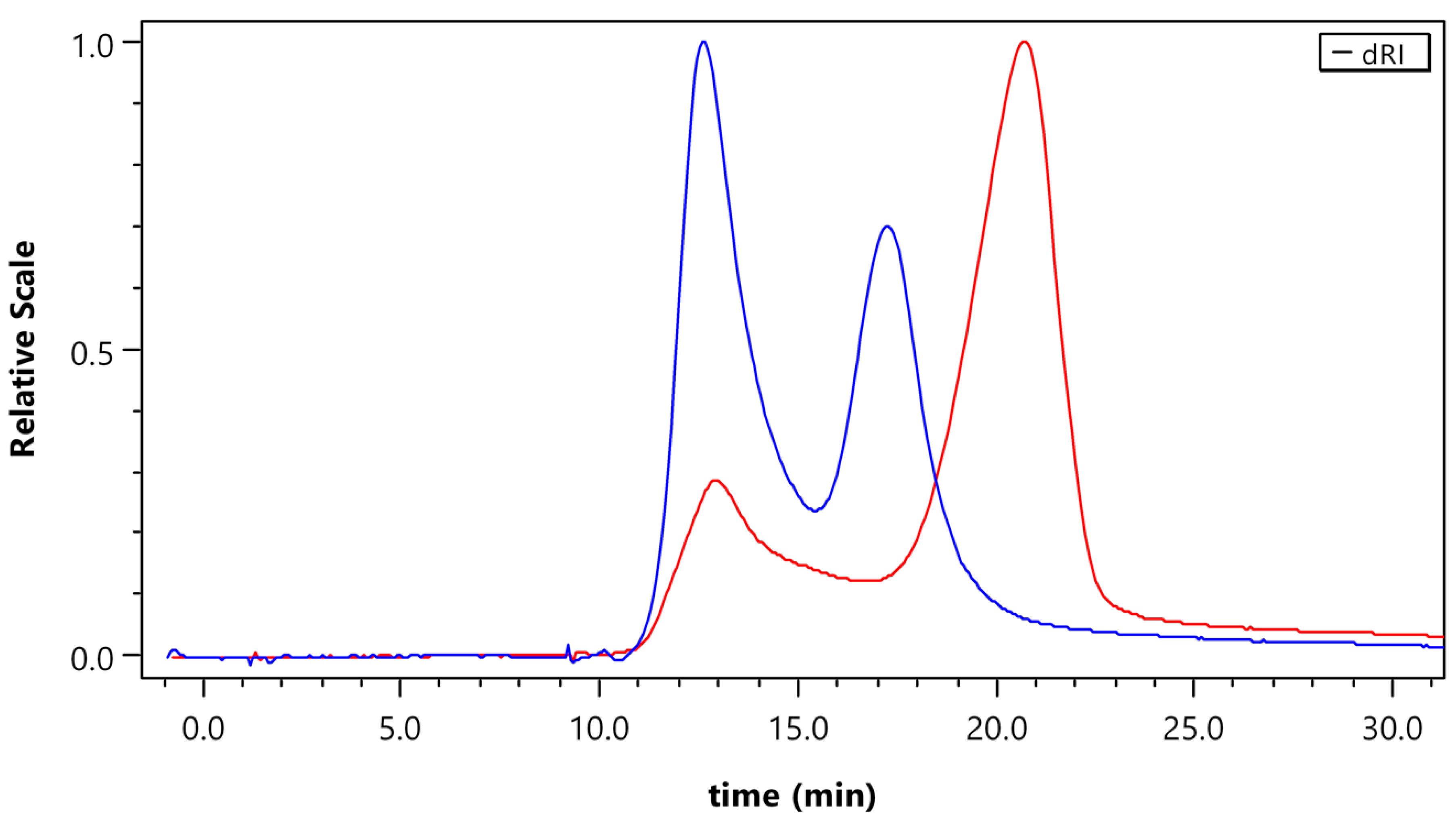
3.2. Size of the Particles (Dh) and Long-Term Stability of Non-Loaded Polymers P1–P6
3.3. CMC of Non-Loaded Polymers P2–P6
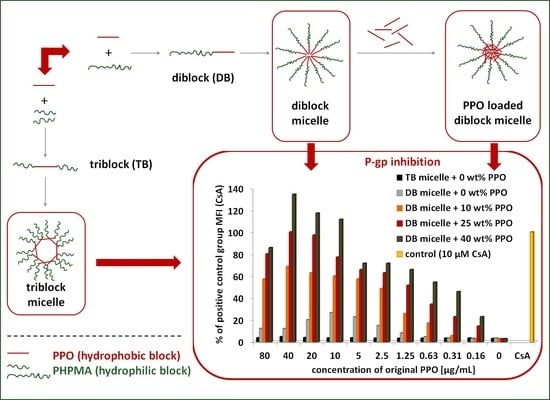
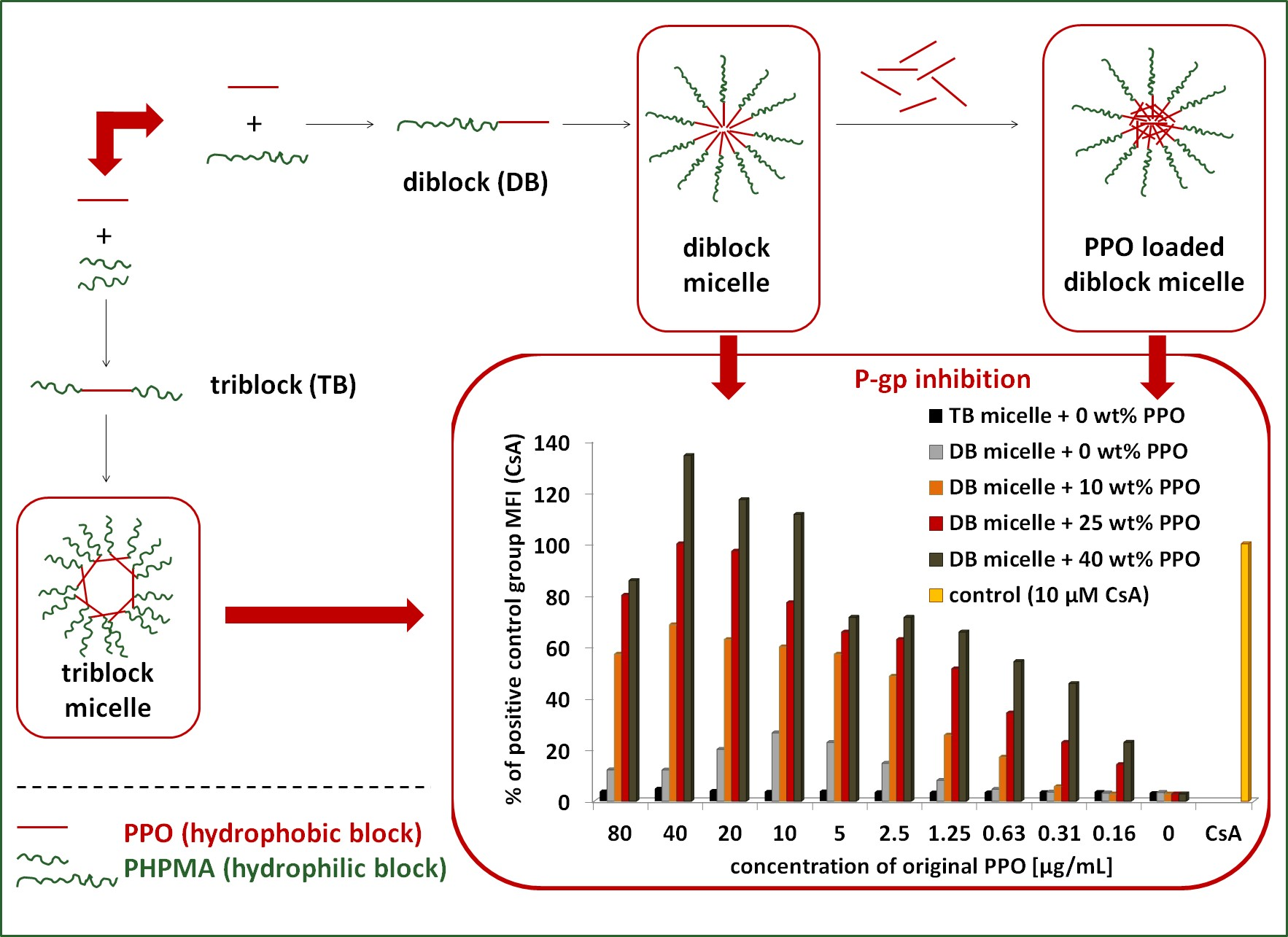
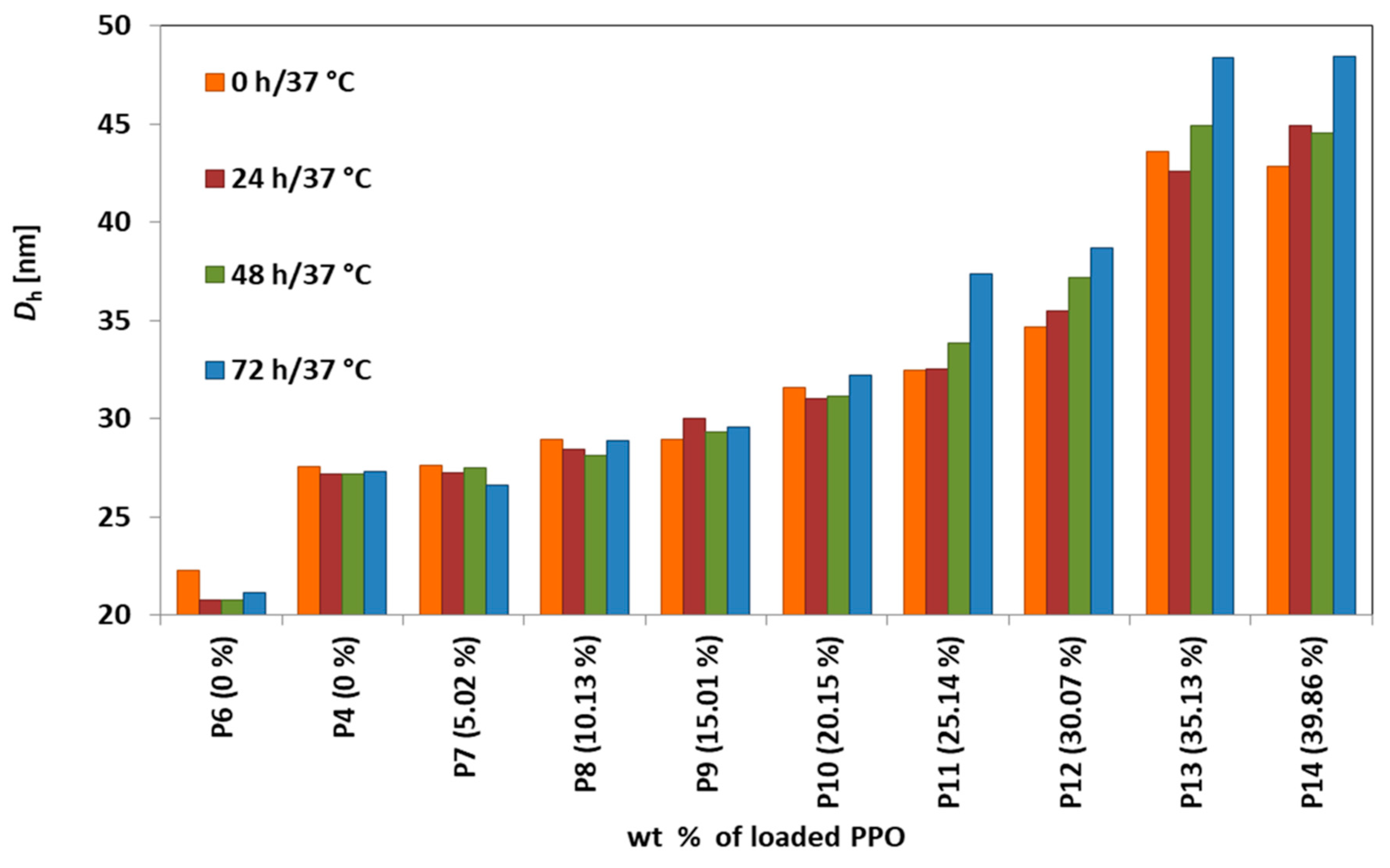
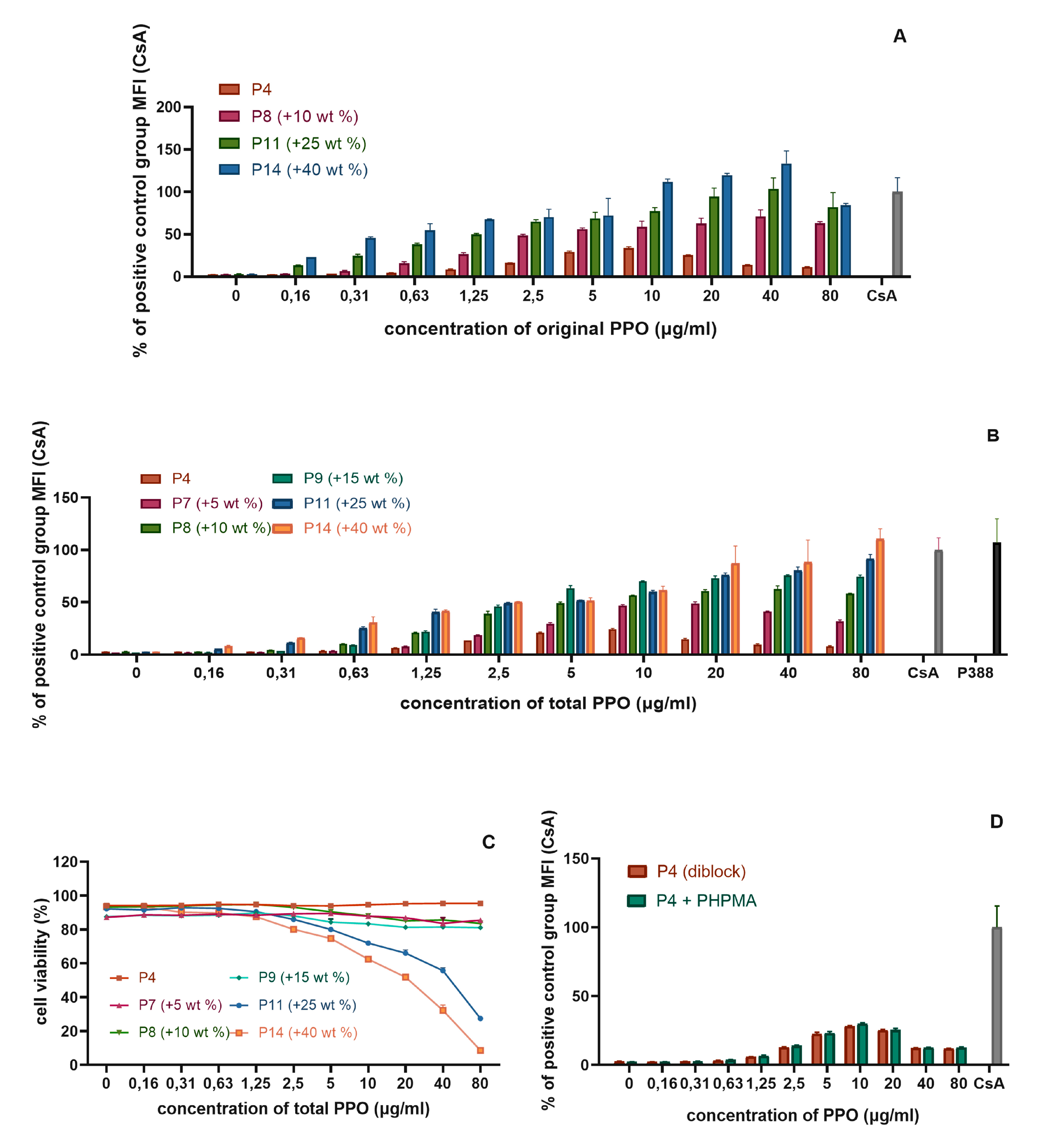
3.4. Diblock Copolymer Micelles Loaded with PPO, Samples P7–P14
3.5. Biological Activity
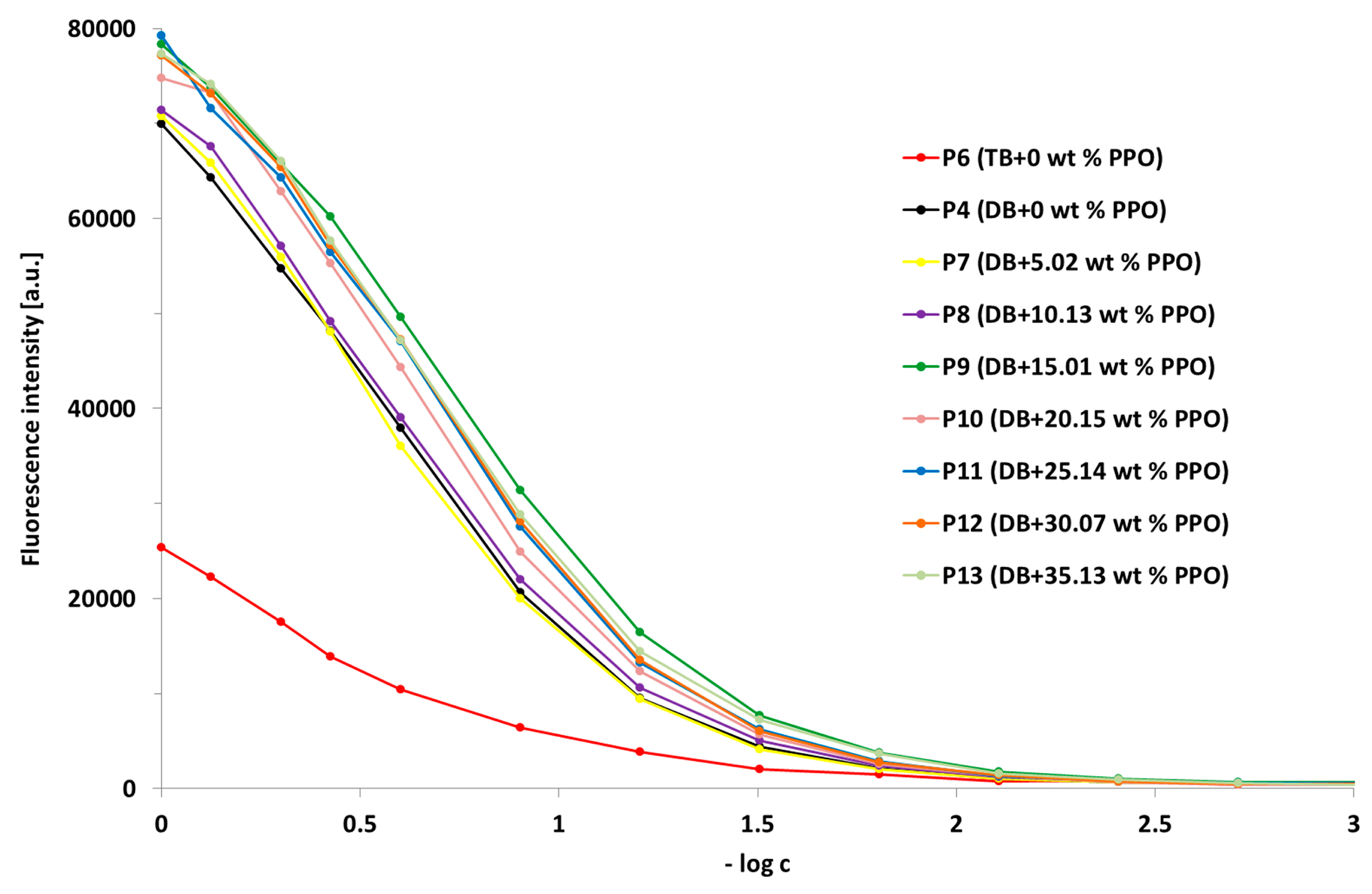
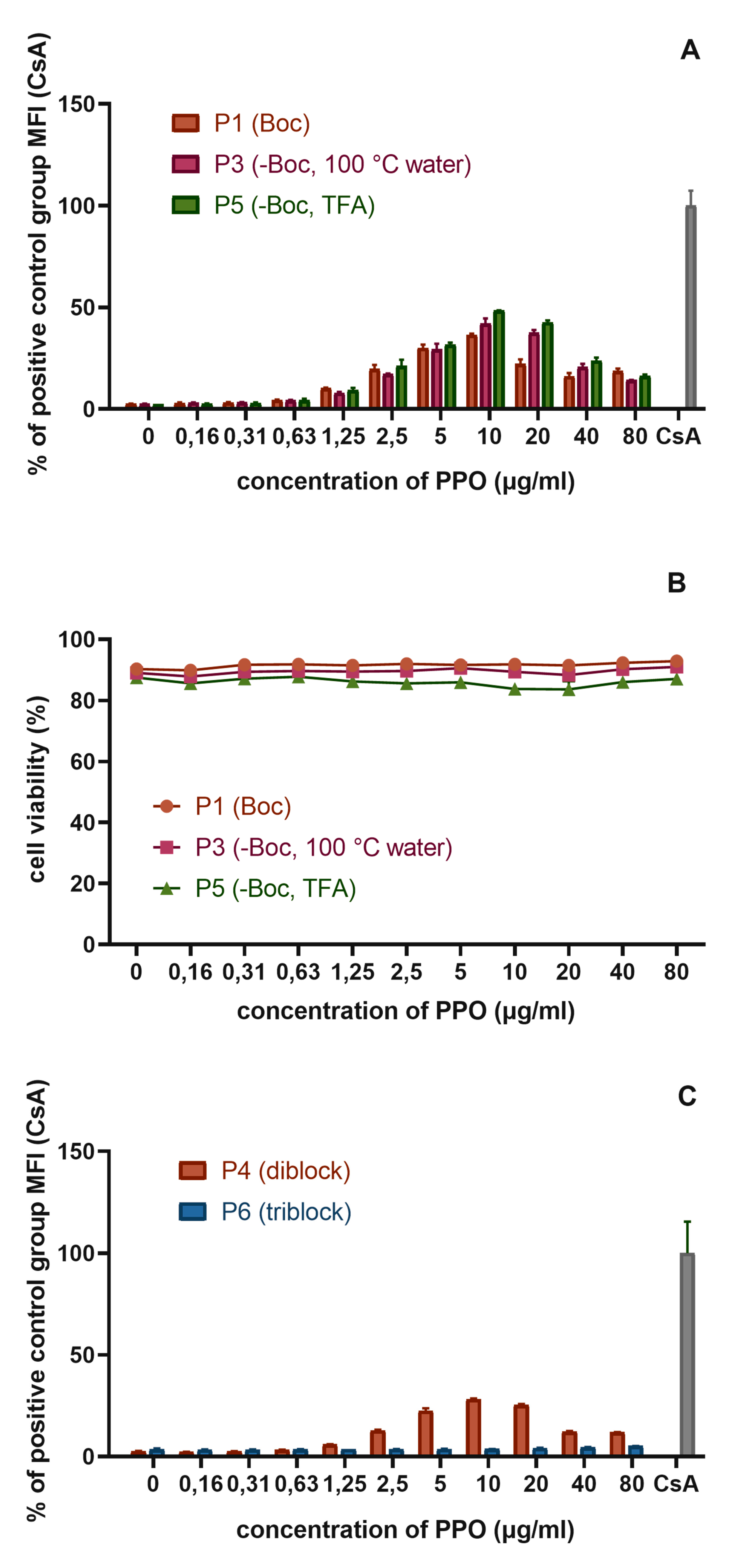
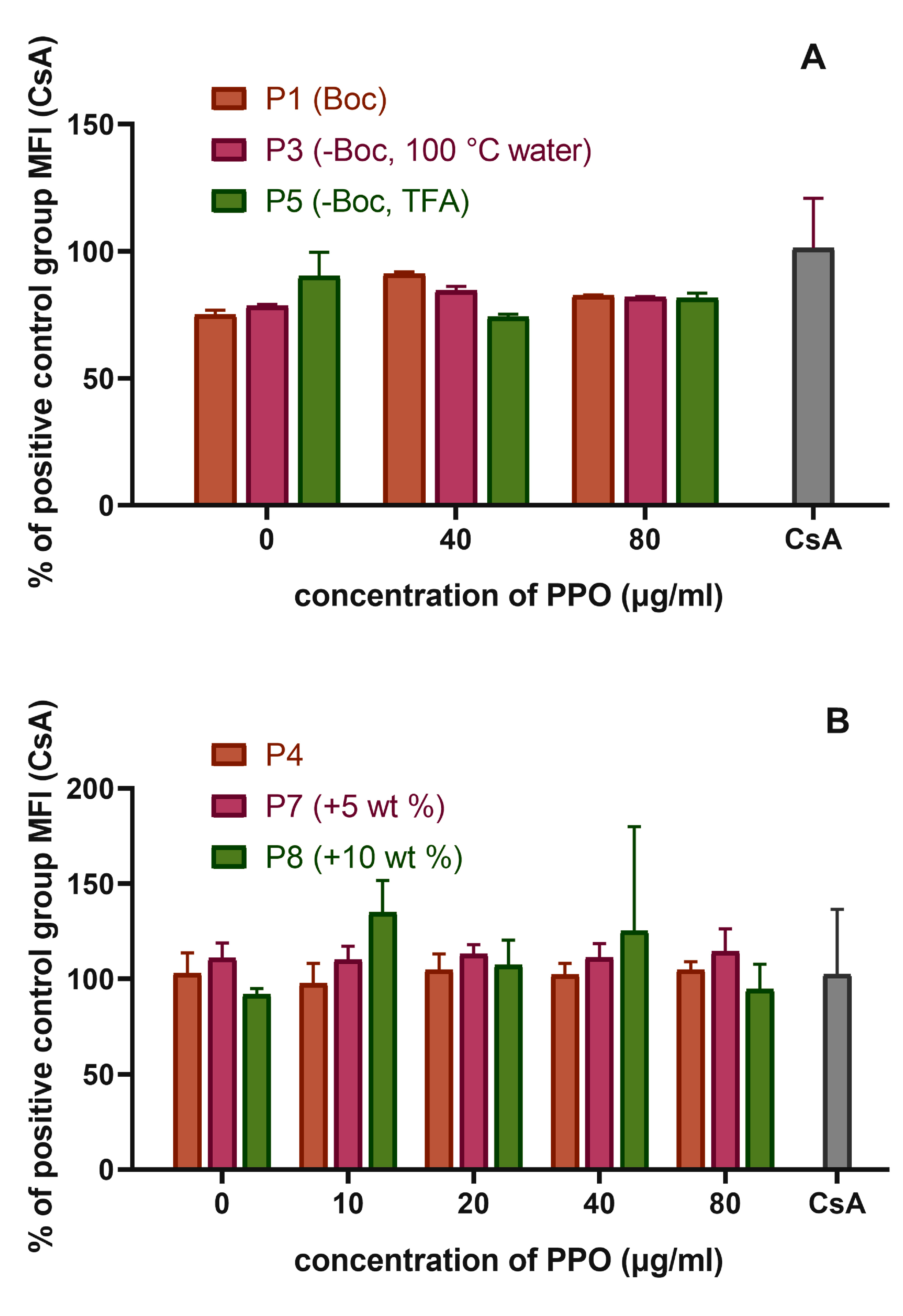
3.5.1. The Influence of the Detailed Structure of the Copolymer Carrier on the P-gp Inhibitory Capacity
3.5.2. The Effect of PPO Loading to the Micelles on P-gp Inhibition
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Ulbrich, K.; Holá, K.; Šubr, V.; Bakandritsos, A.; Tuček, J.; Zbořil, R. Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies. Chem. Rev. 2016, 116, 5338–5431. [Google Scholar] [CrossRef] [PubMed]
- Markman, J.L.; Rekechenetskiy, A.; Holler, E.; Ljubimova, J.Y. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv. Drug Deliv. Rev. 2013, 65, 1866–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Y.; Liang, H.; Bender, A.; Glen, R.C.; Yan, A. P-glycoprotein substrate models using support vector machines based on a comprehensive data set. J. Chem. Inf. Model. 2011, 51, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Y.; Yu, H.; Zhang, L.; Hou, T. Computational models for predicting substrates or inhibitors of P-glycoprotein. Drug Discov. Today 2012, 17, 343–351. [Google Scholar] [CrossRef]
- Koziolová, E.; Janoušková, O.; Cuchalová, L.; Hvězdová, Z.; Hraběta, J.; Eckschlager, T.; Sivák, L.; Ulbrich, K.; Etrych, T.; Šubr, V. Overcoming multidrug resistance in Dox-resistant neuroblastoma cell lines via treatment with HPMA copolymer conjugates containing anthracyclines and P-gp inhibitors. J. Control. Release 2016, 233, 136–146. [Google Scholar] [CrossRef]
- Šubr, V.; Sivák, L.; Koziolová, E.; Braunová, A.; Pechar, M.; Strohalm, J.; Kabešová, M.; Říhová, B.; Ulbrich, K.; Kovář, M. Synthesis of Poly[N-(2-hydroxypropyl)methacrylamide] conjugates of inhibitors of the ABC transporter that overcome multidrug resistance in doxorubicin-resistant P388 cells in vitro. Biomacromolecules 2014, 15, 3030–3043. [Google Scholar] [CrossRef]
- Sivak, L.; Subr, V.; Tomala, J.; Rihova, B.; Strohalm, J.; Etrych, T.; Kovar, M. Overcoming multidrug resistance via simultaneous delivery of cytostatic drug and P-glycoprotein inhibitor to cancer cells by HPMA copolymer conjugate. Biomaterials 2017, 115, 65–80. [Google Scholar] [CrossRef]
- Alakhova, D.Y.; Kabanov, A.V. Pluronics and MDR reversal: An update. Mol. Pharm. 2014, 11, 2566–2578. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Kabanov, A.V. Pluronic block copolymers: Evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J. Control. Release 2008, 130, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabanov, A.V.; Batrakova, E.V.; Alakhov, V.Y. Pluronic® block copolymers for overcoming drug resistance in cancer. Adv. Drug Deliv. Rev. 2002, 54, 759–779. [Google Scholar] [CrossRef]
- Pitto-Barry, A.; Barry, N.P.E. Pluronic® block-copolymers in medicine: From chemical and biological versatility to rationalisation and clinical advances. Polym. Chem. 2014, 5, 3291–3297. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Zhu, J. Pluronic Block Copolymers for Drug and Gene Delivery. In Polymeric Drug Delivery Systems, Series: Drugs and the Pharmaceutical Sciences, Volume 148; Kwon, G.S., Ed.; Taylor and Francis Group, CRC Press: Boca Raton, FL, USA, 2005; pp. 577–613. ISBN 0824725328. [Google Scholar]
- Raval, A.; Pillai, S.A.; Bahadur, A.; Bahadur, P. Systematic characterization of Pluronic® micelles and their application for solubilization and in vitro release of some hydrophobic anticancer drugs. J. Mol. Liq. 2017, 230, 473–481. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Bronich, T.K.; Vetro, J.A.; Kabanov, A.V. Polymer Micelles as Drug Carriers. In Nanoparticulates as Drug Carriers; Imperial College Press: London, UK, 2006; pp. 57–93. [Google Scholar] [Green Version]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Maeda, H.; Sawa, T.; Konno, T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J. Control. Release 2001, 74, 47–61. [Google Scholar] [CrossRef]
- Braunová, A.; Kostka, L.; Sivák, L.; Cuchalová, L.; Hvězdová, Z.; Laga, R.; Filippov, S.; Černoch, P.; Pechar, M.; Janoušková, O.; et al. Tumor-targeted micelle-forming block copolymers for overcoming of multidrug resistance. J. Control. Release 2017, 245, 41–51. [Google Scholar] [CrossRef]
- Nishiyama, N.; Matsumura, Y.; Kataoka, K. Development of polymeric micelles for targeting intractable cancers. Cancer Sci. 2016, 107, 867–874. [Google Scholar] [CrossRef]
- Cabral, H.; Kataoka, K. Progress of drug-loaded polymeric micelles into clinical studies. J. Control. Release 2014, 190, 465–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2001, 47, 113–131. [Google Scholar] [CrossRef]
- Kataoka, K.; Matsumoto, T.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Fukushima, S.; Okamoto, K.; Kwon, G.S. Doxorubicin-loaded poly(ethylene glycol)-poly(β-benzyl-l-aspartate) copolymer micelles: Their pharmaceutical characteristics and biological significance. J. Control. Release 2000, 64, 143–153. [Google Scholar] [CrossRef]
- Dongin, K.; Eun Seong, L.; Kyung Taek, O.; Zhong, G.G.; You Han, B. Doxorubicin-Loaded Polymeric Micelle Overcomes Multidrug Resistance of Cancer by Double-Targeting Folate Receptor and Early Endosomal pH. Small 2009, 4, 2043–2050. [Google Scholar]
- Deshmukh, A.S.; Chauhan, P.N.; Noolvi, M.N.; Chaturvedi, K.; Ganguly, K.; Shukla, S.S.; Nadagouda, M.N.; Aminabhavi, T.M. Polymeric micelles: Basic research to clinical practice. Int. J. Pharm. 2017, 532, 249–268. [Google Scholar] [CrossRef]
- Chytil, P.; Etrych, T.; Kříž, J.; Šubr, V.; Ulbrich, K. N-(2-Hydroxypropyl)methacrylamide-based polymer conjugates with pH-controlled activation of doxorubicin for cell-specific or passive tumour targeting. Synthesis by RAFT polymerisation and physicochemical characterisation. Eur. J. Pharm. Sci. 2010, 41, 473–482. [Google Scholar] [CrossRef]
- Ulbrich, K.; Etrych, T.; Chytil, P.; Jelínková, M.; Říhová, B. Antibody-targeted Polymer–doxorubicin Conjugates with pH-controlled Activation. J. Drug Target. 2004, 12, 477–489. [Google Scholar] [CrossRef]
- Šubr, V.; Koňák, Č.; Laga, R.; Ulbrich, K. Coating of DNA/poly(l-lysine) complexes by covalent attachment of poly[N-(2-hydroxypropyl)methacrylamide]. Biomacromolecules 2006, 7, 122–130. [Google Scholar] [CrossRef]
- Koziolová, E.; Kostka, L.; Kotrchová, L.; Šubr, V.; Konefal, R.; Nottelet, B.; Etrych, T. N-(2-Hydroxypropyl)methacrylamide-Based Linear, Diblock, and Starlike Polymer Drug Carriers: Advanced Process for Their Simple Production. Biomacromolecules 2018, 19, 4003–4013. [Google Scholar] [CrossRef]
- Perrier, S.; Takolpuckdee, P.; Mars, C.A. Reversible addition-fragmentation chain transfer polymerization: End group modification for functionalized polymers and chain transfer agent recovery. Macromolecules 2005, 38, 2033–2036. [Google Scholar] [CrossRef]
- Zinelaabidine, C.; Souad, O.; Zoubir, J.; Malika, B.; Nour-Eddine, A. A Simple and Efficient Green Method for the Deprotection of N-Boc in Various Structurally Diverse Amines under Water-mediated Catalyst-free Conditions. Int. J. Chem. 2012, 4, 73–79. [Google Scholar] [CrossRef]
- Trousil, J.; Syrová, Z.; Dal, N.J.K.; Rak, D.; Konefal, R.; Pavlova, E.; Matějková, J.; Cmarko, D.; Kubíčková, P.; Pavliš, O.; et al. Rifampicin Nanoformulation Enhances Treatment of Tuberculosis in Zebrafish. Biomacromolecules 2019, 20, 1798–1815. [Google Scholar] [CrossRef] [PubMed]
- Kolouchova, K.; Sedlacek, O.; Jirak, D.; Babuka, D.; Blahut, J.; Kotek, J.; Vit, M.; Trousil, J.; Konefał, R.; Janouskova, O.; et al. Self-Assembled Thermoresponsive Polymeric Nanogels for 19F MR Imaging. Biomacromolecules 2018, 19, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Von Richter, O.; Glavinas, H.; Krajcsi, P.; Liehner, S.; Siewert, B.; Zech, K. A novel screening strategy to identify ABCB1 substrates and inhibitors. Naunyn. Schmiedeberg’s Arch. Pharmacol. 2009, 379, 11–26. [Google Scholar] [CrossRef]
- Allen, C.; Maysinger, D.; Eisenberg, A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surfaces B Biointerfaces 1999, 16, 3–27. [Google Scholar] [CrossRef]
- Nagarajan, R.; Ganesh, K. 1989-Nagarajan. J. Chem. Phys. 1989, 90, 5843–5856. [Google Scholar] [CrossRef]
- Klepac, D.; Kostková, H.; Petrova, S.; Chytil, P.; Etrych, T.; Kereïche, S.; Raška, I.; Weitz, D.A.; Filippov, S.K. Interaction of spin-labeled HPMA-based nanoparticles with human blood plasma proteins-the introduction of protein-corona-free polymer nanomedicine. Nanoscale 2018, 10, 6194–6204. [Google Scholar] [CrossRef]
- Tyrrell, Z.L.; Shen, Y.; Radosz, M. Fabrication of micellar nanoparticles for drug delivery through the self-assembly of block copolymers. Prog. Polym. Sci. 2010, 35, 1128–1143. [Google Scholar] [CrossRef]
- Lammers, T.; Kühnlein, R.; Kissel, M.; Subr, V.; Etrych, T.; Pola, R.; Pechar, M.; Ulbrich, K.; Storm, G.; Huber, P.; et al. Effect of physicochemical modification on the biodistribution and tumor accumulation of HPMA copolymers. J. Control. Release 2005, 110, 103–118. [Google Scholar] [CrossRef]
- Sajid, M.S.; Jabeen, F.; Hussain, D.; Ashiq, M.N.; Najam-ul-Haq, M. Hydrazide-functionalized affinity on conventional support materials for glycopeptide enrichment. Anal. Bioanal. Chem. 2017, 409, 3135–3143. [Google Scholar] [CrossRef]
- Roth, J. Protein N-glycosylation along the Secretory Pathway: Relationship to organelle topography and function, protein quality control, and cell interactions. Chem. Rev. 2002, 102, 285–303. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Mn.103 (g/mol) | Ð | Functionality of TT a | Content of NHNH2 (mol%) | Dh, 37 °C (nm) |
|---|---|---|---|---|---|
| A1 | 7.2 | 1.06 | 0.99 | 6.0 | 6.1 |
| A2 | 5.0 | 1.02 | 0.81 | 4.7 | 4.5 |
| Polymer | Sample Code 1 | Mn (SEC) 2 (g/mol) | Ð | % of PPO in Copolymer 3 | Mn (FFF) 4 (g/mol) | % of Micelles | Dh, 37 °C (nm) | CMC5 (g L−1) |
|---|---|---|---|---|---|---|---|---|
| P1 | DB-Boc | 13.20 | 1.05 | 39.7 | - | - | 26.2 | - |
| P2 | DB-Boc-P | 12.90 | 1.18 | 36.6 | 9.63 × 105 | 88% | 23.8 | 0.0503 |
| P3 | DB-H2O | 14.11 | 1.09 | 35.1 | 1.20 × 106 | 88% | 26.6 | 0.0347 |
| P4 | DB-H2O-P | 13.29 | 1.22 | 36.2 | 6.97 × 105 | 76.5% | 25.4 | 0.0558 |
| P5 | DB-TFA | 13.72 | 1.11 | 40.4 | 1.15 × 106 | 88% | 27.4 | 0.0459 |
| P6 | TB-H2O-P | 13.40 | 1.10 | 35.9 | 3.70 × 105 | 45.3% | 22.3 | 0.1307 |
| Sample Code | PPO/mg | Copolymer (P4)/mg | wt % of Loaded PPO * |
|---|---|---|---|
| P4 | 0 | 25 | 0 |
| P7 | 1.35 | 25.55 | 5.02 |
| P8 | 2.63 | 23.33 | 10.13 |
| P9 | 4.51 | 25.54 | 15.01 |
| P10 | 6.11 | 24.21 | 20.15 |
| P11 | 8.31 | 24.75 | 25.14 |
| P12 | 11.53 | 26.82 | 30.07 |
| P13 | 13.52 | 24.97 | 35.13 |
| P14 | 8.26 | 12.46 | 39.86 |
| Sample Code | % of Total PPO in Copolymer 1 | Mn (FFF) 2 (g/mol) | % of Micelle | Dh, 37 °C (nm) | CMC 3 (g L−1) |
|---|---|---|---|---|---|
| P4 | 36.2 | 6.97 × 105 | 76.5% | 25.4 | 0.0558 |
| P7 | 40.3 | - | - | 26.6 | 0.0585 |
| P8 | 44.9 | 5.69 × 105 | 75.3% | 28.4 | 0.0524 |
| P9 | 47.2 | 7.60 × 105 | 80.2% | 27.4 | 0.0339 |
| P10 | 50.8 | 6.19 × 105 | 78.7% | 30.8 | 0.0506 |
| P11 | 55.5 | - | - | 31.6 | 0.0443 |
| P12 | 58.0 | 7.15 × 105 | 71.6% | 33.2 | 0.0438 |
| P13 | 61.3 | - | - | 39.2 | 0.0422 |
| P14 | 64.4 | 5.73 × 105 | 70.2% | 42.0 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braunová, A.; Kaňa, M.; Kudláčová, J.; Kostka, L.; Bouček, J.; Betka, J.; Šírová, M.; Etrych, T. Micelle-Forming Block Copolymers Tailored for Inhibition of P-gp-Mediated Multidrug Resistance: Structure to Activity Relationship. Pharmaceutics 2019, 11, 579. https://doi.org/10.3390/pharmaceutics11110579
Braunová A, Kaňa M, Kudláčová J, Kostka L, Bouček J, Betka J, Šírová M, Etrych T. Micelle-Forming Block Copolymers Tailored for Inhibition of P-gp-Mediated Multidrug Resistance: Structure to Activity Relationship. Pharmaceutics. 2019; 11(11):579. https://doi.org/10.3390/pharmaceutics11110579
Chicago/Turabian StyleBraunová, Alena, Martin Kaňa, Júlia Kudláčová, Libor Kostka, Jan Bouček, Jan Betka, Milada Šírová, and Tomáš Etrych. 2019. "Micelle-Forming Block Copolymers Tailored for Inhibition of P-gp-Mediated Multidrug Resistance: Structure to Activity Relationship" Pharmaceutics 11, no. 11: 579. https://doi.org/10.3390/pharmaceutics11110579