
Two Cytoplasmic Acylation Sites and an Adjacent Hydrophobic Residue, but No Other Conserved Amino Acids in the Cytoplasmic Tail of HA from Influenza A Virus Are Crucial for Virus Replication

 and
and
Abstract
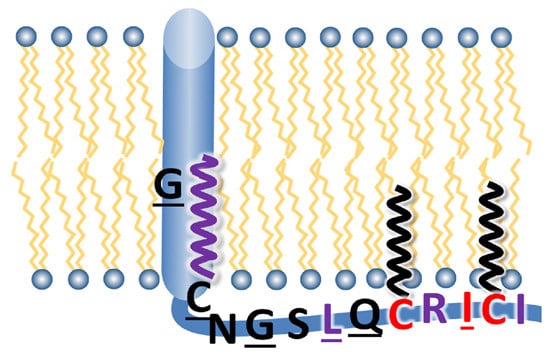
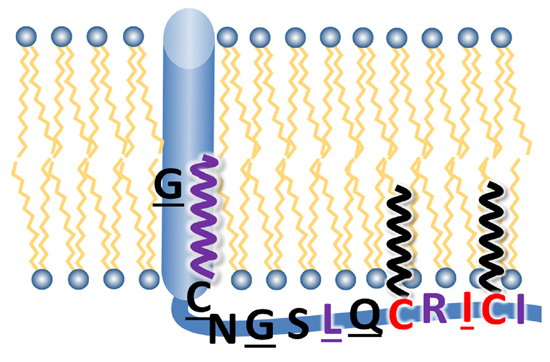
:
1. Introduction
2. Materials and Methods
2.1. Amino Acid Conservation Analysis
2.2. Cells
2.3. Generation of Recombinant Virus
2.4. Sequencing of HA from Virus Particles
2.5. Plaque Assay
2.6. Hemagglutination Assay
2.7. Electron Microscopy
2.8. Growth Kinetics of Recombinant Virus
2.9. Virus Preparation, SDS-PAGE, Western Blotting and Radioactive Labeling
2.10. Quantitative Real-Time RT-PCR
3. Results
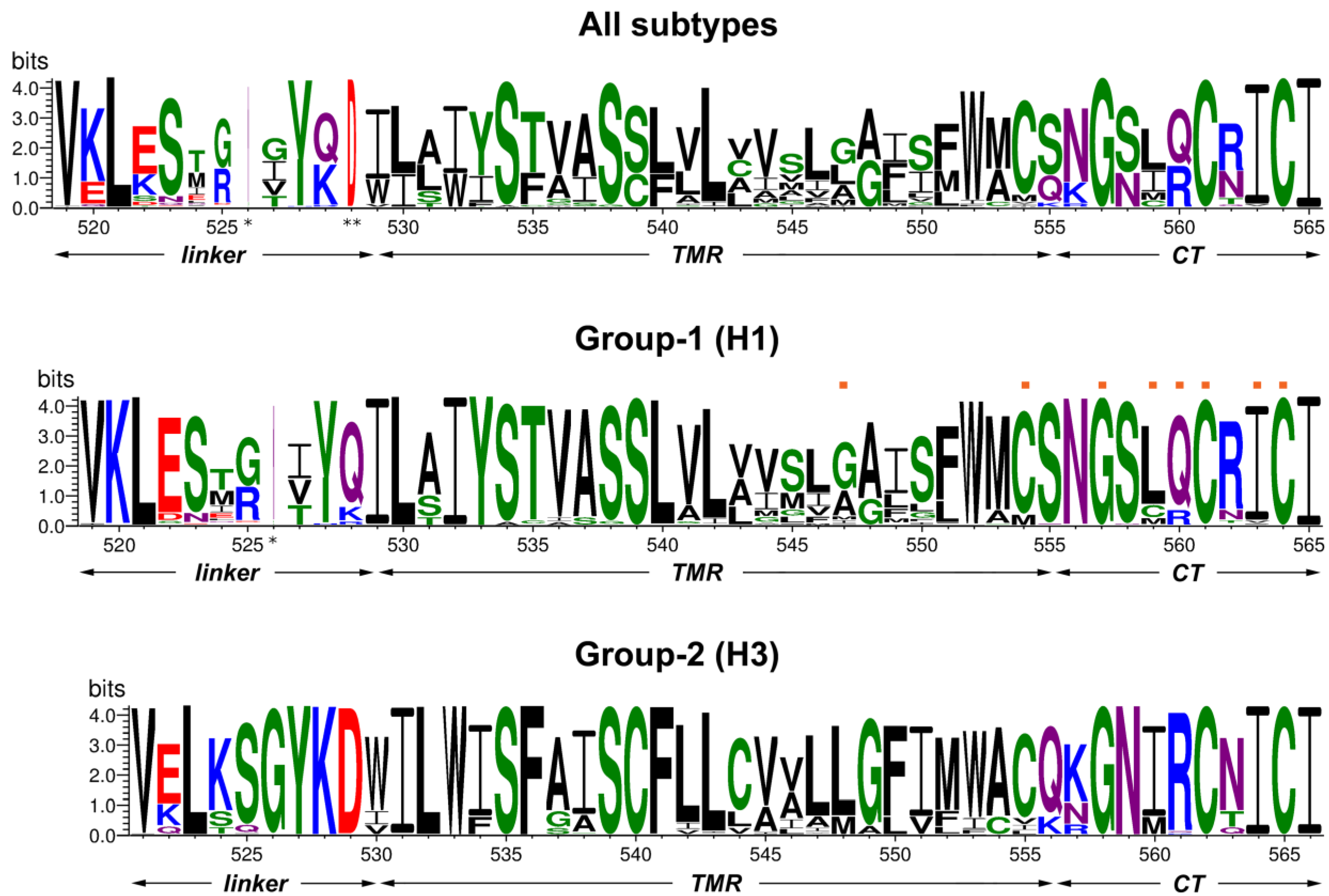
3.1. Sequence Comparison of the Linker Region, Transmembrane Domain and Cytoplasmic Tail between HA Subtypes

3.2. Mutation of Cytoplasmic Palmitoylation Sites and Non-Conservative Substitution of C-Terminal Isoleucine Prevent Virus Rescue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutants | Transmembrane | Cytoplasmic Tail | Virus |
|---|---|---|---|
| Wild type | LGAISFWMS | NGSLQRII | + |
| G557Stop | LGAISFWMS | N * | − |
| Ac2 (C561S) | LGAISFWMS | NGSLQSRII * | − |
| I563Q | LGAISFWMS | NGSLQRQI * | − |
| Ac3 (C564S) | LGAISFWMS | NGSLQRISI * | − |
| G547S | LSAISFWMS | NGSLQRII * | + |
| Ac1 (C554S) | LGAISFWMSS | NGSLQRII * | + |
| Ac1+L559C | LGAISFWMSS | NGSCQRII * | + |
| G557A | LGAISFWMS | NASLQRII * | + |
| G557E | LGAISFWMS | NESLQRII * | + |
| Q560E | LGAISFWMS | NGSLERII * | + |
| I563L | LGAISFWMS | NGSLQRLI * | + |
3.3. Reversion at Three Codons Causing Amino Acid Exchanges during Amplification of Recombinant Virus

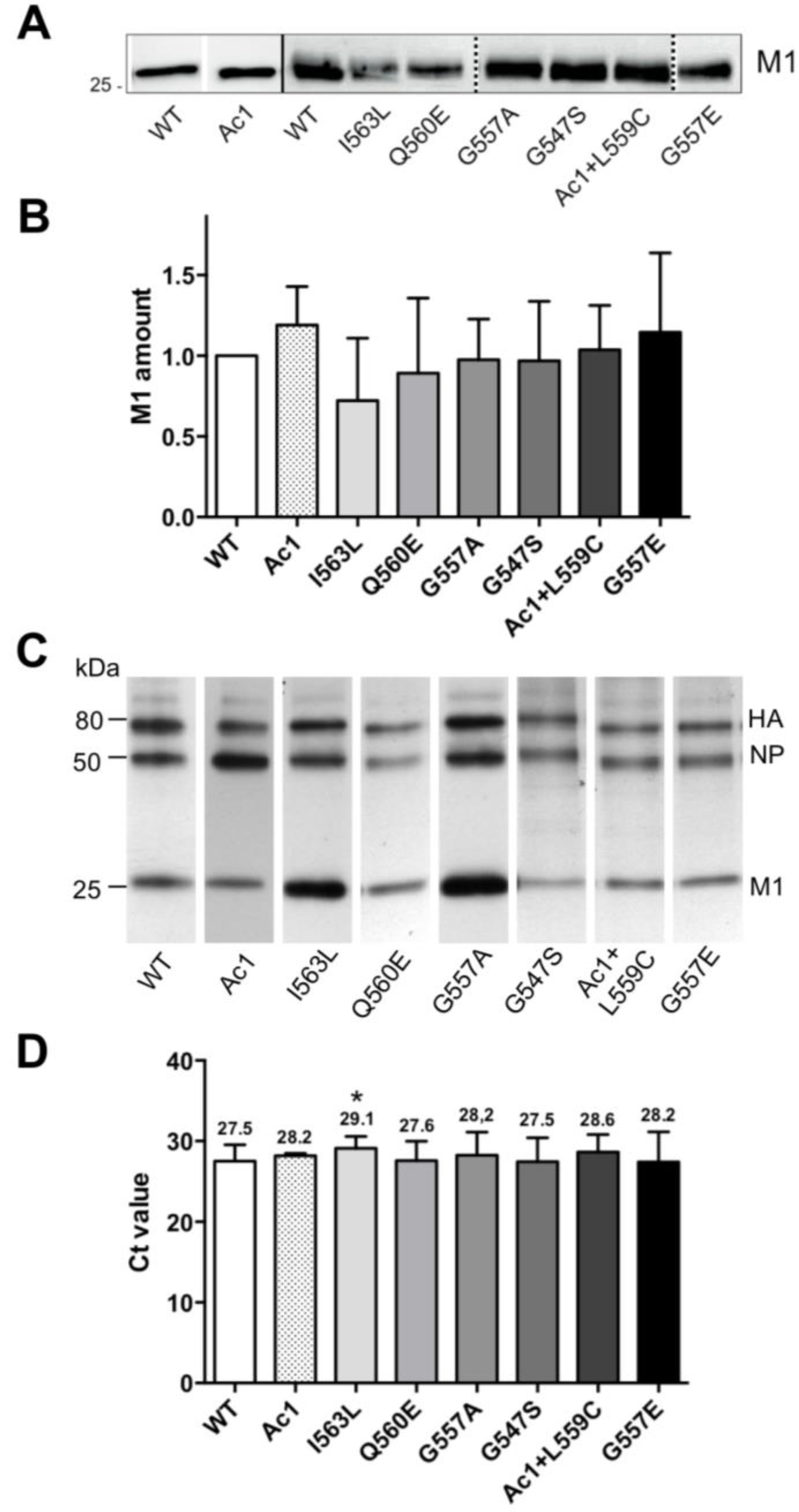
3.4. Growth Kinetics of Recombinant Viruses
 1.5 logs. Calculating the plaque forming unit (PFU) to HA titer ratio revealed that the relative infectivity of the mutants was reduced to 0.8 (G547S), 0.7 (G557A), 0.2 (AC1) and 0.01 (G557E) relative to wild type virus (1.0). The other mutants showed (almost) identical titers to wild type virus, no reduction in specific infectivity (I563L and Q560E = 1.8); and no difference in plaque size was obvious between these mutants and wild type WSN.
1.5 logs. Calculating the plaque forming unit (PFU) to HA titer ratio revealed that the relative infectivity of the mutants was reduced to 0.8 (G547S), 0.7 (G557A), 0.2 (AC1) and 0.01 (G557E) relative to wild type virus (1.0). The other mutants showed (almost) identical titers to wild type virus, no reduction in specific infectivity (I563L and Q560E = 1.8); and no difference in plaque size was obvious between these mutants and wild type WSN.

4. Discussion


Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barman, S.; Ali, A.; Hui, E.K.; Adhikary, L.; Nayak, D.P. Transport of viral proteins to the apical membranes and interaction of matrix protein with glycoproteins in the assembly of influenza viruses. Virus Res. 2001, 77, 61–69. [Google Scholar] [CrossRef]
- Nayak, D.P.; Hui, E.K.; Barman, S. Assembly and budding of influenza virus. Virus Res. 2004, 106, 147–165. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Lamb, R.A. Influenza virus assembly and budding. Virology 2011, 411, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Thaa, B.; Herrmann, A.; Veit, M. The polybasic region is not essential for membrane binding of the matrix protein M1 of influenza virus. Virology 2009, 383, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Enami, M.; Enami, K. Influenza virus hemagglutinin and neuraminidase glycoproteins stimulate the membrane association of the matrix protein. J. Virol. 1996, 70, 6653–6657. [Google Scholar] [PubMed]
- Kretzschmar, E.; Bui, M.; Rose, J.K. Membrane association of influenza virus matrix protein does not require specific hydrophobic domains or the viral glycoproteins. Virology 1996, 220, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lamb, R.A. Characterization of the membrane association of the influenza virus matrix protein in living cells. Virology 1996, 225, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Avalos, R.T.; Ponimaskin, E.; Nayak, D.P. Influenza virus assembly: Effect of influenza virus glycoproteins on the membrane association of m1 protein. J. Virol. 2000, 74, 8709–8719. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Leser, G.P.; Morita, E.; Lamb, R.A. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 2007, 81, 7111–7123. [Google Scholar] [CrossRef] [PubMed]
- Naeve, C.W.; Williams, D. Fatty acids on the a/Japan/305/57 influenza virus hemagglutinin have a role in membrane fusion. EMBO J. 1990, 9, 3857–3866. [Google Scholar] [PubMed]
- Veit, M.; Kretzschmar, E.; Kuroda, K.; Garten, W.; Schmidt, M.F.; Klenk, H.D.; Rott, R. Site-specific mutagenesis identifies three cysteine residues in the cytoplasmic tail as acylation sites of influenza virus hemagglutinin. J. Virol. 1991, 65, 2491–2500. [Google Scholar] [PubMed]
- Kordyukova, L.V.; Serebryakova, M.V.; Baratova, L.A.; Veit, M. S acylation of the hemagglutinin of influenza viruses: Mass spectrometry reveals site-specific attachment of stearic acid to a transmembrane cysteine. J. Virol. 2008, 82, 9288–9292. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Takeda, M.; Lamb, R.A. Influenza virus hemagglutinin (H3 subtype) requires palmitoylation of its cytoplasmic tail for assembly: M1 proteins of two subtypes differ in their ability to support assembly. J. Virol. 2005, 79, 13673–13684. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.; Herwig, A.; Azzouz, N.; Klenk, H.D. Acylation-mediated membrane anchoring of avian influenza virus hemagglutinin is essential for fusion pore formation and virus infectivity. J. Virol. 2005, 79, 6449–6458. [Google Scholar] [CrossRef] [PubMed]
- Zurcher, T.; Luo, G.; Palese, P. Mutations at palmitylation sites of the influenza virus hemagglutinin affect virus formation. J. Virol. 1994, 68, 5748–5754. [Google Scholar] [PubMed]
- Engel, S.; Scolari, S.; Thaa, B.; Krebs, N.; Korte, T.; Herrmann, A.; Veit, M. FLIM-FRET and FRAP reveal association of influenza virus haemagglutinin with membrane rafts. Biochem. J. 2010, 425, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.T.; Gould, T.J.; Gudheti, M.V.; Maas, S.A.; Mills, K.D.; Zimmerberg, J. Dynamic clustered distribution of hemagglutinin resolved at 40 nm in living cell membranes discriminates between raft theories. Proc. Natl. Acad. Sci. USA 2007, 104, 17370–17375. [Google Scholar] [CrossRef] [PubMed]
- Leser, G.P.; Lamb, R.A. Influenza virus assembly and budding in raft-derived microdomains: A quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology 2005, 342, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Veit, M.; Serebryakova, M.V.; Kordyukova, L.V. Palmitoylation of influenza virus proteins. Biochem. Soc. Trans. 2013, 41, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Leser, G.P.; Lamb, R.A. The influenza virus hemagglutinin cytoplasmic tail is not essential for virus assembly or infectivity. EMBO J. 1994, 13, 5504–5515. [Google Scholar] [PubMed]
- Jin, H.; Leser, G.P.; Zhang, J.; Lamb, R.A. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 1997, 16, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Brett, K.; Kordyukova, L.V.; Serebryakova, M.V.; Mintaev, R.R.; Alexeevski, A.V.; Veit, M. Site-specific S-acylation of influenza virus hemagglutinin: The location of the acylation site relative to the membrane border is the decisive factor for attachment of stearate. J. Biol. Chem. 2014, 289, 34978–34989. [Google Scholar] [CrossRef] [PubMed]
- Influenza Research Database. Available online: http://www.fludb.org/ (accessed on 20 March 2014).
- MAFFT multiple sequence alignment program. Available online: http://align.bmr.kyushu-u.ac.jp/mafft/software/ (accessed on 12 April 2014). moved to http://mafft.cbrc.jp/alignment/server/ (MAFFT version 7).
- Katoh, K.; Toh, H. Recent developments in the mafft multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- WebLogo 3. Available online: http://weblogo.threeplusone.com/ (accessed on 18 November 2014).
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Neumann, G.; Kawaoka, Y.; Hobom, G.; Webster, R.G. A DNA transfection system for generation of influenza a virus from eight plasmids. Proc. Natl. Acad. Sci. USA 2000, 97, 6108–6113. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.; Chen, L.M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza a virus from bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza a viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [PubMed]
- Brand, C.M.; Skehel, J.J. Crystalline antigen from the influenza virus envelope. Nat. New Biol. 1972, 238, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Serebryakova, M.V.; Kordyukova, L.V.; Semashko, T.A.; Ksenofontov, A.L.; Rudneva, I.A.; Kropotkina, E.A.; Filippova, I.Y.; Veit, M.; Baratova, L.A. Influenza virus hemagglutinin spike neck architectures and interaction with model enzymes evaluated by MALDI-TOF mass spectrometry and bioinformatics tools. Virus Res. 2011, 160, 294–304. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.; Herrmann, A.; Veit, M. A cholesterol consensus motif is required for efficient intracellular transport and raft association of a group 2 HA from influenza virus. Biochem. J. 2015, 465, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Kordyukova, L.V.; Serebryakova, M.V.; Polyansky, A.A.; Kropotkina, E.A.; Alexeevski, A.V.; Veit, M.; Efremov, R.G.; Filippova, I.Y.; Baratova, L.A. Linker and/or transmembrane regions of influenza A/group-1, A/group-2, and type B virus hemagglutinins are packed differently within trimers. Biochim. Biophys. Acta 2011, 1808, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza A virus ribonucleoproteins. Nat. Rev. Microbiol. 2015, 13, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Watanabe, S.; Kawaoka, Y. The cytoplasmic tail domain of influenza b virus hemagglutinin is important for its incorporation into virions but is not essential for virus replication in cell culture in the presence of compensatory mutations. J. Virol. 2012, 86, 11633–11644. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martinez-Alonso, M.; Fodor, E. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 2014, 5, 4816. [Google Scholar] [CrossRef] [PubMed]
- Noda, T. Native morphology of influenza virions. Front. Microbiol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Mineev, K.S.; Lyukmanova, E.N.; Krabben, L.; Serebryakova, M.V.; Shulepko, M.A.; Arseniev, A.S.; Kordyukova, L.V.; Veit, M. Structural investigation of influenza virus hemagglutinin membrane-anchoring peptide. Protein Eng. Des. Sel. 2013, 26, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Veit, M. Palmitoylation of virus proteins. Biol. Cell. 2012, 104, 493–515. [Google Scholar] [CrossRef] [PubMed]
- Javadpour, M.M.; Eilers, M.; Groesbeek, M.; Smith, S.O. Helix packing in polytopic membrane proteins: Role of glycine in transmembrane helix association. Biophys. J. 1999, 77, 1609–1618. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell. Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Bucher, D.J.; Kharitonenkov, I.G.; Zakomirdin, J.A.; Grigoriev, V.B.; Klimenko, S.M.; Davis, J.F. Incorporation of influenza virus M-protein into liposomes. J. Virol. 1980, 36, 586–590. [Google Scholar] [PubMed]
- Gregoriades, A.; Frangione, B. Insertion of influenza m protein into the viral lipid bilayer and localization of site of insertion. J. Virol. 1981, 40, 323–328. [Google Scholar] [PubMed]
- Ruigrok, R.W.; Barge, A.; Durrer, P.; Brunner, J.; Ma, K.; Whittaker, G.R. Membrane interaction of influenza virus M1 protein. Virology 2000, 267, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Chlanda, P.; Schraidt, O.; Kummer, S.; Riches, J.; Oberwinkler, H.; Prinz, S.; Krausslich, H.G.; Briggs, J.A. Structural analysis of the roles of influenza a virus membrane-associated proteins in assembly and morphology. J. Virol. 2015, 89, 8957–8966. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siche, S.; Brett, K.; Möller, L.; Kordyukova, L.V.; Mintaev, R.R.; Alexeevski, A.V.; Veit, M. Two Cytoplasmic Acylation Sites and an Adjacent Hydrophobic Residue, but No Other Conserved Amino Acids in the Cytoplasmic Tail of HA from Influenza A Virus Are Crucial for Virus Replication. Viruses 2015, 7, 6458-6475. https://doi.org/10.3390/v7122950
Siche S, Brett K, Möller L, Kordyukova LV, Mintaev RR, Alexeevski AV, Veit M. Two Cytoplasmic Acylation Sites and an Adjacent Hydrophobic Residue, but No Other Conserved Amino Acids in the Cytoplasmic Tail of HA from Influenza A Virus Are Crucial for Virus Replication. Viruses. 2015; 7(12):6458-6475. https://doi.org/10.3390/v7122950
Chicago/Turabian StyleSiche, Stefanie, Katharina Brett, Lars Möller, Larisa V. Kordyukova, Ramil R. Mintaev, Andrei V. Alexeevski, and Michael Veit. 2015. "Two Cytoplasmic Acylation Sites and an Adjacent Hydrophobic Residue, but No Other Conserved Amino Acids in the Cytoplasmic Tail of HA from Influenza A Virus Are Crucial for Virus Replication" Viruses 7, no. 12: 6458-6475. https://doi.org/10.3390/v7122950