
Regulation of the Host Antiviral State by Intercellular Communications

Abstract
:1. Introduction
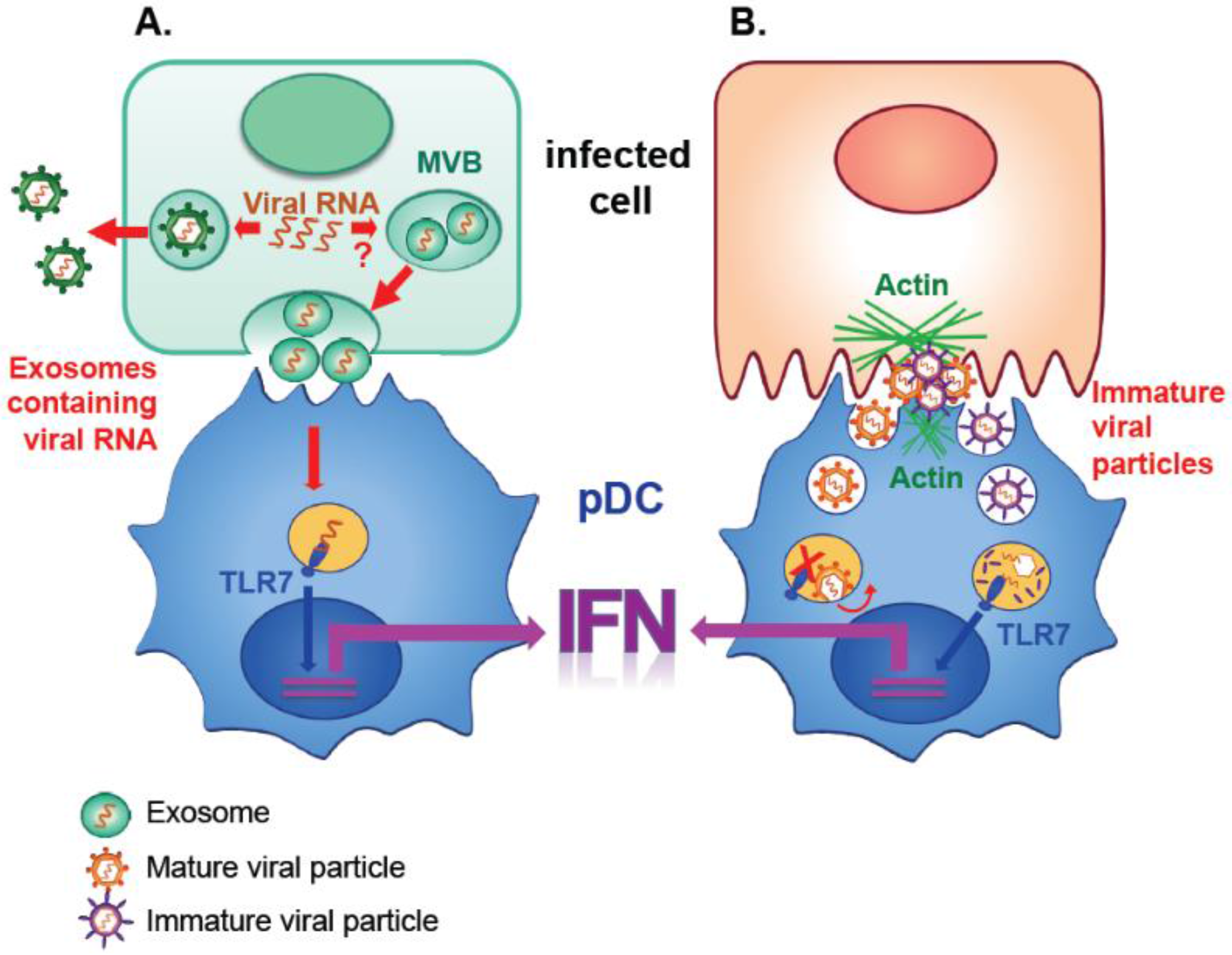
2. Transmission of Replicating Viral Genomes by Exosomal Transfer
3. pDC Activation by Vesicle-Mediated Transfer of Viral RNA
3.1. pDC Activation by Exosomal Transfer

3.2. pDC Activation Independently of Exosomal Transfer
3.3. Common Features of pDC Activation
4. Immune Activation by Cell-to-Cell Transfer of Viral Elements from Infected Cells
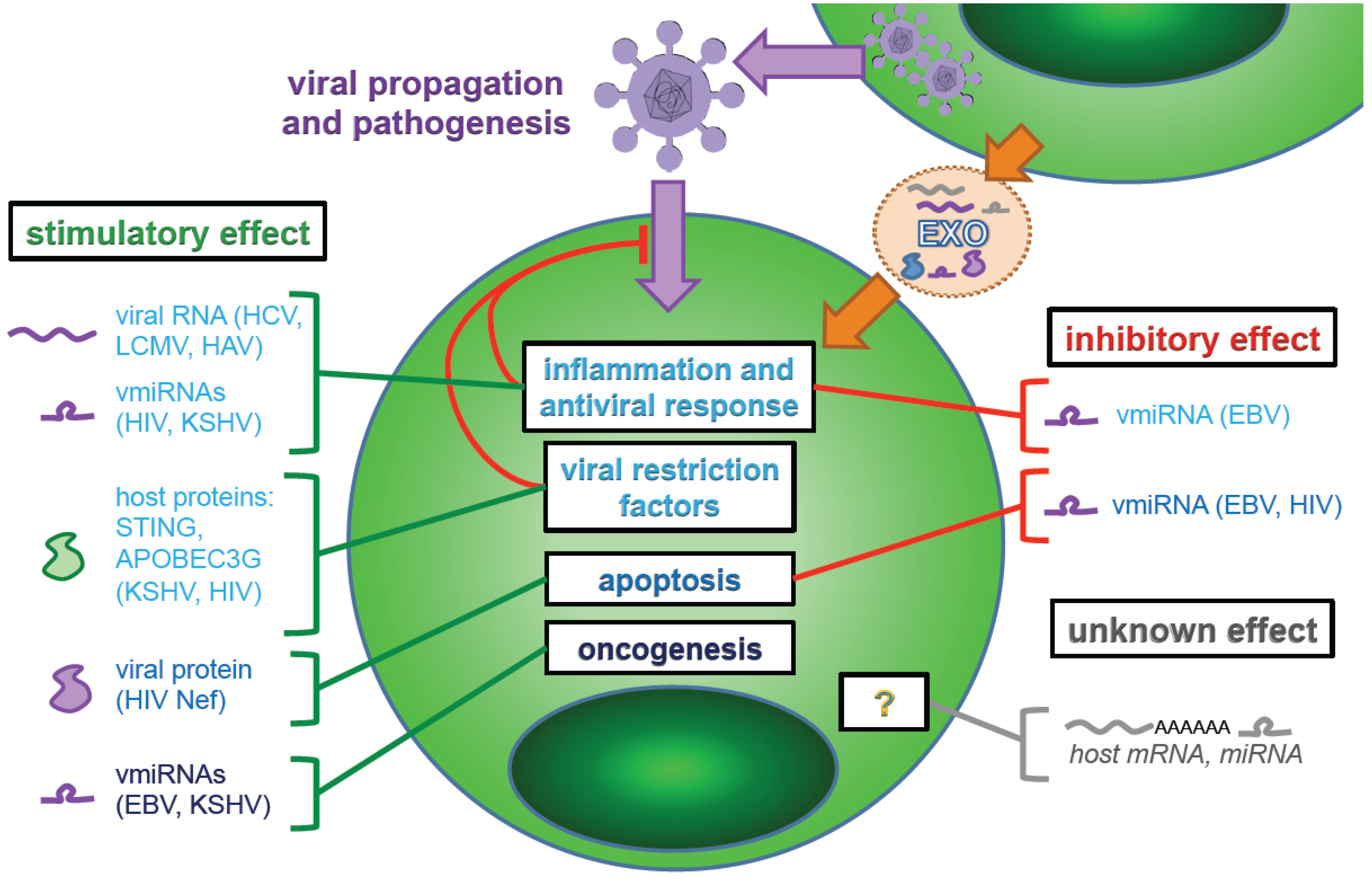
5. Transfer of Regulatory Components by Exosomes
5.1. Virally-Derived MicroRNAs


{kind=link}
{kind=link}
| Virus | Name of Viral miRNA(s) | Source of Exosome-Resident vmiRNAs | vmiRNA Effect | vmiRNA Target(s) | Exosome/Virion Separation Validation | Exosome/Other Microvesicle Separation Validation |
|---|---|---|---|---|---|---|
| EBV | miR-BART15 | Namalwa B cell line | Anti-inflammatory | NLRP3 mRNA [86] | Cells do not produce virions | N.P. |
| AGS gastric cancer cell line | Anti-apoptotic | BRUCE mRNA [95] | N.P. | CD9, CD81 western blot | ||
| BHRF-1-3 | LCL lymphoblastoid B cell line | Anti-inflammatory | CXCL11 mRNA [84] | EM; no large RNAs in exosomes | EM; CD63 western blot | |
| miR-BART3 | NPC nasopharyngeal carcinoma cell line | Anti-inflammatory (N.D.) | IPO7 mRNA (IL-6 inducer) [92,93] | N.P. | Exosome Co-IP with CD9, MHC II | |
| miR-BART1 | NPC nasopharyngeal carcinoma cell line | Immune evasion (N.D) | LMP1 mRNA (viral membrane protein) [93,96] | N.P. | Exosome Co-IP with CD9, MHC II | |
| miR-BART2 | NPC nasopharyngeal carcinoma cell line | Immune evasion (N.D) | MICB mRNA [89,90,93] | N.P. | Exosome Co-IP with CD9, MHC II | |
| NPC nasopharyngeal carcinoma cell line | Latency promotion (N.D) | BALF5 mRNA (viral polymerase) [93,94] | N.P. | Exosome Co-IP with CD9, MHC II | ||
| KSHV | miR-K12-(4-5p, 4-3p, 5, 6-5p, 10a, 11) | Patient serum, pleural fluid | Pro-inflammatory, cell migration | Targets unknown (increased IL-6 expression) [97] | No viral DNA (QPCR); no viral protein (Western blotting); EM | Exosome Co-IP with CD63; EM; Flotillin, HSP90, CD9, CD63 western blot |
| HIV | miR-TAR | Jurkat lymphoblastoid T cell line | Anti-apoptotic, increased susceptibility to HIV-1 infection | Bim/CDK9 mRNA [98] | No viral DNA (QPCR) | EM; CD45, HSP70, β-actin, Alix, CD63 western blot |
| vmiR88, vmiR99 | Macrophage-differentiated THP-1 promonocytic cell line | Pro-inflammatory | Direct TLR8 stimulation (TNF-α release) [99] | N.P. | CD63 western blot |
5.2. Transfer of Host-Derived miRNAs, mRNAs and Proteins by Exosomes
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, D.; Marzesco, A.M.; Wilsch-Brauninger, M.; Huttner, W.B. The intriguing links between prominin-1 (CD133), cholesterol-based membrane microdomains, remodeling of apical plasma membrane protrusions, extracellular membrane particles, and (neuro)epithelial cell differentiation. FEBS Lett. 2010, 584, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.W.; Lichty, B.D.; Bowdish, D.M. Microvesicles: Ubiquitous contributors to infection and immunity. J. Leukoc. Biol. 2015, 97, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.; Levy, M.A.; Stahl, P. Morphological analysis of ligand uptake and processing: The role of multivesicular endosomes and CURL in receptor-ligand processing. Eur. J. Cell Biol. 1985, 36, 230–238. [Google Scholar] [PubMed]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.M.; Fang, Y.; Fallon, J.K.; Yang, J.M.; Hildreth, J.E.; Gould, S.J. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J. Cell Biol. 2006, 172, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Decembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Columba Cabezas, S.; Federico, M. Sequences within RNA coding for HIV-1 Gag p17 are efficiently targeted to exosomes. Cell Microbiol. 2013, 15, 412–429. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Useros, N.; Naranjo-Gomez, M.; Archer, J.; Hatch, S.C.; Erkizia, I.; Blanco, J.; Borras, F.E.; Puertas, M.C.; Connor, J.H.; Fernandez-Figueras, M.T.; et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 2009, 113, 2732–2741. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Useros, N.; Naranjo-Gomez, M.; Erkizia, I.; Puertas, M.C.; Borras, F.E.; Blanco, J.; Martinez-Picado, J. HIV and mature dendritic cells: Trojan exosomes riding the Trojan horse? PLoS Pathog. 2010, 6, e1000740. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014, 10, e1004424. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Boyd, B.; Chisari, F.V. Virion-independent transfer of replication competent HCV RNA between permissive cells. J. Virol. 2014, 89, 2956–2961. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, J.K.; Katz, A.A.; Schafer, G. Dynamic reciprocity: The role of annexin A2 in tissue integrity. J. Cell Commun. Signal. 2014, 8, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Mayran, N.; Parton, R.G.; Gruenberg, J. Annexin II regulates multivesicular endosome biogenesis in the degradation pathway of animal cells. EMBO J. 2003, 22, 3242–3253. [Google Scholar] [CrossRef] [PubMed]
- Grabski, E.; Wappler, I.; Pfaender, S.; Steinmann, E.; Haid, S.; Dzionek, A.; Pietschmann, T.; Kalinke, U. Efficient virus assembly, but not infectivity, determines the magnitude of hepatitis C virus-induced interferon alpha responses of plasmacytoid dendritic cells. J. Virol. 2015, 89, 3200–3208. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; D'Asti, E.; Magnus, N.; Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles as mediators of intercellular communication in cancer—The emerging science of cellular “debris”. Semin. Immunopathol. 2011, 33, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Colombo, M.; Raposo, G.; Thery, C. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic 2011, 12, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M.Z. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Chahar, H.S.; Bao, X.; Casola, A. Exosomes and their role in the life cycle and pathogenesis of RNA viruses. Viruses 2015, 7, 3204–3225. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Cao, W.; Liu, Y.J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.F.; Takahashi, K.; Boyd, B.; Whitten-Bauer, C.; Ngo, N.; de la Torre, J.C.; Chisari, F.V. Human plasmacytoid dendritic cells sense lymphocytic choriomeningitis virus-infected cells in vitro. J. Virol. 2014, 88, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Li, Y.; McKnight, K.L.; Hensley, L.; Lanford, R.E.; Walker, C.M.; Lemon, S.M. Human pDCs preferentially sense enveloped hepatitis A virions. J. Clin. Investig. 2014, 125, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Odorizzi, G. Get on the exosome bus with ALIX. Nat. Cell Biol. 2012, 14, 654–655. [Google Scholar] [CrossRef] [PubMed]
- Florentin, J.; Aouar, B.; Dental, C.; Thumann, C.; Firaguay, G.; Gondois-Rey, F.; Soumelis, V.; Baumert, T.F.; Nunes, J.A.; Olive, D.; et al. HCV glycoprotein E2 is a novel BDCA-2 ligand and acts as an inhibitor of IFN production by plasmacytoid dendritic cells. Blood 2012, 120, 4544–4551. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, Y.; Shi, B.; Zhang, X.; Wang, J.; Zhang, Z.; Shen, F.; Zhang, Q.; Sun, S.; Yuan, Z. HBsAg inhibits TLR9-mediated activation and IFN-α production in plasmacytoid dendritic cells. Mol. Immunol. 2009, 46, 2640–2646. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Asabe, S.; Wieland, S.; Garaigorta, U.; Gastaminza, P.; Isogawa, M.; Chisari, F.V. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. USA 2010, 107, 7431–7436. [Google Scholar] [CrossRef] [PubMed]
- Python, S.; Gerber, M.; Suter, R.; Ruggli, N.; Summerfield, A. Efficient sensing of infected cells in absence of virus particles by plasmacytoid dendritic cells is blocked by the viral ribonuclease E(rns.). PLoS Pathog. 2013, 9, e1003412. [Google Scholar] [CrossRef] [PubMed]
- Decembre, E.; Assil, S.; Hillaire, M.L.; Dejnirattisai, W.; Mongkolsapaya, J.; Screaton, G.R.; Davidson, A.D.; Dreux, M. Sensing of immature particles produced by dengue virus infected cells induces an antiviral response by plasmacytoid dendritic cells. PLoS Pathog. 2014, 10, e1004434. [Google Scholar] [CrossRef] [PubMed]
- Pasquato, A.; Ramos da Palma, J.; Galan, C.; Seidah, N.G.; Kunz, S. Viral envelope glycoprotein processing by proprotein convertases. Antivir. Res. 2013, 99, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Rodenhuis-Zybert, I.A.; van der Schaar, H.M.; da Silva Voorham, J.M.; van der Ende-Metselaar, H.; Lei, H.Y.; Wilschut, J.; Smit, J.M. Immature dengue virus: A veiled pathogen? PLoS Pathog. 2010, 6, e1000718. [Google Scholar] [CrossRef] [PubMed]
- Rodenhuis-Zybert, I.A.; Moesker, B.; da Silva Voorham, J.M.; van der Ende-Metselaar, H.; Diamond, M.S.; Wilschut, J.; Smit, J.M. A fusion-loop antibody enhances the infectious properties of immature flavivirus particles. J. Virol. 2011, 85, 11800–11808. [Google Scholar] [CrossRef] [PubMed]
- Keelapang, P.; Sriburi, R.; Supasa, S.; Panyadee, N.; Songjaeng, A.; Jairungsri, A.; Puttikhunt, C.; Kasinrerk, W.; Malasit, P.; Sittisombut, N. Alterations of pr-M cleavage and virus export in pr-M junction chimeric dengue viruses. J. Virol. 2004, 78, 2367–2381. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; He, R.; Anderson, R. PrM- and cell-binding domains of the dengue virus E protein. J. Virol. 1999, 73, 2547–2551. [Google Scholar] [PubMed]
- Van der Schaar, H.M.; Rust, M.J.; Waarts, B.L.; van der Ende-Metselaar, H.; Kuhn, R.J.; Wilschut, J.; Zhuang, X.; Smit, J.M. Characterization of the early events in dengue virus cell entry by biochemical assays and single-virus tracking. J. Virol. 2007, 81, 12019–12028. [Google Scholar] [CrossRef] [PubMed]
- Junjhon, J.; Lausumpao, M.; Supasa, S.; Noisakran, S.; Songjaeng, A.; Saraithong, P.; Chaichoun, K.; Utaipat, U.; Keelapang, P.; Kanjanahaluethai, A.; et al. Differential modulation of prM cleavage, extracellular particle distribution, and virus infectivity by conserved residues at nonfurin consensus positions of the dengue virus pr-M junction. J. Virol. 2008, 82, 10776–10791. [Google Scholar] [CrossRef] [PubMed]
- Rodenhuis-Zybert, I.A.; Wilschut, J.; Smit, J.M. Partial maturation: An immune-evasion strategy of dengue virus? Trends Microbiol. 2011, 19, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. Degrees of maturity: The complex structure and biology of flaviviruses. Curr. Opin. Virol. 2012, 2, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.M.; Prestwood, T.R.; Shresta, S. Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease. Cell Host Microbe 2010, 7, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Cherrier, M.V.; Kaufmann, B.; Nybakken, G.E.; Lok, S.M.; Warren, J.T.; Chen, B.R.; Nelson, C.A.; Kostyuchenko, V.A.; Holdaway, H.A.; Chipman, P.R.; et al. Structural basis for the preferential recognition of immature flaviviruses by a fusion-loop antibody. EMBO J. 2009, 28, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Jumnainsong, A.; Onsirisakul, N.; Fitton, P.; Vasanawathana, S.; Limpitikul, W.; Puttikhunt, C.; Edwards, C.; Duangchinda, T.; Supasa, S.; et al. Cross-reacting antibodies enhance dengue virus infection in humans. Science 2010, 328, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.Y.; Feng, J.J.; Zhou, J.M.; Yu, Z.Z.; Fang, D.Y.; Yan, H.J.; Zeng, G.C.; Jiang, L.F. Identification of a novel infection-enhancing epitope on dengue prM using a dengue cross-reacting monoclonal antibody. BMC Microbiol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.M.; Moesker, B.; Rodenhuis-Zybert, I.; Wilschut, J. Flavivirus cell entry and membrane fusion. Viruses 2011, 3, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, D.A. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 1999, 258, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Galloway, S.E.; Reed, M.L.; Russell, C.J.; Steinhauer, D.A. Influenza HA subtypes demonstrate divergent phenotypes for cleavage activation and pH of fusion: Implications for host range and adaptation. PLoS Pathog. 2013, 9, e1003151. [Google Scholar] [CrossRef] [PubMed]
- Moesker, B.; Rodenhuis-Zybert, I.A.; Meijerhof, T.; Wilschut, J.; Smit, J.M. Characterization of the functional requirements of West Nile virus membrane fusion. J. Gen. Virol. 2010, 91, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Bruni, D.; Chazal, M.; Sinigaglia, L.; Chauveau, L.; Schwartz, O.; Despres, P.; Jouvenet, N. Viral entry route determines how human plasmacytoid dendritic cells produce type I interferons. Sci. Signal. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Lepelley, A.; Louis, S.; Sourisseau, M.; Law, H.K.; Pothlichet, J.; Schilte, C.; Chaperot, L.; Plumas, J.; Randall, R.E.; Si-Tahar, M.; et al. Innate sensing of HIV-infected cells. PLoS Pathog. 2011, 7, e1001284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megjugorac, N.J.; Jacobs, E.S.; Izaguirre, A.G.; George, T.C.; Gupta, G.; Fitzgerald-Bocarsly, P. Image-based study of interferongenic interactions between plasmacytoid dendritic cells and HSV-infected monocyte-derived dendritic cells. Immunol. Investig. 2007, 36, 739–761. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.T.; Fish, P.M.; Sinha, M.; Owen, D.M.; Lemon, S.M.; Gale, M., Jr. Interferon regulatory factor-3 activation, hepatic interferon-stimulated gene expression, and immune cell infiltration in hepatitis C virus patients. Hepatology 2008, 47, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kodys, K.; Li, K.; Szabo, G. Human type 2 myeloid dendritic cells produce interferon-lambda and amplify interferon-α in response to hepatitis C virus infection. Gastroenterology 2013, 144, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Thitithanyanont, A.; Engering, A.; Ekchariyawat, P.; Wiboon-ut, S.; Limsalakpetch, A.; Yongvanitchit, K.; Kum-Arb, U.; Kanchongkittiphon, W.; Utaisincharoen, P.; Sirisinha, S.; et al. High susceptibility of human dendritic cells to avian influenza H5N1 virus infection and protection by IFN- α and TLR ligands. J. Immunol. 2007, 179, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Baranek, T.; Zucchini, N.; Dalod, M. Plasmacytoid dendritic cells and the control of herpesvirus infections. Viruses 2009, 1, 383–419. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B.; Bunin, A.; Ghosh, H.S.; Lewis, K.L.; Sisirak, V. Plasmacytoid dendritic cells: Recent progress and open questions. Annu. Rev. Immunol. 2011, 29, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Yoshio, S.; Kanto, T.; Kuroda, S.; Matsubara, T.; Higashitani, K.; Kakita, N.; Ishida, H.; Hiramatsu, N.; Nagano, H.; Sugiyama, M.; et al. Human blood dendritic cell antigen 3 (BDCA3)+ dendritic cells are a potent producer of interferon-lambda in response to hepatitis C virus. Hepatology 2013, 57, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Dansako, H.; Yamane, D.; Welsch, C.; McGivern, D.R.; Hu, F.; Kato, N.; Lemon, S.M. Class A scavenger receptor 1 (MSR1) restricts hepatitis C virus replication by mediating toll-like receptor 3 recognition of viral RNAs produced in neighboring cells. PLoS Pathog. 2013, 9, e1003345. [Google Scholar] [CrossRef] [PubMed]
- DeWitte-Orr, S.J.; Collins, S.E.; Bauer, C.M.; Bowdish, D.M.; Mossman, K.L. An accessory to the “Trinity”: SR-As are essential pathogen sensors of extracellular dsRNA, mediating entry and leading to subsequent type I IFN responses. PLoS Pathog. 2010, 6, e1000829. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Varin, A.; Chen, Y.; Liu, B.; Tryggvason, K.; Gordon, S. SR-A/MARCO-mediated ligand delivery enhances intracellular TLR and NLR function, but ligand scavenging from cell surface limits TLR4 response to pathogens. Blood 2011, 117, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Rice, C.M. Structures of hepatitis C virus nonstructural proteins required for replicase assembly and function. Curr. Opin. Virol. 2013, 3, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef] [PubMed]
- Rosen, H.R. Emerging concepts in immunity to hepatitis C virus infection. J. Clin. Investig. 2013, 123, 4121–4130. [Google Scholar] [CrossRef] [PubMed]
- Chattergoon, M.A.; Latanich, R.; Quinn, J.; Winter, M.E.; Buckheit, R.W., 3rd; Blankson, J.N.; Pardoll, D.; Cox, A.L. HIV and HCV activate the inflammasome in monocytes and macrophages via endosomal Toll-like receptors without induction of type 1 interferon. PLoS Pathog. 2014, 10, e1004082. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr. Exosomal communication goes viral. J. Virol. 2015, 89, 5200–5203. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009, 11, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Pressman, S.; Andress, A.P.; Kim, K.; White, J.L.; Cassidy, J.J.; Li, X.; Lubell, K.; Lim do, H.; Cho, I.S.; et al. Silencing by small RNAs is linked to endosomal trafficking. Nat. Cell Biol. 2009, 11, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, S.; Li, L.; Li, M.; Guo, C.; Yao, J.; Mi, S. Exosome and exosomal microRNA: Trafficking, sorting, and function. Genom. Proteom. Bioinf. 2015, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Wurdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; van de Garde, M.D.; Middeldorp, J.M. Viral miRNAs exploiting the endosomal-exosomal pathway for intercellular cross-talk and immune evasion. Biochim. Biophys. Acta 2011, 1809, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.; Masters, S.L. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1β production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [PubMed]
- Rechavi, O.; Erlich, Y.; Amram, H.; Flomenblit, L.; Karginov, F.V.; Goldstein, I.; Hannon, G.J.; Kloog, Y. Cell contact-dependent acquisition of cellular and viral nonautonomously encoded small RNAs. Genes Dev. 2009, 23, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Zomer, A.; Vendrig, T.; Hopmans, E.S.; van Eijndhoven, M.; Middeldorp, J.M.; Pegtel, D.M. Exosomes: Fit to deliver small RNA. Commun. Integr. Biol. 2010, 3, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Lankry, D.; Wolf, D.G.; Mandelboim, O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat. Immunol. 2010, 11, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; To, K.F.; Lo, K.W.; Lung, R.W.; Hui, J.W.; Liao, G.; Hayward, S.D. Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc. Natl. Acad. Sci. USA 2007, 104, 16164–16169. [Google Scholar] [CrossRef] [PubMed]
- Dolken, L.; Malterer, G.; Erhard, F.; Kothe, S.; Friedel, C.C.; Suffert, G.; Marcinowski, L.; Motsch, N.; Barth, S.; Beitzinger, M.; et al. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human γ-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe 2010, 7, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Canitano, A.; Venturi, G.; Borghi, M.; Ammendolia, M.G.; Fais, S. Exosomes released in vitro from Epstein-Barr virus (EBV)-infected cells contain EBV-encoded latent phase mRNAs. Cancer Lett. 2013, 337, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Pfuhl, T.; Mamiani, A.; Ehses, C.; Roemer, K.; Kremmer, E.; Jaker, C.; Hock, J.; Meister, G.; Grasser, F.A. Epstein-Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic Acids Res. 2008, 36, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Lee, H.; Kim, S.R.; Gho, Y.S.; Lee, S.K. Epstein-Barr virus-encoded microRNA BART15-3p promotes cell apoptosis partially by targeting BRUCE. J. Virol. 2013, 87, 8135–8144. [Google Scholar] [CrossRef] [PubMed]
- Antonyak, M.A.; Cerione, R.A. Microvesicles as mediators of intercellular communication in cancer. Methods Mol. Biol. 2014, 1165, 147–173. [Google Scholar] [PubMed]
- Chugh, P.E.; Sin, S.H.; Ozgur, S.; Henry, D.H.; Menezes, P.; Griffith, J.; Eron, J.J.; Damania, B.; Dittmer, D.P. Systemically circulating viral and tumor-derived microRNAs in KSHV-associated malignancies. PLoS Pathog. 2013, 9, e1003484. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Iordanskiy, S.; Das, R.; van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [PubMed]
- Bernard, M.A.; Zhao, H.; Yue, S.C.; Anandaiah, A.; Koziel, H.; Tachado, S.D. Novel HIV-1 miRNAs stimulate TNFα release in human macrophages via TLR8 signaling pathway. PLoS ONE 2014, 9, e106006. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Jones, K.D.; Tosato, G. Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. J. Hematother. Stem Cell Res. 2000, 9, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Molden, J.; Chang, Y.; You, Y.; Moore, P.S.; Goldsmith, M.A. A Kaposi’s sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J. Biol. Chem. 1997, 272, 19625–19631. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, J.L.; Weinerman, B.; Basole, C.; Salazar, J.C. TLR8: The forgotten relative revindicated. Cell. Mol. Immunol. 2012, 9, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Giffin, L.; Yan, F.; Ben Major, M.; Damania, B. Modulation of Kaposi’s sarcoma-associated herpesvirus interleukin-6 function by hypoxia-upregulated protein 1. J. Virol. 2014, 88, 9429–9441. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Jaffe, E.S.; Chang, Y.; Jones, K.; Teruya-Feldstein, J.; Moore, P.S.; Tosato, G. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood 1999, 93, 4034–4043. [Google Scholar] [PubMed]
- Zhao, J.; Punj, V.; Matta, H.; Mazzacurati, L.; Schamus, S.; Yang, Y.; Yang, T.; Hong, Y.; Chaudhary, P.M. K13 blocks KSHV lytic replication and deregulates vIL6 and hIL6 expression: A model of lytic replication induced clonal selection in viral oncogenesis. PLoS ONE 2007, 2, e1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagana, A.; Russo, F.; Veneziano, D.; Bella, S.D.; Giugno, R.; Pulvirenti, A.; Croce, C.M.; Ferro, A. Extracellular circulating viral microRNAs: Current knowledge and perspectives. Front. Genet. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Aliotta, J.M.; Asara, J.M.; Tucker, L.; Quesenberry, P.; Lally, M.; Ramratnam, B. Quantitative proteomic analysis of exosomes from HIV-1-infected lymphocytic cells. Proteomics 2012, 12, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Gunawardena, H.P.; Dekroon, R.M.; Heaton, P.R.; Edwards, R.H.; Ozgur, S.; Griffith, J.D.; Damania, B.; Raab-Traub, N. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proc. Natl. Acad. Sci. USA 2013, 110, E2925–E2933. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, Y.; Duan, J.; Ma, Y.; Shen, Z.; Wei, L.; Cui, X.; Zhang, J.; Xie, Y.; Liu, J. Quantitative proteomic analysis of exosome protein content changes induced by hepatitis B virus in Huh-7 cells using SILAC labeling and LC-MS/MS. J. Proteome Res. 2014, 13, 5391–5402. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Du, T.; Roizman, B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc. Natl. Acad. Sci. USA 2014, 111, E4991–E4996. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Roizman, B. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc. Natl. Acad. Sci. USA 2014, 111, E611–E617. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Vance, R.E. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 2013, 14, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Sandri-Goldin, R.M. Initiation of transcription and RNA synthesis, processing and transport in HSV and VZV infected cells. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Knipe, D.M.; Howley, P.M. Fields Virology, 6th ed.; Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2013; pp. 1823–1897. [Google Scholar]
- Zhu, X.; He, Z.; Yuan, J.; Wen, W.; Huang, X.; Hu, Y.; Lin, C.; Pan, J.; Li, R.; Deng, H.; et al. IFITM3-containing exosome as a novel mediator for anti-viral response in dengue virus infection. Cell. Microbiol. 2015, 17, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Khatua, A.K.; Taylor, H.E.; Hildreth, J.E.; Popik, W. Exosomes packaging APOBEC3G confer human immunodeficiency virus resistance to recipient cells. J. Virol. 2009, 83, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Belanger, K.; Savoie, M.; Rosales Gerpe, M.C.; Couture, J.F.; Langlois, M.A. Binding of RNA by APOBEC3G controls deamination-independent restriction of retroviruses. Nucleic Acids Res. 2013, 41, 7438–7452. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.N.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005, 15, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, K.; Liu, Y.; Xu, Y.; Zhang, F.; Yang, H.; Liu, J.; Pan, T.; Chen, J.; Wu, M.; et al. Exosomes mediate the cell-to-cell transmission of IFN-α-induced antiviral activity. Nat. Immunol. 2013, 14, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Loveday, E.K.; Svinti, V.; Diederich, S.; Pasick, J.; Jean, F. Temporal- and strain-specific host microRNA molecular signatures associated with swine-origin H1N1 and avian-origin H7N7 influenza A virus infection. J. Virol. 2012, 86, 6109–6122. [Google Scholar] [CrossRef] [PubMed]
- Honegger, A.; Schilling, D.; Bastian, S.; Sponagel, J.; Kuryshev, V.; Sultmann, H.; Scheffner, M.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Dependence of intracellular and exosomal microRNAs on viral E6/E7 oncogene expression in HPV-positive tumor cells. PLoS Pathog. 2015, 11, e1004712. [Google Scholar] [CrossRef] [PubMed]
- Madison, M.N.; Roller, R.J.; Okeoma, C.M. Human semen contains exosomes with potent anti-HIV-1 activity. Retrovirology 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Madison, M.N.; Jones, P.H.; Okeoma, C.M. Exosomes in human semen restrict HIV-1 transmission by vaginal cells and block intravaginal replication of LP-BM5 murine AIDS virus complex. Virology 2015, 482, 189–201. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Assil, S.; Webster, B.; Dreux, M. Regulation of the Host Antiviral State by Intercellular Communications. Viruses 2015, 7, 4707-4733. https://doi.org/10.3390/v7082840
Assil S, Webster B, Dreux M. Regulation of the Host Antiviral State by Intercellular Communications. Viruses. 2015; 7(8):4707-4733. https://doi.org/10.3390/v7082840
Chicago/Turabian StyleAssil, Sonia, Brian Webster, and Marlène Dreux. 2015. "Regulation of the Host Antiviral State by Intercellular Communications" Viruses 7, no. 8: 4707-4733. https://doi.org/10.3390/v7082840