
Genomic Characterization and Molecular Evolution of Sapovirus in Children under 5 Years of Age

Abstract
:1. Introduction
2. Materials and Methods
2.1. Source of the Specimen
2.2. RNA Extraction and SaV RT-qPCR Detection
2.3. High-Throughput Sequencing, Generation of Virus Genome Sequences
2.4. Genotyping of the SaV
2.5. Analysis of Sequence Identity, Phylogeny, and Genetic Evolution
3. Results
3.1. Prevalence of SaV
3.2. SaV Sequencing Results and Genotype Identification
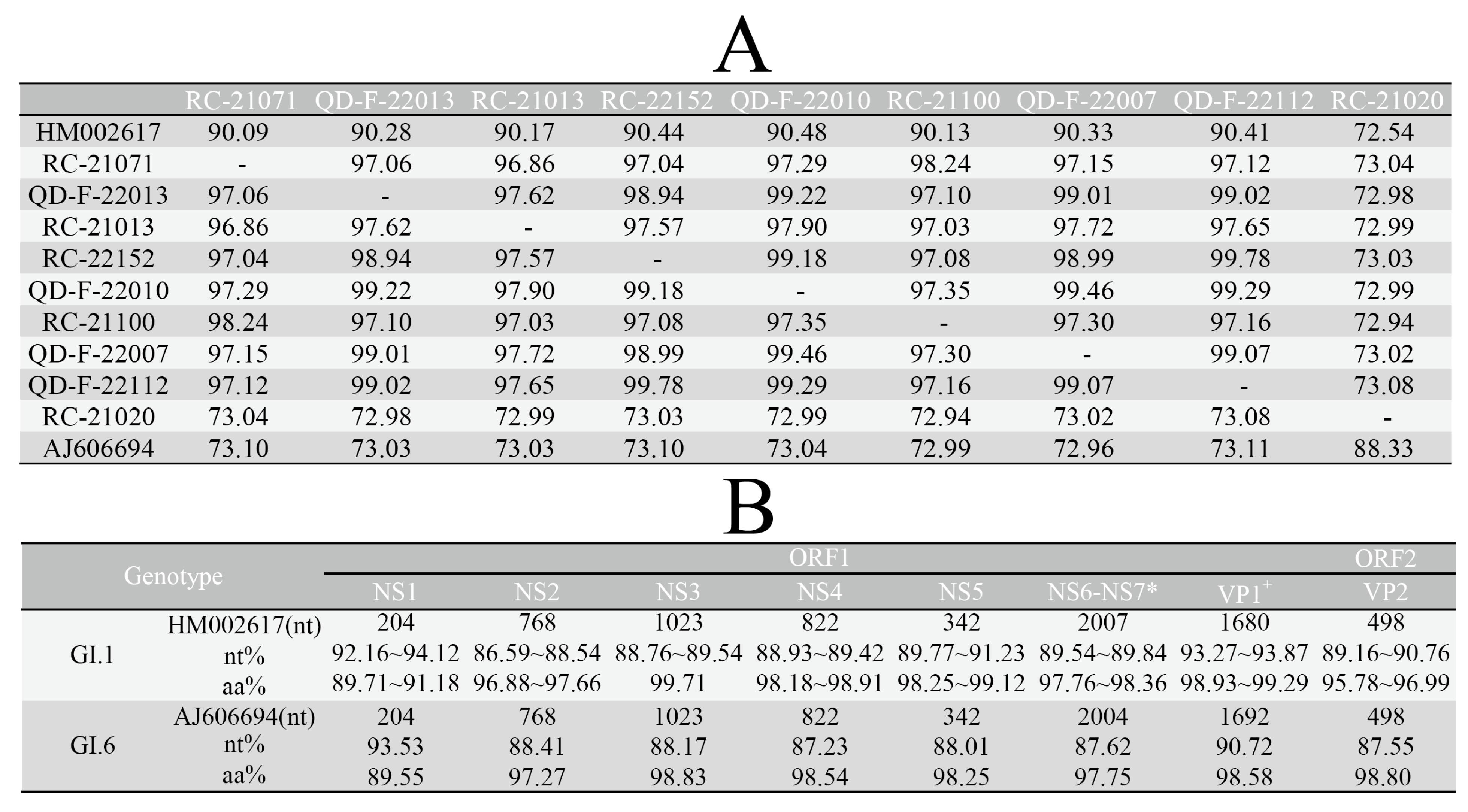
3.3. Analysis of Sequence Identity and Amino Acid Variation Characteristics
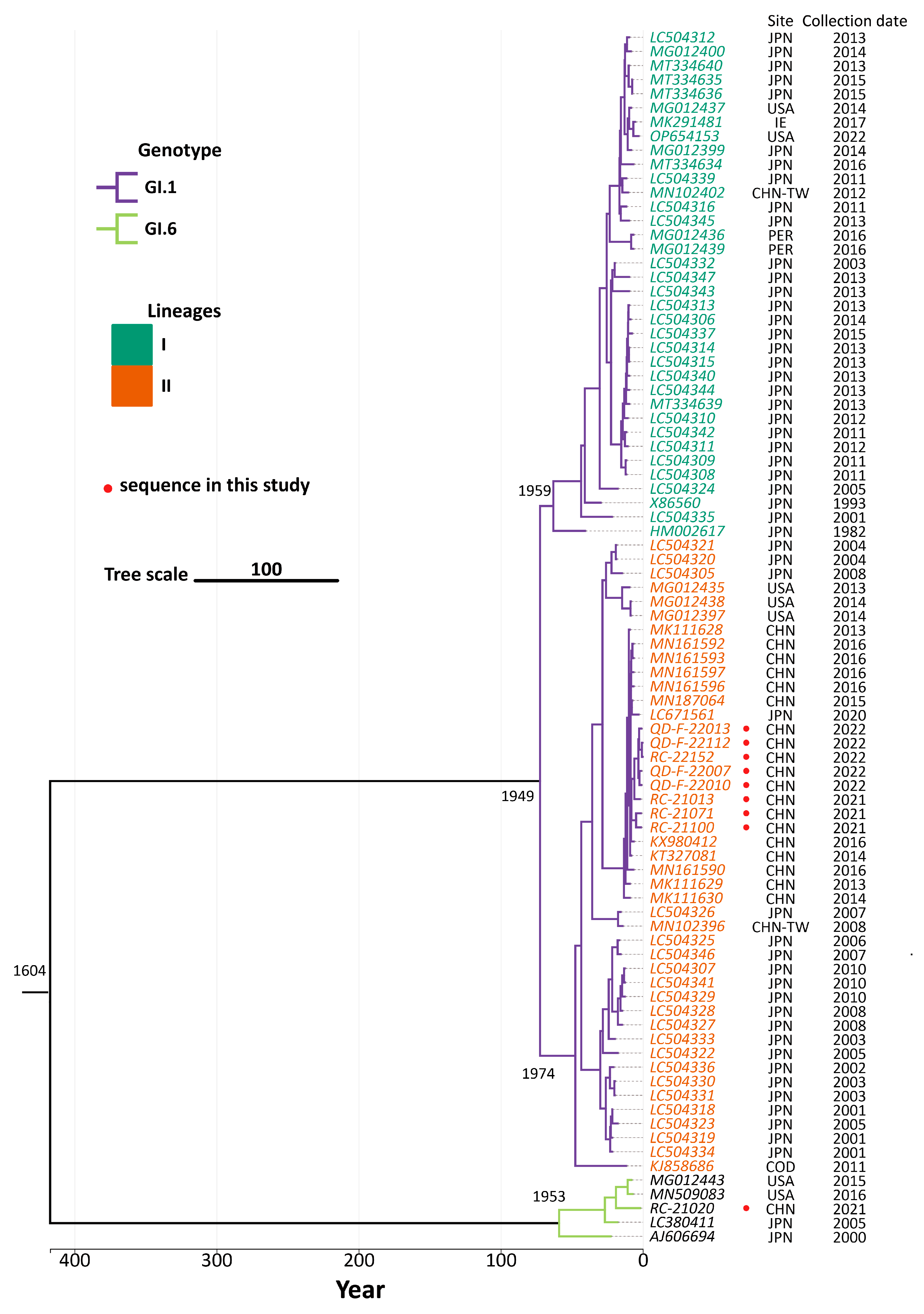
3.4. Phylogenetic Analysis
3.5. Genetic Evolutionary Analysis
3.6. Analysis of Amino Acid Variations between Two Lineages of GI.1 SaV
3.7. Analysis of Population Dynamics
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Becker-Dreps, S.; González, F.; Bucardo, F. Sapovirus: An emerging cause of childhood diarrhea. Curr. Opin. Infect. Dis. 2020, 33, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Biscaro, V.; Piccinelli, G.; Gargiulo, F.; Ianiro, G.; Caruso, A.; Caccuri, F.; De Francesco, M.A. Detection and molecular characterization of enteric viruses in children with acute gastroenteritis in Northern Italy. Infect. Genet. Evol. 2018, 60, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Okitsu, S.; Khamrin, P.; Takanashi, S.; Thongprachum, A.; Hoque, S.A.; Takeuchi, H.; Khan, M.A.; Hasan, S.M.T.; Iwata, T.; Shimizu, H.; et al. Molecular detection of enteric viruses in the stool samples of children without diarrhea in Bangladesh. Infect. Genet. Evol. 2020, 77, 104055. [Google Scholar] [CrossRef] [PubMed]
- Kumthip, K.; Khamrin, P.; Ushijima, H.; Chen, L.; Li, S.; Maneekarn, N. Genetic recombination and diversity of sapovirus in pediatric patients with acute gastroenteritis in Thailand, 2010-2018. PeerJ 2020, 8, e8520. [Google Scholar] [CrossRef] [PubMed]
- Zintz, C.; Bok, K.; Parada, E.; Barnes-Eley, M.; Berke, T.; Staat, M.A.; Azimi, P.; Jiang, X.; Matson, D.O. Prevalence and genetic characterization of caliciviruses among children hospitalized for acute gastroenteritis in the United States. Infect. Genet. Evol. 2005, 5, 281–290. [Google Scholar] [CrossRef]
- Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Wang, Q.; Saif, L.J.; Green, K.Y. Reverse genetics system for porcine enteric calicivirus, a prototype sapovirus in the Caliciviridae. J. Virol. 2005, 79, 1409–1416. [Google Scholar] [CrossRef]
- Neill, J.D. Nucleotide sequence of a region of the feline calicivirus genome which encodes picornavirus-like RNA-dependent RNA polymerase, cysteine protease and 2C polypeptides. Virus Res. 1990, 17, 145–160. [Google Scholar] [CrossRef]
- Lambden, P.R.; Clarke, I.N. Genome organization in the caliciviridae. Trends Microbiol. 1995, 3, 261–265. [Google Scholar] [CrossRef]
- Clarke, I.N.; Lambden, P.R. The molecular biology of caliciviruses. J. Gen. Virol. 1997, 78 Pt 2, 291–301. [Google Scholar] [CrossRef]
- Smertina, E.; Hall, R.N.; Urakova, N.; Strive, T.; Frese, M. Calicivirus Non-structural Proteins: Potential Functions in Replication and Host Cell Manipulation. Front. Microbiol. 2021, 12, 712710. [Google Scholar] [CrossRef]
- Yinda, C.K.; Conceição-Neto, N.; Zeller, M.; Heylen, E.; Maes, P.; Ghogomu, S.M.; Van Ranst, M.; Matthijnssens, J. Novel highly divergent sapoviruses detected by metagenomics analysis in straw-colored fruit bats in Cameroon. Emerg. Microbes Infect. 2017, 6, e38. [Google Scholar] [CrossRef]
- Oka, T.; Wang, Q.; Katayama, K.; Saif, L.J. Comprehensive review of human sapoviruses. Clin. Microbiol. Rev. 2015, 28, 32–53. [Google Scholar] [CrossRef]
- Xue, L.; Cai, W.; Gao, J.; Jiang, Y.; Wu, H.; Zhang, L.; Zuo, Y.; Dong, R.; Pang, R.; Zeng, H.; et al. Genome characteristics and molecular evolution of the human sapovirus variant GII.8. Infect. Genet. Evol. 2019, 73, 362–367. [Google Scholar] [CrossRef]
- Oka, T.; Mori, K.; Iritani, N.; Harada, S.; Ueki, Y.; Iizuka, S.; Mise, K.; Murakami, K.; Wakita, T.; Katayama, K. Human sapovirus classification based on complete capsid nucleotide sequences. Arch. Virol. 2012, 157, 349–352. [Google Scholar] [CrossRef] [PubMed]
- van Maarseveen, N.M.; Wessels, E.; de Brouwer, C.S.; Vossen, A.C.; Claas, E.C. Diagnosis of viral gastroenteritis by simultaneous detection of Adenovirus group F, Astrovirus, Rotavirus group A, Norovirus genogroups I and II, and Sapovirus in two internally controlled multiplex real-time PCR assays. J. Clin. Virol. 2010, 49, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Chhabra, P.; Diez-Valcarce, M.; Barclay, L.; Cannon, J.L.; Vinjé, J. Human Calicivirus Typing tool: A web-based tool for genotyping human norovirus and sapovirus sequences. J. Clin. Virol. 2021, 134, 104718. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.J.; Qiu, Y.; Pu, Y.; Huang, X.; Ge, X.Y. BioAider: An efficient tool for viral genome analysis and its application in tracing SARS-CoV-2 transmission. Sustain. Cities Soc. 2020, 63, 102466. [Google Scholar] [CrossRef]
- Nakanishi, K.; Tatsumi, M.; Kinoshita-Numata, K.; Tsugawa, T.; Nakata, S.; Tsutsumi, H. Full sequence analysis of the original Sapporo virus. Microbiol. Immunol. 2011, 55, 657–660. [Google Scholar] [CrossRef]
- Oka, T.; Iritani, N.; Okada, M.; Ogawa, T.; Iizuka, S.; Tatsumi, C.; Harada, S.; Haga, K.; Doan, Y.H. First Complete Genome Sequences of Human Sapovirus Strains Classified as GI.3, GI.4, GI.6, GI.7, and GII.7. Genome Announc. 2018, 6, e00168-18. [Google Scholar] [CrossRef]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Yong-kang, Z.; Miao, J.; Xiang-yu, K.; Hui-ying, L.; Qing, Z.; Xiao-man, S.; Yu-ning, L.; Zhao-jun, D. The analysis of sapovirus infection in children under 5 years old with diarrhea in eight provinces of China, 2012–2014. Int. J. Virol. 2016, 23, 81–84. [Google Scholar]
- Xiaochun Shang, H.S. Xueqin Zhao, Xiaohong Zhou, Etiological and epidemiological characteristics of viral diarrhea in children under 5 years old in a sentinel hospital in Hangzhou, Zhejiang, 2012–2019. Dis. Surveill. 2020, 35, 809–814. [Google Scholar]
- de Oliveira-Tozetto, S.; Santiso-Bellón, C.; Ferrer-Chirivella, J.M.; Navarro-Lleó, N.; Vila-Vicent, S.; Rodríguez-Díaz, J.; Buesa, J. Epidemiological and Genetic Characterization of Sapovirus in Patients with Acute Gastroenteritis in Valencia (Spain). Viruses 2021, 13, 184. [Google Scholar] [CrossRef] [PubMed]
- Sabiremu Tuohetamu, A.Y. LIU Hong-bin, WU Yue-wen, LI Xin-lan, Surveillance of viral diarrhea among hospitalized children in Urumqi, 2010. Dis. Surveill. 2013, 28, 439–442. [Google Scholar]
- Phan, T.G.; Trinh, Q.D.; Yagyu, F.; Okitsu, S.; Ushijima, H. Emergence of rare sapovirus genotype among infants and children with acute gastroenteritis in Japan. Eur. J. Clin. Microbiol. Infect. Dis. 2007, 26, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Mann, P.; Pietsch, C.; Liebert, U.G. Genetic Diversity of Sapoviruses among Inpatients in Germany, 2008ߝ22018. Viruses 2019, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Han, T.; Qi, X.; Gao, Y.; Zhao, J.; Zhang, Y.; Li, B.; Zhang, Z.; Du, J.; Sun, L. Genotypes Diversity of Acute Gastroenteritis Outbreaks Caused by Human Sapovirus—Beijing Municipality, China, 2015ߝ2021. China CDC Wkly. 2023, 5, 625–631. [Google Scholar] [CrossRef]
- Cilli, A.; Luchs, A.; Morillo, S.G.; Carmona, R.C.C.; Dos Santos, F.C.P.; Maeda, A.Y.; Primo, D.; Pacheco, G.T.; Souza, E.V.; Medeiros, R.S.; et al. Surveillance and molecular characterization of human sapovirus in patients with acute gastroenteritis in Brazil, 2010 to 2017. J. Clin. Virol. 2021, 140, 104844. [Google Scholar] [CrossRef]
- Murray, T.Y.; Nadan, S.; Page, N.A.; Taylor, M.B. Diverse sapovirus genotypes identified in children hospitalised with gastroenteritis in selected regions of South Africa. J. Clin. Virol. 2016, 76, 24–29. [Google Scholar] [CrossRef]
- Mahar, J.E.; Jenckel, M.; Huang, N.; Smertina, E.; Holmes, E.C.; Strive, T.; Hall, R.N. Frequent intergenotypic recombination between the non-structural and structural genes is a major driver of epidemiological fitness in caliciviruses. Virus Evol. 2021, 7, veab080. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Clade | Nucleotide Substitution Models |
|---|---|---|
| Whole genome | GI.1-Lineage I | GTR + F + I + G4 |
| GI.1-Lineage II | GTR + F + I + G4 | |
| GI.1 | GTR + F + G4 | |
| GI.6 | GTR + F + G4 | |
| Total | GTR + F + I + G4 |
| Clade | Local Strain | Reference Strain | Nucleotide Substitution Rate [Substitutions/Site/Year (95%HPD)] | Date of MRCA |
|---|---|---|---|---|
| Lineage I | 0 | 36 | 1.603 × 10−3 (6.597 × 10−4~2.737 × 10−3) | 1959 |
| Lineage II | 8 | 37 | 2.082 × 10−3 (1.569 × 10−3~2.653 × 10−3) | 1974 |
| GI.1 | 8 | 73 | 2.092 × 10−3 (1.484 × 10−3~2.739 × 10−3) | 1949 |
| GI.6 | 1 | 4 | 6.881 × 10−3 (1.874 × 10−5~2.930 × 10−2) | 1953 |
| total | 9 | 86 | 2.099 × 10−3 (1.143 × 10−3~2.761 × 10−3) | 1604 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, X.; Guo, C.; Dai, Y.; Chen, L.; Chen, Y.; Wang, S.; Sun, Y. Genomic Characterization and Molecular Evolution of Sapovirus in Children under 5 Years of Age. Viruses 2024, 16, 146. https://doi.org/10.3390/v16010146
Ji X, Guo C, Dai Y, Chen L, Chen Y, Wang S, Sun Y. Genomic Characterization and Molecular Evolution of Sapovirus in Children under 5 Years of Age. Viruses. 2024; 16(1):146. https://doi.org/10.3390/v16010146
Chicago/Turabian StyleJi, Xiaolei, Chen Guo, Yaoyao Dai, Lu Chen, Yujia Chen, Shifang Wang, and Yihua Sun. 2024. "Genomic Characterization and Molecular Evolution of Sapovirus in Children under 5 Years of Age" Viruses 16, no. 1: 146. https://doi.org/10.3390/v16010146