
U-CAN-seq: A Universal Competition Assay by Nanopore Sequencing
 , , , and
, , , and
Abstract
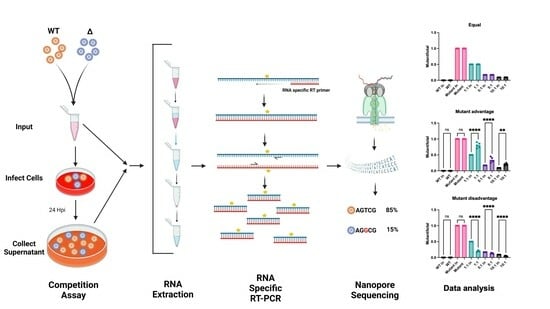
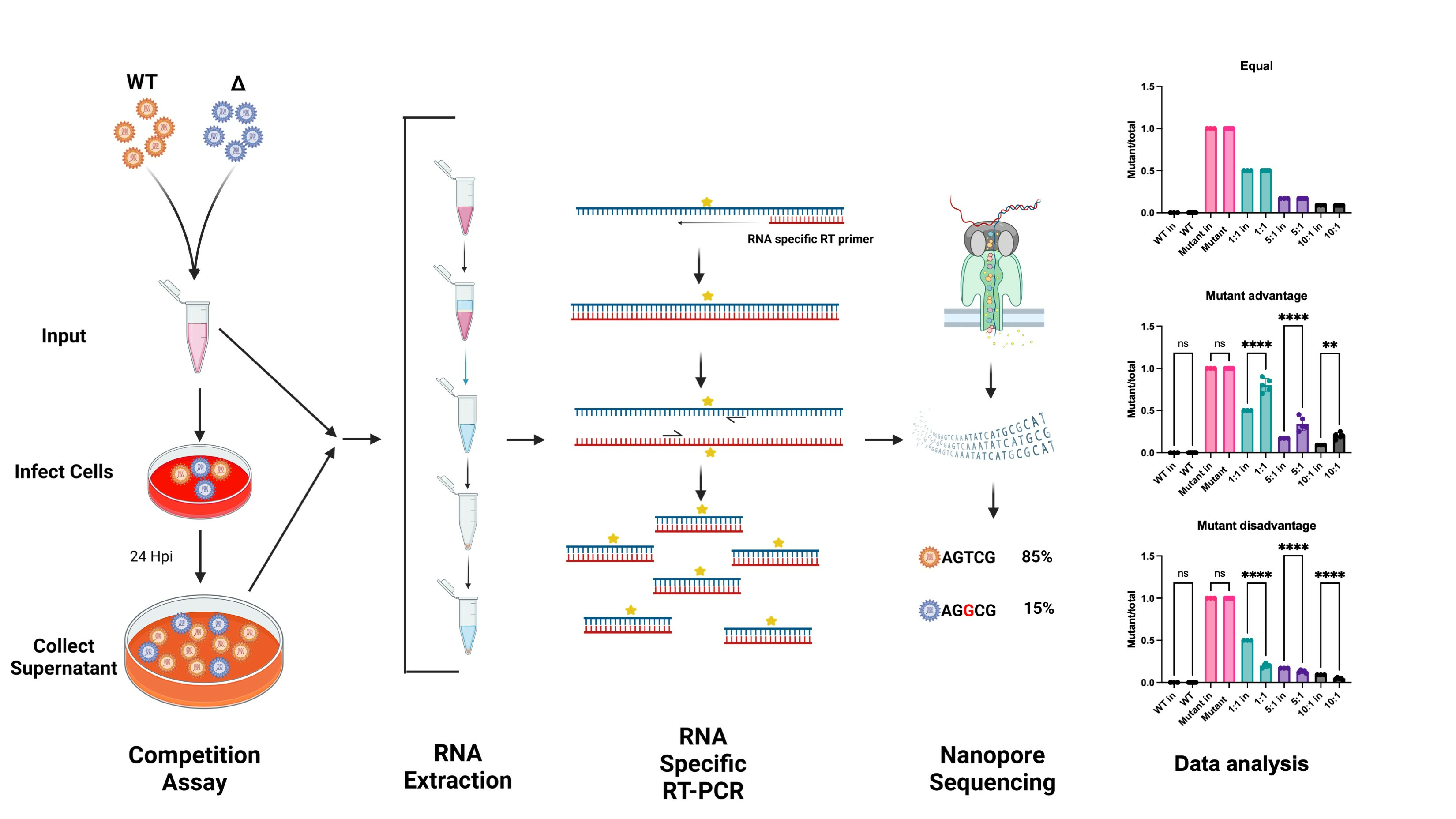
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viruses
2.2. Growth Curves
2.3. Competition Assays
3. Results
3.1. U-CAN-seq Detects a Previously Described Growth Defect in the CHIKV nsp3 N24A Genotype
3.2. U-CAN-seq Can Detect Previously Unrecognized Fitness Differences Due to Mutations in an RNA Structure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Domingo, E.; García-Crespo, C.; Lobo-Vega, R.; Perales, C. Mutation Rates, Mutation Frequencies, and Proofreading-Repair Activities in RNA Virus Genetics. Viruses 2021, 13, 1882. [Google Scholar] [CrossRef]
- Mandary, M.B.; Masomian, M.; Poh, C.L. Impact of RNA Virus Evolution on Quasispecies Formation and Virulence. Int. J. Mol. Sci. 2019, 20, 4657. [Google Scholar] [CrossRef]
- Szemiel, A.M.; Merits, A.; Orton, R.J.; MacLean, O.A.; Pinto, R.M.; Wickenhagen, A.; Lieber, G.; Turnbull, M.L.; Wang, S.; Furnon, W.; et al. In Vitro Selection of Remdesivir Resistance Suggests Evolutionary Predictability of SARS-CoV-2. PLoS Pathog. 2021, 17, e1009929. [Google Scholar] [CrossRef]
- Prado, J.G.; Franco, S.; Matamoros, T.; Ruiz, L.; Clotet, B.; Menéndez-Arias, L.; Martínez, M.Á.; Martinez-Picado, J. Relative Replication Fitness of Multi-Nucleoside Analogue-Resistant HIV-1 Strains Bearing a Dipeptide Insertion in the Fingers Subdomain of the Reverse Transcriptase and Mutations at Codons 67 and 215. Virology 2004, 326, 103–112. [Google Scholar] [CrossRef]
- Collins, J.A.; Thompson, M.G.; Paintsil, E.; Ricketts, M.; Gedzior, J.; Alexander, L. Competitive Fitness of Nevirapine-Resistant Human Immunodeficiency Virus Type 1 Mutants. J. Virol. 2004, 78, 603–611. [Google Scholar] [CrossRef]
- Lee, L.Y.; Zhou, J.; Koszalka, P.; Frise, R.; Farrukee, R.; Baba, K.; Miah, S.; Shishido, T.; Galiano, M.; Hashimoto, T.; et al. Evaluating the Fitness of PA/I38T-Substituted Influenza A Viruses with Reduced Baloxavir Susceptibility in a Competitive Mixtures Ferret Model. PLoS Pathog. 2021, 17, e1009527. [Google Scholar] [CrossRef]
- Carrasco, P.; Daròs, J.A.; Agudelo-Romero, P.; Elena, S.F. A Real-Time RT-PCR Assay for Quantifying the Fitness of Tobacco Etch Virus in Competition Experiments. J. Virol. Methods 2007, 139, 181–188. [Google Scholar] [CrossRef]
- Manocheewa, S.; Lanxon-Cookson, E.C.; Liu, Y.; Swain, J.V.; McClure, J.; Rao, U.; Maust, B.; Deng, W.; Sunshine, J.E.; Kim, M.; et al. Pairwise Growth Competition Assay for Determining the Replication Fitness of Human Immunodeficiency Viruses. JoVE J. Vis. Exp. 2015, 99, e52610. [Google Scholar] [CrossRef]
- Neumann, T.; Hagmann, I.; Lohrengel, S.; Heil, M.L.; Derdeyn, C.A.; Kräusslich, H.-G.; Dittmar, M.T. T20-Insensitive HIV-1 from Naïve Patients Exhibits High Viral Fitness in a Novel Dual-Color Competition Assay on Primary Cells. Virology 2005, 333, 251–262. [Google Scholar] [CrossRef]
- Petrie, S.M.; Butler, J.; Barr, I.G.; McVernon, J.; Hurt, A.C.; McCaw, J.M. Quantifying Relative Within-Host Replication Fitness in Influenza Virus Competition Experiments. J. Theor. Biol. 2015, 382, 259–271. [Google Scholar] [CrossRef]
- Baker, C.; Shi, P.-Y. Construction of Stable Reporter Flaviviruses and Their Applications. Viruses 2020, 12, 1082. [Google Scholar] [CrossRef]
- Kanai, Y.; Kawagishi, T.; Nouda, R.; Onishi, M.; Pannacha, P.; Nurdin, J.A.; Nomura, K.; Matsuura, Y.; Kobayashi, T. Development of Stable Rotavirus Reporter Expression Systems. J. Virol. 2019, 93, e01774-18. [Google Scholar] [CrossRef]
- Ren, L.; Peng, Z.; Chen, X.; Ouyang, H. Live Cell Reporter Systems for Positive-Sense Single Strand RNA Viruses. Appl. Biochem. Biotechnol. 2016, 178, 1567–1585. [Google Scholar] [CrossRef]
- Suphatrakul, A.; Duangchinda, T.; Jupatanakul, N.; Prasittisa, K.; Onnome, S.; Pengon, J.; Siridechadilok, B. Multi-Color Fluorescent Reporter Dengue Viruses with Improved Stability for Analysis of a Multi-Virus Infection. PLoS ONE 2018, 13, e0194399. [Google Scholar] [CrossRef]
- Todd, E.V.; Black, M.A.; Gemmell, N.J. The Power and Promise of RNA-Seq in Ecology and Evolution. Mol. Ecol. 2016, 25, 1224–1241. [Google Scholar] [CrossRef]
- Takele Assefa, A.; Vandesompele, J.; Thas, O. On the Utility of RNA Sample Pooling to Optimize Cost and Statistical Power in RNA Sequencing Experiments. BMC Genom. 2020, 21, 312. [Google Scholar] [CrossRef]
- Arora, S.; Pattwell, S.S.; Holland, E.C.; Bolouri, H. Variability in Estimated Gene Expression among Commonly Used RNA-Seq Pipelines. Sci. Rep. 2020, 10, 2734. [Google Scholar] [CrossRef]
- Madden, E.A.; Plante, K.S.; Morrison, C.R.; Kutchko, K.M.; Sanders, W.; Long, K.M.; Taft-Benz, S.; Cruz Cisneros, M.C.; White, A.M.; Sarkar, S.; et al. Using SHAPE-MaP To Model RNA Secondary Structure and Identify 3′UTR Variation in Chikungunya Virus. J. Virol. 2020, 94, e00701-20. [Google Scholar] [CrossRef]
- Eckei, L.; Krieg, S.; Bütepage, M.; Lehmann, A.; Gross, A.; Lippok, B.; Grimm, A.R.; Kümmerer, B.M.; Rossetti, G.; Lüscher, B.; et al. The Conserved Macrodomains of the Non-Structural Proteins of Chikungunya Virus and Other Pathogenic Positive Strand RNA Viruses Function as Mono-ADP-Ribosylhydrolases. Sci. Rep. 2017, 7, 41746. [Google Scholar] [CrossRef]
- Gadea, G.; Bos, S.; Krejbich-Trotot, P.; Clain, E.; Viranaicken, W.; El-Kalamouni, C.; Mavingui, P.; Desprès, P. A Robust Method for the Rapid Generation of Recombinant Zika Virus Expressing the GFP Reporter Gene. Virology 2016, 497, 157–162. [Google Scholar] [CrossRef]
- Funk, A.; Truong, K.; Nagasaki, T.; Torres, S.; Floden, N.; Balmori Melian, E.; Edmonds, J.; Dong, H.; Shi, P.-Y.; Khromykh, A.A. RNA Structures Required for Production of Subgenomic Flavivirus RNA. J. Virol. 2010, 84, 11407–11417. [Google Scholar] [CrossRef]
- Jaafar, Z.A.; Kieft, J.S. Viral RNA Structure-Based Strategies to Manipulate Translation. Nat. Rev. Microbiol. 2019, 17, 110–123. [Google Scholar] [CrossRef]
- Robertson, M.P.; Igel, H.; Baertsch, R.; Haussler, D.; Ares Jr, M.; Scott, W.G. The Structure of a Rigorously Conserved RNA Element within the SARS Virus Genome. PLoS Biol. 2004, 3, e5. [Google Scholar] [CrossRef]
- Simmonds, P.; Smith, D.B. Structural Constraints on RNA Virus Evolution. J. Virol. 1999, 73, 5787–5794. [Google Scholar] [CrossRef]
- Ranasinghe, D.; Jayadas, T.T.P.; Jayathilaka, D.; Jeewandara, C.; Dissanayake, O.; Guruge, D.; Ariyaratne, D.; Gunasinghe, D.; Gomes, L.; Wijesinghe, A.; et al. Comparison of Different Sequencing Techniques for Identification of SARS-CoV-2 Variants of Concern with Multiplex Real-Time PCR. PLoS ONE 2022, 17, e0265220. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Zheng, P.; Zhou, C.; Ding, Y.; Liu, B.; Lu, L.; Zhu, F.; Duan, S. Nanopore Sequencing Technology and Its Applications. MedComm 2023, 4, e316. [Google Scholar] [CrossRef]
- Abraham, R.; McPherson, R.L.; Dasovich, M.; Badiee, M.; Leung, A.K.L.; Griffin, D.E. Both ADP-Ribosyl-Binding and Hydrolase Activities of the Alphavirus nsP3 Macrodomain Affect Neurovirulence in Mice. mBio 2020, 11, e03253-19. [Google Scholar] [CrossRef]
- Park, E.; Griffin, D.E. The nsP3 Macro Domain Is Important for Sindbis Virus Replication in Neurons and Neurovirulence in Mice. Virology 2009, 388, 305–314. [Google Scholar] [CrossRef]
- Zhang, S.; Garzan, A.; Haese, N.; Bostwick, R.; Martinez-Gzegozewska, Y.; Rasmussen, L.; Streblow, D.N.; Heise, M.T.; Pathak, A.K.; Augelli-Szafran, C.E.; et al. Pyrimidone Inhibitors Targeting Chikungunya Virus nsP3 Macrodomain by Fragment-Based Drug Design. PLoS ONE 2021, 16, e0245013. [Google Scholar] [CrossRef]
- Adams, R.H.; Brown, D.T. BHK Cells Expressing Sindbis Virus-Induced Homologous Interference Allow the Translation of Nonstructural Genes of Superinfecting Virus. J. Virol. 1985, 54, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.E.; Wan, K.; Bose, H.R. Homologous Interference Induced by Sindbis Virus. J. Virol. 1974, 14, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Karpf, A.R.; Lenches, E.; Strauss, E.G.; Strauss, J.H.; Brown, D.T. Superinfection Exclusion of Alphaviruses in Three Mosquito Cell Lines Persistently Infected with Sindbis Virus. J. Virol. 1997, 71, 7119–7123. [Google Scholar] [CrossRef] [PubMed]
- Singer, Z.S.; Ambrose, P.M.; Danino, T.; Rice, C.M. Quantitative Measurements of Early Alphaviral Replication Dynamics in Single Cells Reveals the Basis for Superinfection Exclusion. Cell Syst. 2021, 12, 210–219.e3. [Google Scholar] [CrossRef]
- Singh, I.R.; Suomalainen, M.; Varadarajan, S.; Garoff, H.; Helenius, A. Multiple Mechanisms for the Inhibition of Entry and Uncoating of Superinfecting Semliki Forest Virus. Virology 1997, 231, 59–71. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| nsp3 N24A | scrSL3 | scr5′ CSE | |
|---|---|---|---|
| Mutation location (base pairs in CHIKV genome) | 4145–4147 | 91–145 | 175–217 |
| RT primer | CGCAGTCTATGGAGATGTGCTCATC | CATGAACGGGGTTGTGTCGAACCC | CATGAACGGGGTTGTGTCGAACCC |
| PCR forward primer | GGTACTGGTGGGCGACTTGACTAATCCGCCC | GCAAAGCAAGAGATTAATAACCC | GCAAAGCAAGAGATTAATAACCC |
| PCR reverse primer | CGCAGTCTATGGAGATGTGCTCATC | CATGAACGGGGTTGTGTCGAACCC | CATGAACGGGGTTGTGTCGAACCC |
| WT k-mer | AGAGTGCGTGGTTAACGCCGCCAAC | ACGTGGACATAGACGCTGACAGCGCCTTTTT | ATCGAATGACCATGCTAATGCTAGAGCGTTCTCGCATCTAGCC |
| Mutant k-mer | AGAGTGCGTGGTTAACGCCGCCGCC | ATGTAGACATAGACGCTGACAGCGCCTTTCT | GTCGAATGACCATGCCAACGCCAGAGCGTTTTCGCATCTGGCT |
| Step | Reagent | Supplier | Product Number | Reactions | Cost of Kit | Reactions/Kit | Cost |
|---|---|---|---|---|---|---|---|
| RT | Superscript III first-strand synthesis kit | Invitrogen, Carlsbad, CA, USA | 18080051 | 33 | 558 | 50 | 368.28 |
| Reverse primer | Invitrogen, Carlsbad, CA, USA | 14 | 25 nmol | 14 | |||
| PCR tubes | Greiner, Kremsmünster, Austria | 07-000-717 | 33 | 254.5 | 960 | 8.75 | |
| PCR (20 µL reaction) | Phusion kit | NEB, Ipswich, MA, USA | M0530L | 33 | 480 | 1250 | 12.67 |
| Forward primer | Invitrogen, Carlsbad, CA, USA | 14 | 25 nmol | 14 | |||
| dNTPs | NEB, Ipswich, MA, USA | N0447L | 33 | 274 | 2000 | 4.52 | |
| PCR tubes | Greiner, Kremsmünster, Austria | 07-000-717 | 33 | 254.5 | 960 | 8.75 | |
| Clean up | Zymo DNA Clean & Concentrator-5 | Zymo, Irvine, CA, USA | D4004 | 33 | 323 | 200 | 53.30 |
| Sequencing | Amplicon sequencing | Plasmidsaurus, Louisville, KY, USA | 33 | 15 | 1 | 495 | |
| SUM | 979.27 |
| St.Dev = 0.0358 | St.Dev = 0.0214 | |
|---|---|---|
| n = 3 | 11.73% | 7.97% |
| n = 5 | 7.67% | 5.21% |
| n = 10 | 5.01% | 3.4% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz, J.; Sears, J.; Chang, C.-K.; Burdick, J.; Law, I.; Sanders, W.; Linnertz, C.; Sylvester, P.; Moorman, N.; Ferris, M.T.; et al. U-CAN-seq: A Universal Competition Assay by Nanopore Sequencing. Viruses 2024, 16, 636. https://doi.org/10.3390/v16040636
Diaz J, Sears J, Chang C-K, Burdick J, Law I, Sanders W, Linnertz C, Sylvester P, Moorman N, Ferris MT, et al. U-CAN-seq: A Universal Competition Assay by Nanopore Sequencing. Viruses. 2024; 16(4):636. https://doi.org/10.3390/v16040636
Chicago/Turabian StyleDiaz, Jennifer, John Sears, Che-Kang Chang, Jane Burdick, Isabella Law, Wes Sanders, Colton Linnertz, Paul Sylvester, Nathaniel Moorman, Martin T. Ferris, and et al. 2024. "U-CAN-seq: A Universal Competition Assay by Nanopore Sequencing" Viruses 16, no. 4: 636. https://doi.org/10.3390/v16040636