Amorphous Nanosuspensions Aggregated from Paclitaxel–Hemoglobulin Complexes with Enhanced Cytotoxicity

Abstract
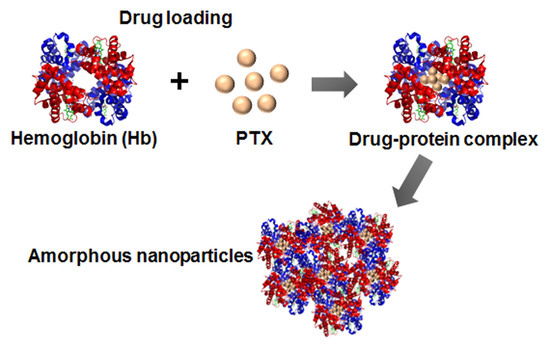
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation
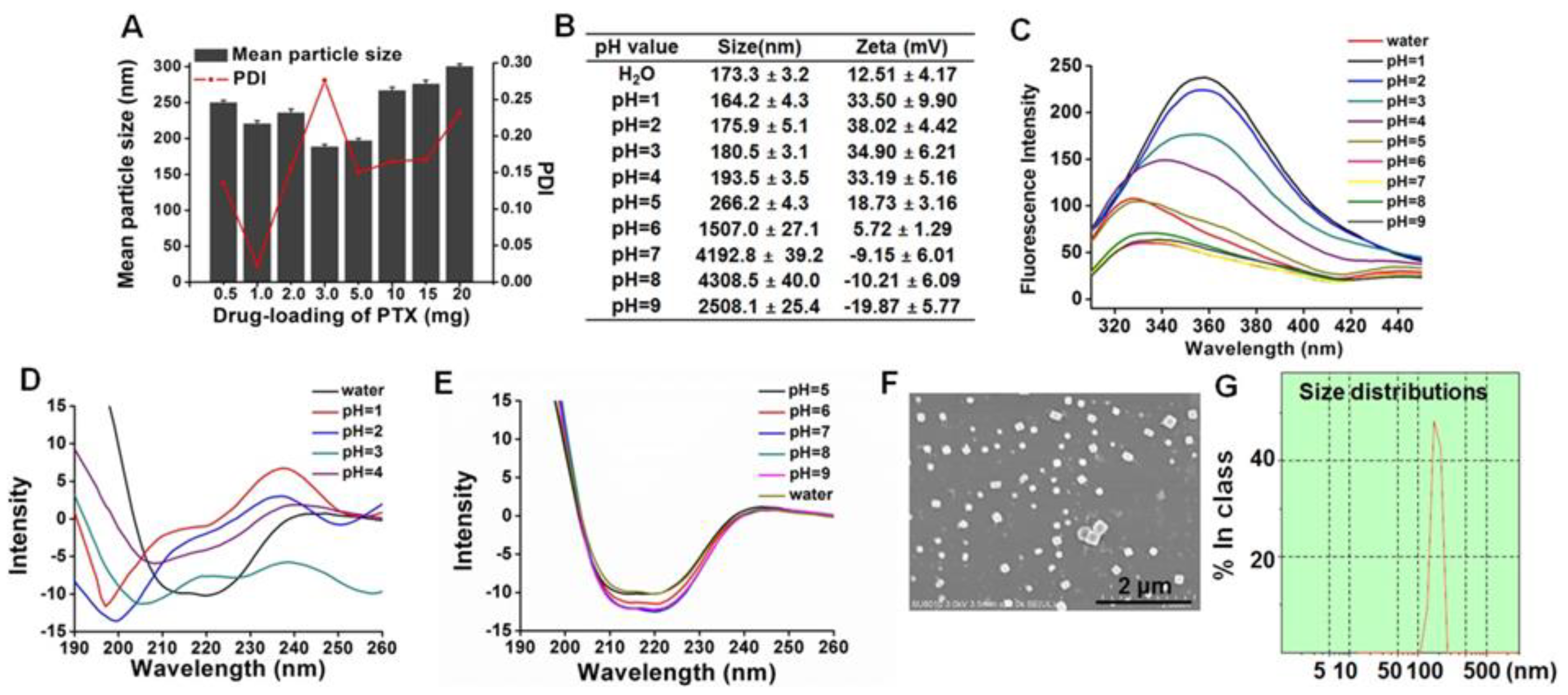
2.3. Characterization
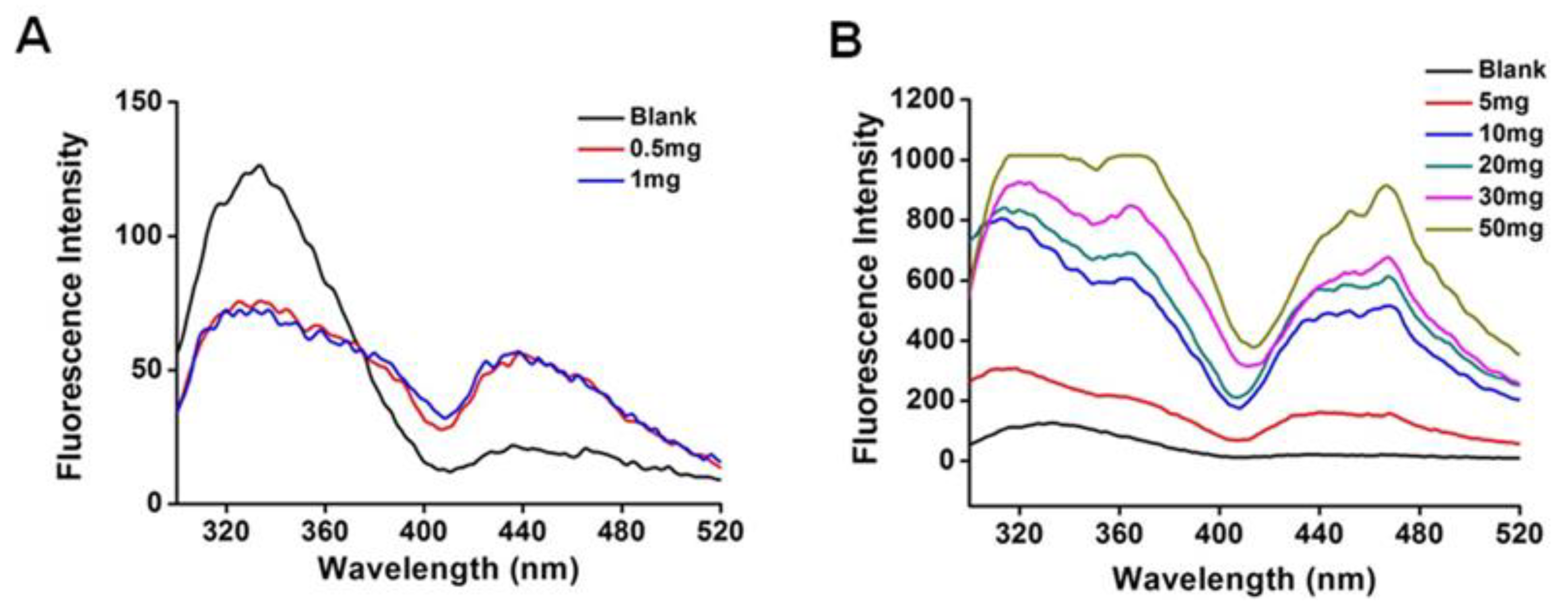
2.4. Fluorescence Spectra
2.5. Circular Dichroism (CD) Spectra
2.6. Affinity Study
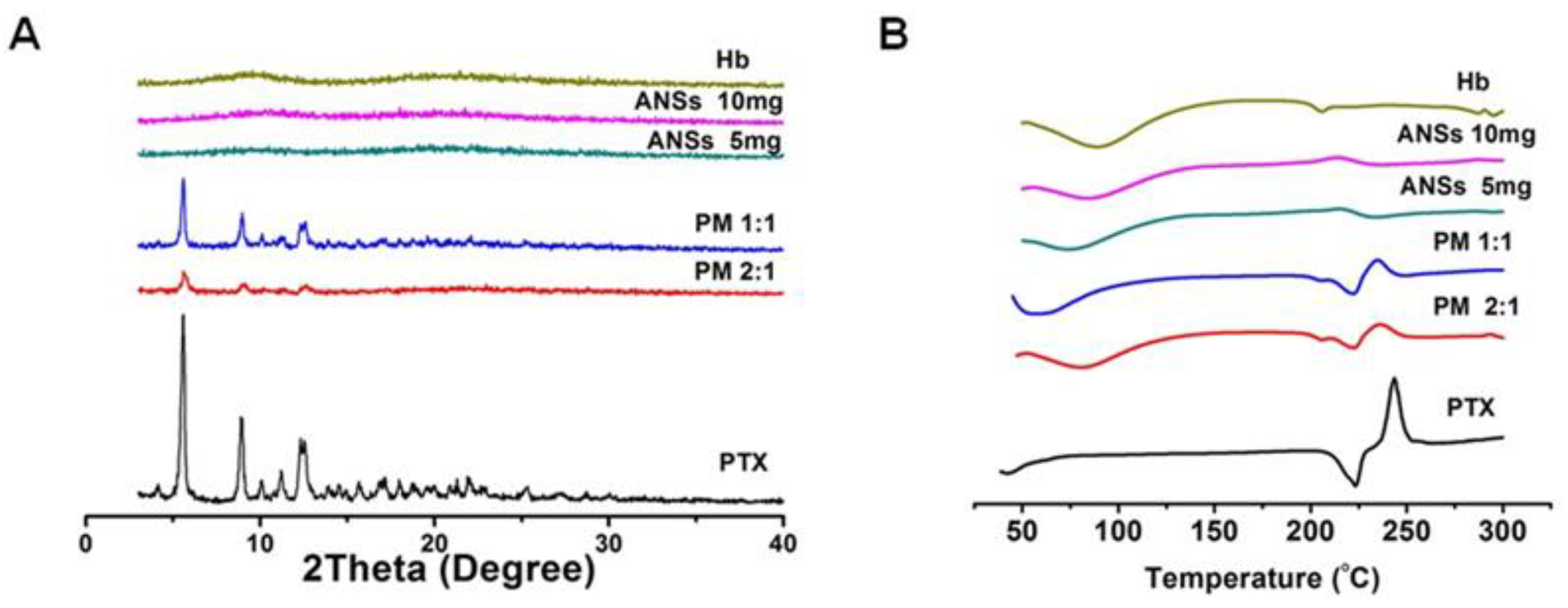
2.7. Powder X-Ray Diffraction (PXRD) and Differential Scanning Calorimetry (DSC)
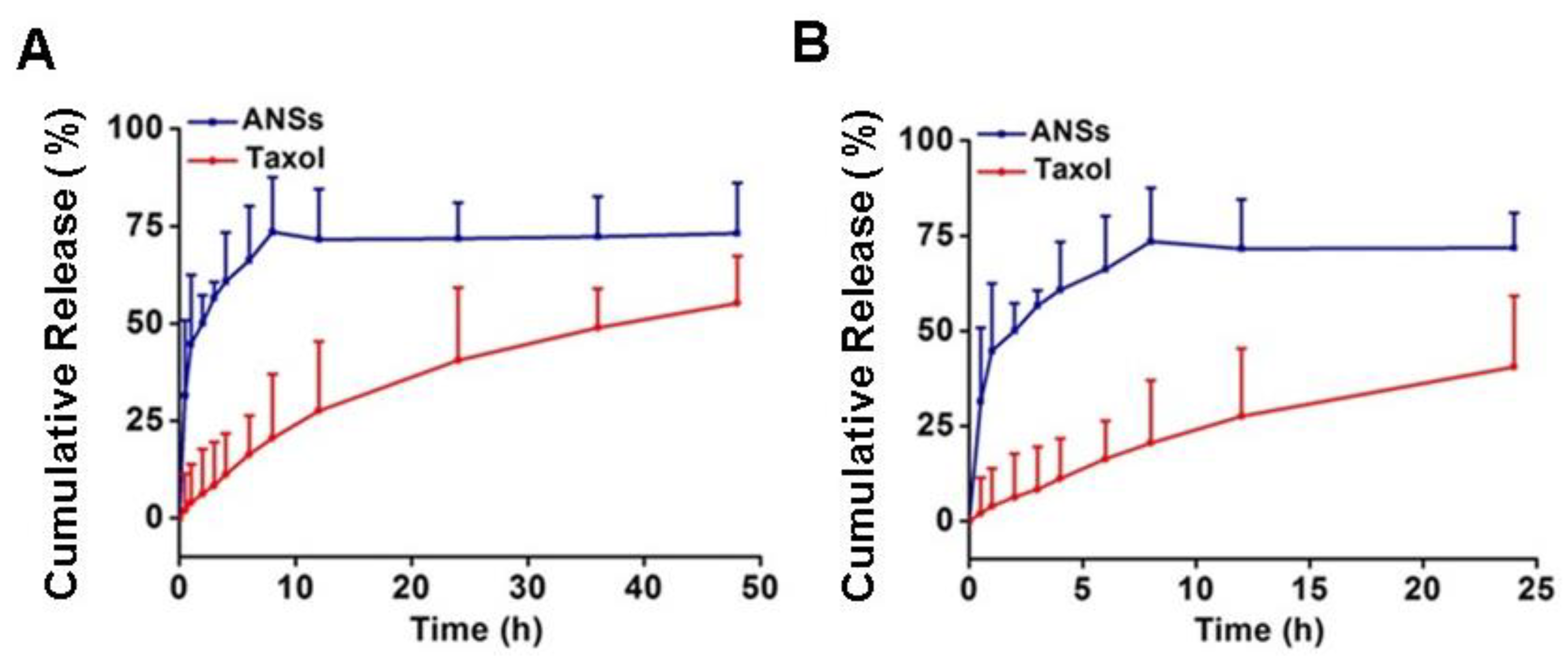
2.8. In Vitro Release
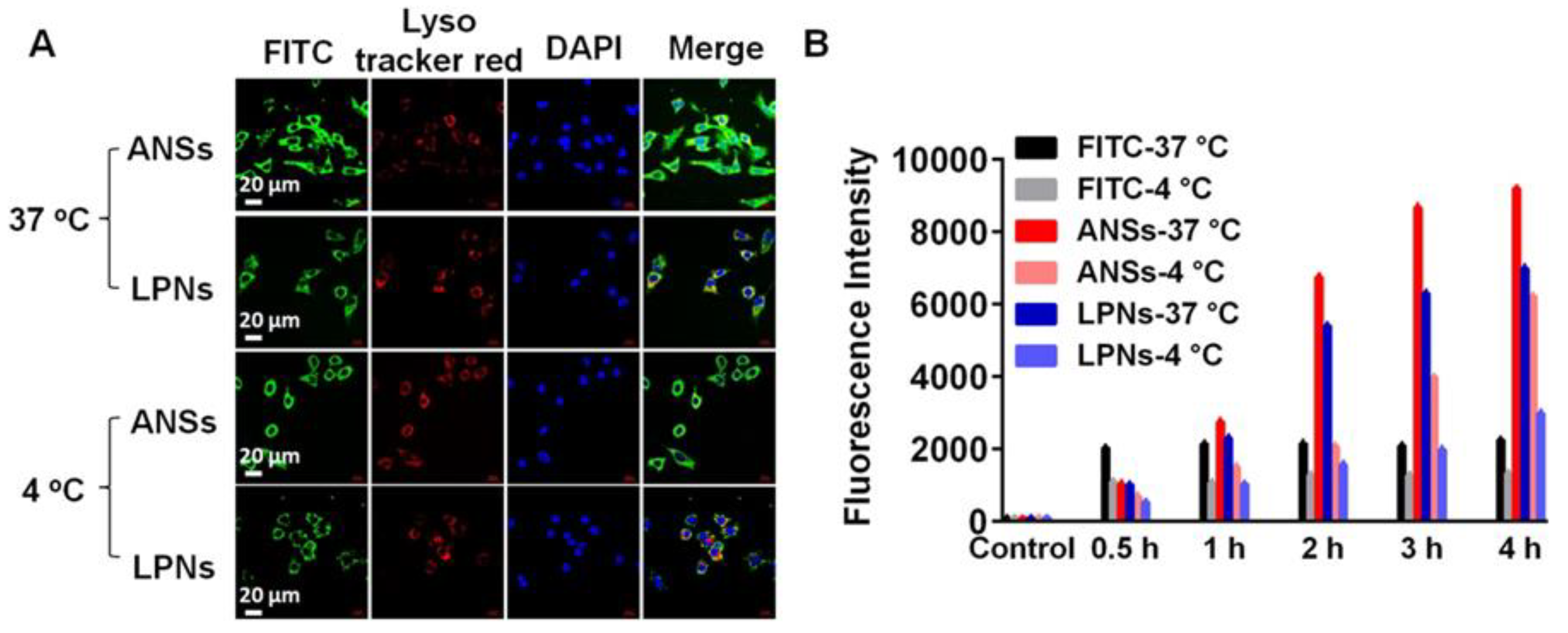
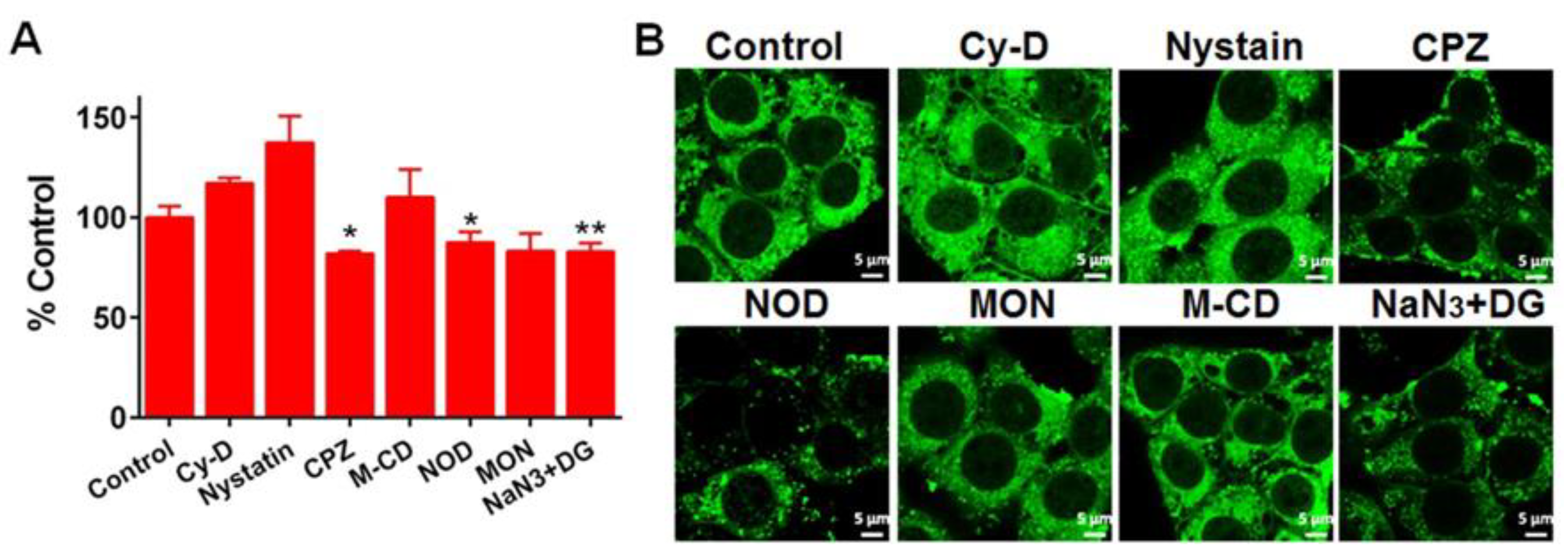
2.9. Flow Cytometry (FCM)
2.10. Confocal Imaging
2.11. Cytotoxicity
2.12. Statistical Analysis
3. Results and Discussion
3.1. Drug-Protein Complex
3.2. Preparation and Characterization of ANSs
3.3. Amorphous State and In Vitro Drug Release
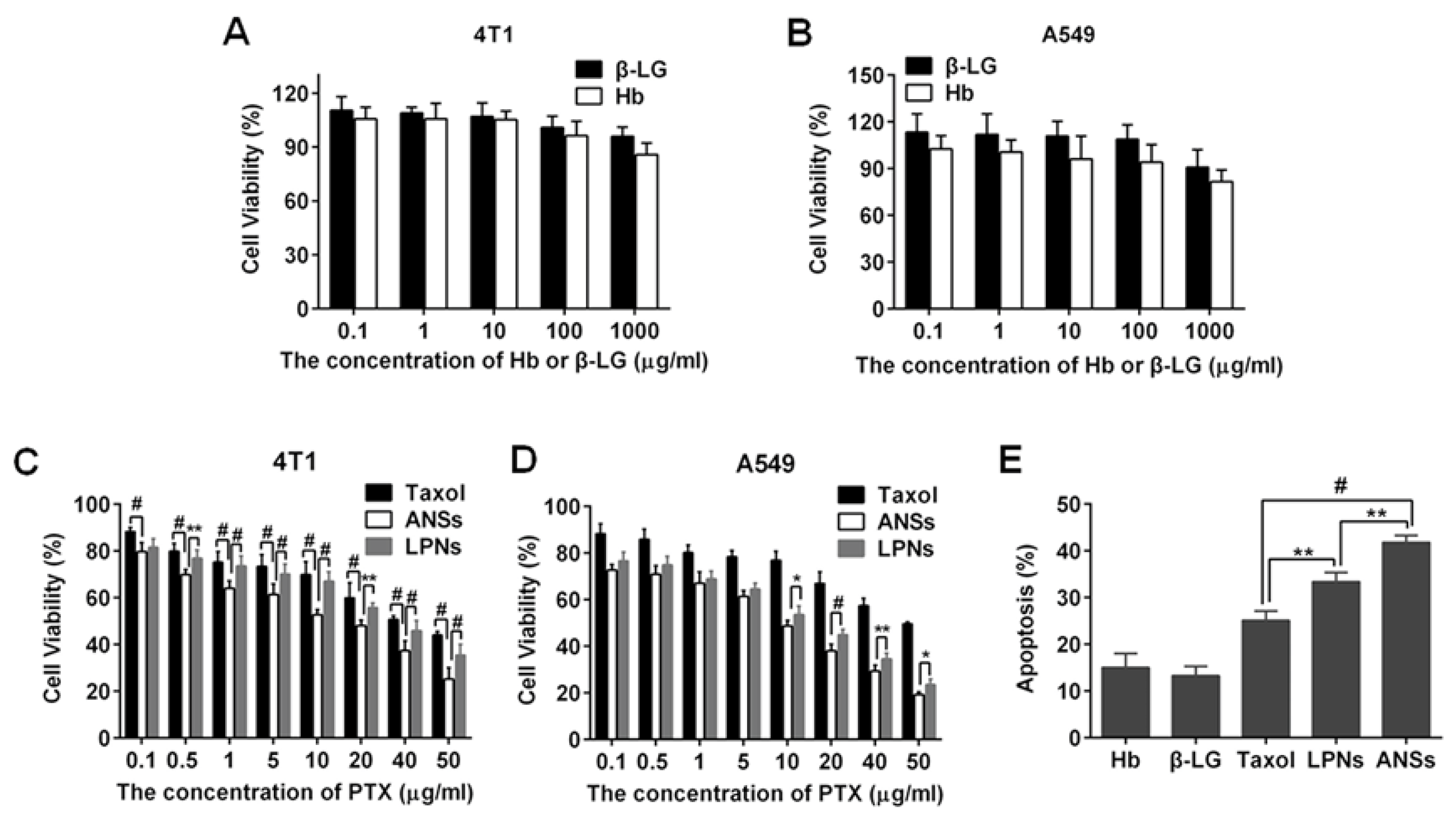
3.4. Cellular Uptake and Cytotoxicity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Miriyala, N.; Ouyang, D.; Perrie, Y.; Lowry, D.; Kirby, D.J. Activated carbon as a carrier for amorphous drug delivery: Effect of drug characteristics and carrier wettability. Eur. J. Pharm. Biopharm. 2017, 115, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Gao, H. Nanoparticles for modulating tumor microenvironment to improve drug delivery and tumor therapy. Pharm. Res. 2017, 126, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Gao, H. Shaping tumor microenvironment for improving nanoparticle delivery. Curr. Drug Metab. 2016, 17, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, Y.; Zhang, L.; Wang, Q.; Zhang, D. Stability of nanosuspensions in drug delivery. J. Control. Release 2013, 172, 1126–1141. [Google Scholar] [CrossRef] [PubMed]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Dis. 2004, 3, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, K.; Gauthier, M.A.; Leroux, J.C. Targeting of injectable drug nanocrystals. Mol. Pharm. 2014, 11, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, M.E.; Brettmann, B.K.; Rogers, T.L.; Elder, E.J.; Williams, R.O.; Johnston, K.P. Design of potent amorphous drug nanoparticles for rapid generation of highly supersaturated media. Mol. Pharm. 2007, 4, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Cheow, W.S.; Kiew, T.Y.; Yang, Y.; Hadinoto, K. Amorphization strategy affects the stability and supersaturation profile of amorphous drug nanoparticles. Mol. Pharm. 2014, 11, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Yonashiro, H.; Higashi, K.; Morikawa, C.; Ueda, K.; Itoh, T.; Ito, M.; Masu, H.; Noguchi, S.; Moribe, K. Morphological and physicochemical evaluation of two distinct glibenclamide/hypromellose amorphous nanoparticles prepared by the antisolvent method. Mol. Pharm. 2018, 15, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Lee, D.; Meng, Z.; Li, T. Exploring intracellular fate of drug nanocrystals with crystal-integrated and environment-sensitive fluorophores. J. Control. Release 2017, 267, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Qi, J.; Dong, X.; Zhao, W.; Wu, W. The in vivo fate of nanocrystals. Drug Dis. Today 2017, 22, 744–750. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wang, Y.; Lv, Y.; Xiao, Q.; Ye, L.; Cai, B.; Qin, C.; Han, X.; Cai, T.; Yin, L. Denatured protein stabilized drug nanoparticles: Tunable drug state and penetration across the intestinal barrier. J. Mater. Chem. B 2017, 5, 1081–1097. [Google Scholar] [CrossRef]
- Zhang, L.; Xiao, Q.; Wang, Y.; Zhang, C.; He, W.; Yin, L. Denatured protein-coated docetaxel nanoparticles: Alterable drug state and cytosolic delivery. Int. J. Pharm. 2017, 523, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Duan, L.; Li, J. Hemoglobin-based nanoarchitectonic assemblies as oxygen carriers. Adv. Mater. 2016, 28, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K.; Onetto, N.; Canetta, R.M.; Arbuck, S.G. Taxol: The first of the taxanes, an important new class of antitumor agents. Semin. Oncol. 1992, 19, 646–662. [Google Scholar] [PubMed]
- Koudelka, Š.; Turánek, J. Liposomal paclitaxel formulations. J. Control. Release 2012, 163, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Pei, X.; Yang, X.; Lv, Y.; Zhang, L.; He, W.; Yin, L. Rod-shaped active drug particles enable efficient and safe gene delivery. Adv. Sci. 2017, 4, 1700324. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Xin, X.; Li, Y.; Han, X.; Qin, C.; Yin, L. Rod-shaped drug particles for cancer therapy: The importance of particle size and participation of caveolae pathway. Part. Part. Syst. Charact. 2017, 34, 1600371. [Google Scholar] [CrossRef]
- Liang, L.; Tajmir-Riahi, H.A.; Subirade, M. Interaction of beta-lactoglobulin with resveratrol and its biological implications. Biomacromolecules 2008, 9, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Field, C.J.; Vine, D.; Chen, L. Intestinal uptake and transport of vitamin b12-loaded soy protein nanoparticles. Pharm. Res. 2015, 32, 1288–1303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, Z.; Liang, L.; Subirade, M.; Chen, L. Binding interactions of beta-conglycinin and glycinin with vitamin b12. J. Phys. Chem. B 2013, 117, 14018–14028. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, S.; Joshi, P.; Chakravarty, D.; Shanker, V.; Ansari, Z.A.; Singh, S.P.; Chakrabarti, P. Interaction of polyethyleneimine-functionalized ZnO nanoparticles with bovine serum albumin. Langmuir 2012, 28, 11142–11152. [Google Scholar] [CrossRef] [PubMed]
- Diarrassouba, F.; Liang, L.; Remondetto, G.; Subirade, M. Nanocomplex formation between riboflavin and β-lactoglobulin: Spectroscopic investigation and biological characterization. Food Res. Int. 2013, 52, 557–567. [Google Scholar] [CrossRef]
- Tanford, C.; Buzzell, J.G.; Rands, D.G.; Swanson, S.A. The reversible expansion of bovine serum albumin in acid solutions1. J. Am. Chem. Soc. 1955, 77, 6421–6428. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, T.; Wang, J.; Wang, Q.; Lv, X.; Ke, H.; Guo, Z.; Shen, J.; Wang, Y.; Xing, C.; et al. Size-tunable Gd2O3@albumin nanoparticles conjugating chlorin e6 for magnetic resonance imaging-guided photo-induced therapy. Theranostics 2017, 7, 764–774. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Lv, Y.; Zhao, Y.; Xu, C.; Jin, Z.; Qin, C.; Yin, L. Core–shell structured gel-nanocarriers for sustained drug release and enhanced antitumor effect. Int. J. Pharm. 2015, 484, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Junghanns, J.U.; Müller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–309. [Google Scholar] [Green Version]
- Li, Y.; Wu, Z.; He, W.; Qin, C.; Yao, J.; Zhou, J.; Yin, L. Globular protein-coated paclitaxel nanosuspensions: Interaction mechanism, direct cytosolic delivery, and significant improvement in pharmacokinetics. Mol. Pharm. 2015, 12, 1485–1500. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhen, X.; Wang, X.; Wu, W.; Jiang, X. Cellular entry fashion of hollow milk protein spheres. Soft Matter 2011, 7, 11526–11534. [Google Scholar] [CrossRef]
- Mohammad, I.S.; He, W.; Yin, L. Smart paclitaxel-disulfiram nanococrystals for efficient MDR reversal and enhanced apoptosis. Pharm. Res. 2018, 35, 77. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T(K) | Ksv/(M−1) | Kq/(M−1·s−1) | n | Ka/(M−1) |
|---|---|---|---|---|
| 288 | 3.5 × 109 | 3.5 × 1017 | 0.8 | 7.2 × 107 |
| 298 | 2.7 × 109 | 2.7 × 1017 | 0.7 | 1.6 × 106 |
| 308 | 1.5 × 109 | 1.5 × 1017 | 0.5 | 1.7 × 104 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, C.; Xin, X.; Pei, X.; Yin, L.; He, W. Amorphous Nanosuspensions Aggregated from Paclitaxel–Hemoglobulin Complexes with Enhanced Cytotoxicity. Pharmaceutics 2018, 10, 92. https://doi.org/10.3390/pharmaceutics10030092
Qin C, Xin X, Pei X, Yin L, He W. Amorphous Nanosuspensions Aggregated from Paclitaxel–Hemoglobulin Complexes with Enhanced Cytotoxicity. Pharmaceutics. 2018; 10(3):92. https://doi.org/10.3390/pharmaceutics10030092
Chicago/Turabian StyleQin, Chao, Xiaofei Xin, Xue Pei, Lifang Yin, and Wei He. 2018. "Amorphous Nanosuspensions Aggregated from Paclitaxel–Hemoglobulin Complexes with Enhanced Cytotoxicity" Pharmaceutics 10, no. 3: 92. https://doi.org/10.3390/pharmaceutics10030092