
Not Asian Anymore: Reconstruction of the History, Evolution, and Dispersal of the “Asian” Lineage of CPV-2c


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
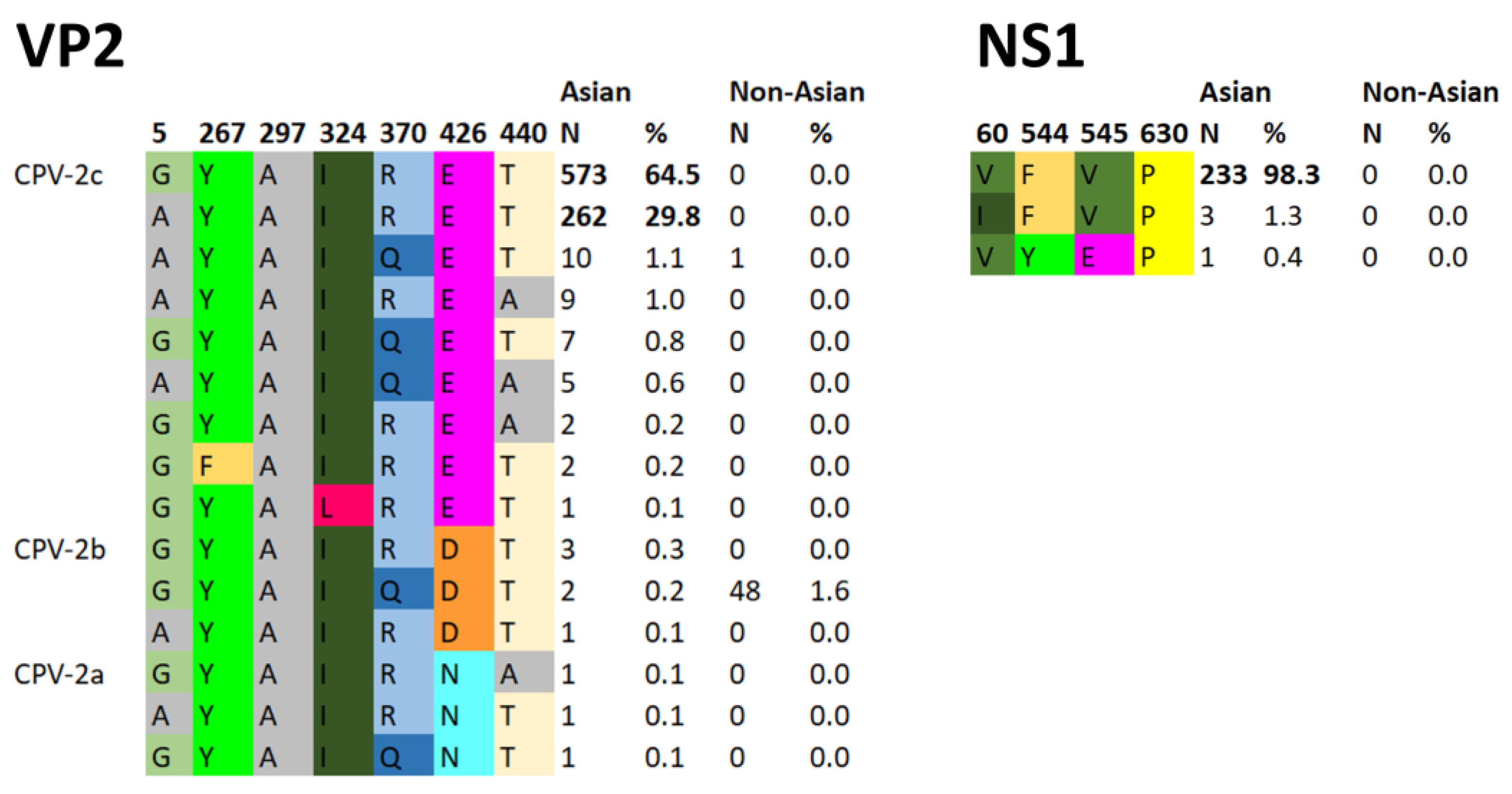
2.2. Asian CPV-2c Lineage Definition
- (1)
- The presence of the amino acid motif 5A/G, 267Y, 297A, 324I, 370R, 426E, and 440T in VP2 and 630P in NS1. Motif identification was automatically performed using specifically designed R scripts.
- (2)
- Being part of a monophyletic clade, including the majority of strains featured in the above-mentioned markers. This choice was necessary to classify as part of the “Asian CPV-2c” clade some strains that, although originating from an Asian ancestor, lost, because of amino acid toggling and reversion, the peculiar phenotypic pattern.
- (3)
- The strains were identified based on different trees and expert opinions were compared to classify them based on a majority consensus rule: strains fulfilling the second rule in at least two out of three trees built with at least one of the three alignments were classified as “Asian CPV-2c”.
2.3. Phylodynamic Analyses
3. Results
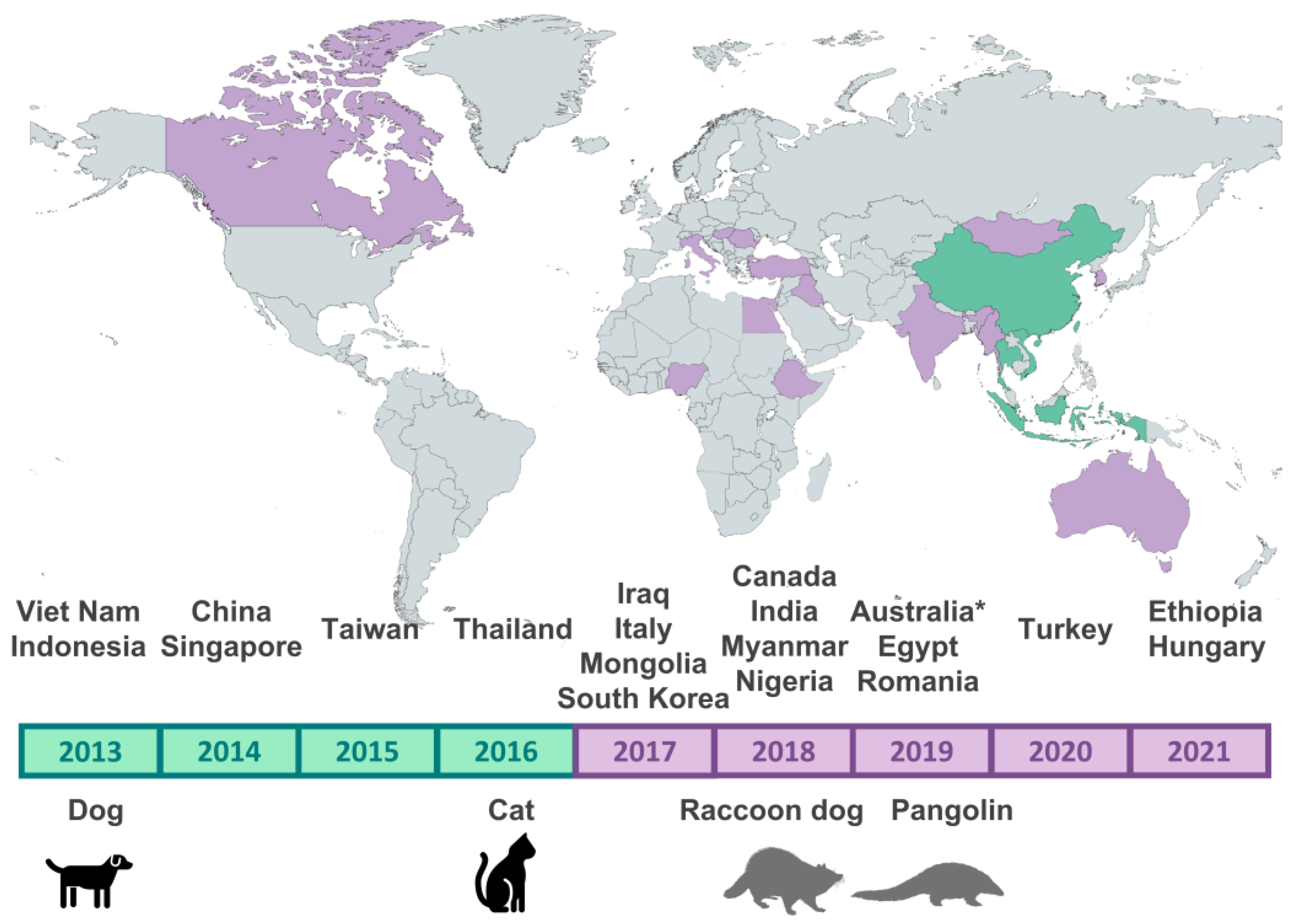
3.1. Details of the Sequences Identified as Belonging to the Asian CPV-2c Lineage
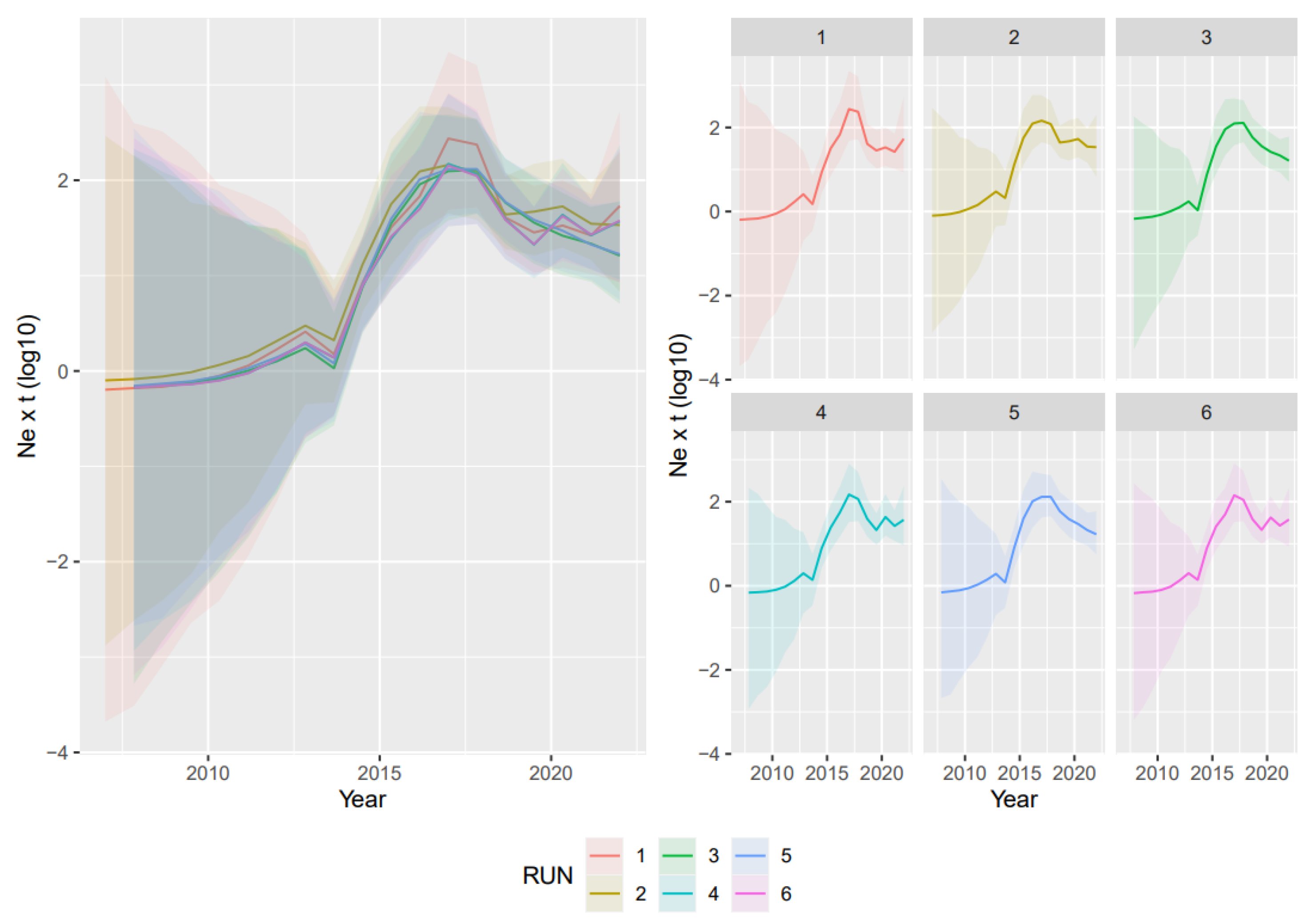
3.2. Phylodynamic Analyses
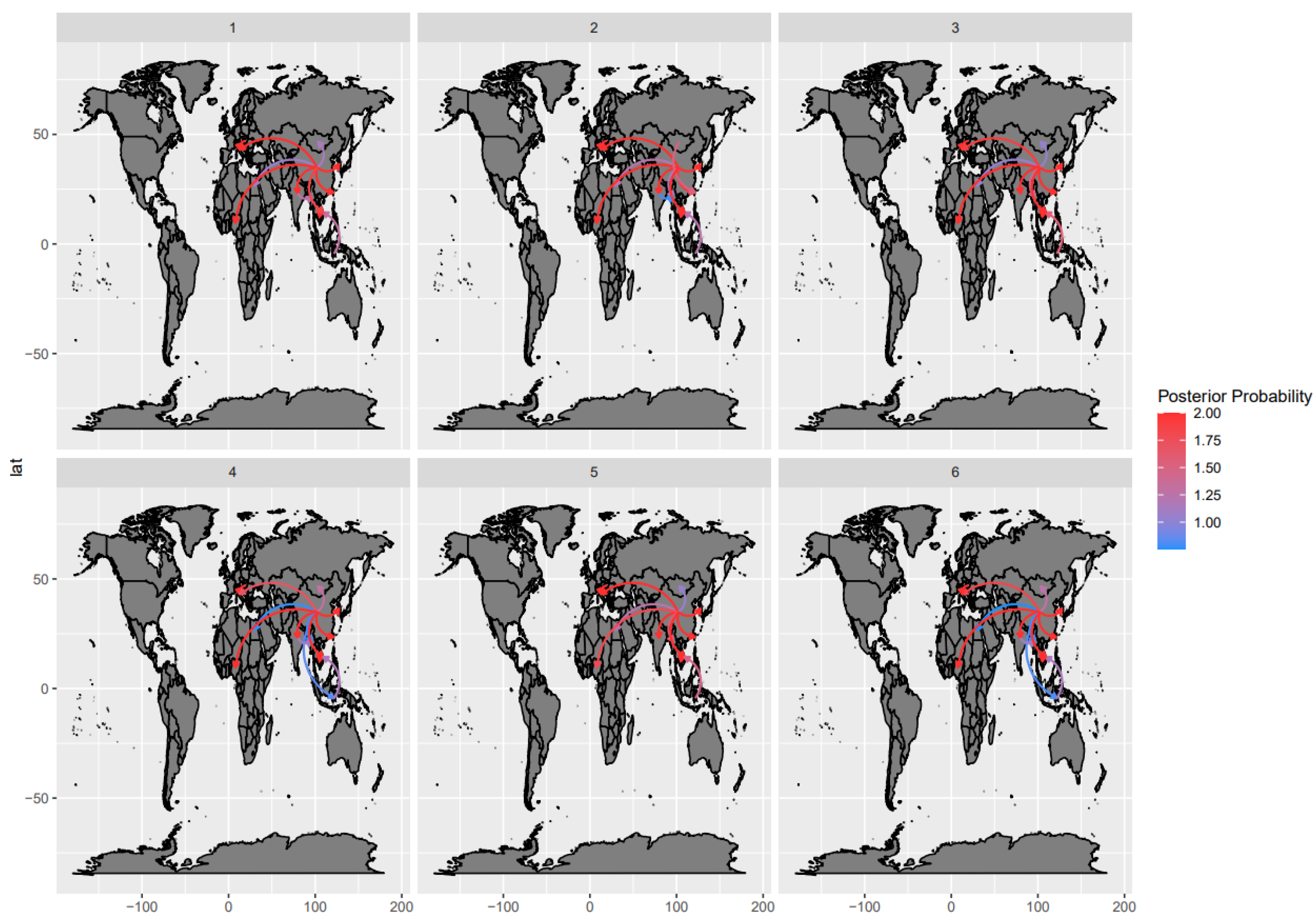
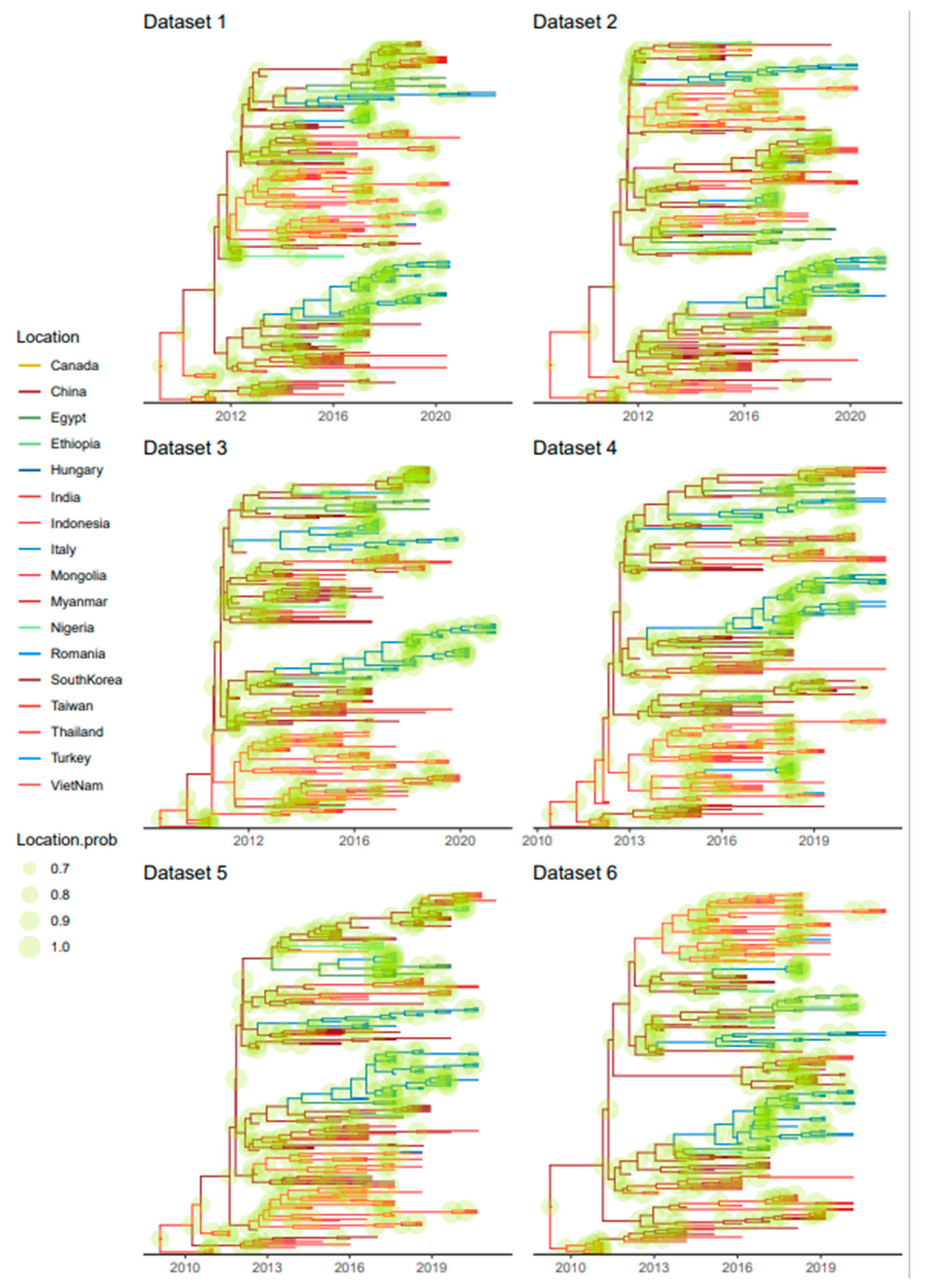
3.3. Phylogeographic Analyses
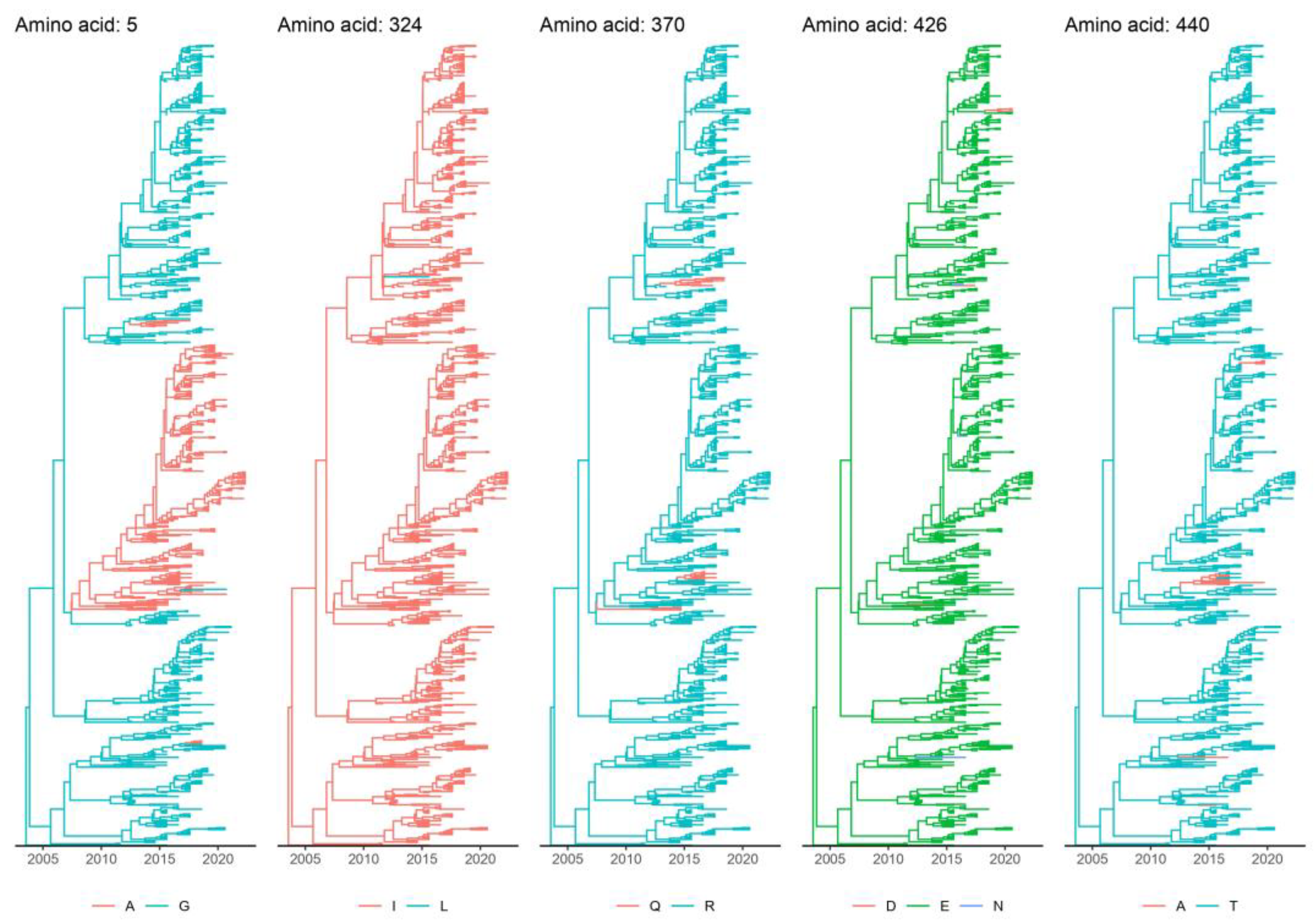
3.4. Marker Amino Acid Evolution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parrish, C.R.; Have, P.; Foreyt, W.J.; Evermann, J.F.; Senda, M.; Carmichael, L.E. The Global Spread and Replacement of Canine Parvovirus Strains. J. Gen. Virol. 1988, 69, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.; Thompson, G. Canine Parvovirus: The Worldwide Occurrence of Antigenic Variants. J. Gen. Virol. 2016, 97, 2043–2057. [Google Scholar] [CrossRef]
- Parrish, C.R. Pathogenesis of Feline Panleukopenia Virus and Canine Parvovirus. Baillière’s Clin. Haematol. 1995, 8, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Buonavoglia, C. Canine Parvovirus—A Review of Epidemiological and Diagnostic Aspects, with Emphasis on Type 2c. Vet. Microbiol. 2012, 155, 1–12. [Google Scholar] [CrossRef]
- Miranda, C.; Parrish, C.R.; Thompson, G. Canine Parvovirus 2c Infection in a Cat with Severe Clinical Disease. J. Vet. Diagn. Investig. 2014, 26, 462–464. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef]
- Pénzes, J.J.; Söderlund-Venermo, M.; Canuti, M.; Eis-Hübinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. Reorganizing the Family Parvoviridae: A Revised Taxonomy Independent of the Canonical Approach Based on Host Association. Arch. Virol. 2020, 165, 2133–2146. [Google Scholar] [CrossRef]
- Allison, A.B.; Kohler, D.J.; Ortega, A.; Hoover, E.A.; Grove, D.M.; Holmes, E.C.; Parrish, C.R. Host-Specific Parvovirus Evolution in Nature Is Recapitulated by in Vitro Adaptation to Different Carnivore Species. PLOS Pathog. 2014, 10, e1004475. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Todd, M.; Monteiro, P.; Van Osch, K.; Weir, R.; Schwantje, H.; Britton, A.P.; Lang, A.S. Ecology and Infection Dynamics of Multi-Host Amdoparvoviral and Protoparvoviral Carnivore Pathogens. Pathogens 2020, 9, 124. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Parrish, C.R.; Truyen, U.; Holmes, E.C. High Rate of Viral Evolution Associated with the Emergence of Carnivore Parvovirus. Proc. Natl. Acad. Sci. USA 2005, 102, 379–384. [Google Scholar] [CrossRef]
- Hueffer, K.; Parker, J.S.L.; Weichert, W.S.; Geisel, R.E.; Sgro, J.-Y.; Parrish, C.R. The Natural Host Range Shift and Subsequent Evolution of Canine Parvovirus Resulted from Virus-Specific Binding to the Canine Transferrin Receptor. J. Virol. 2003, 77, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Mira, F.; Sorensen, R.G.; Rodrigues, B.; Bouchard, É.; Walzthoni, N.; Hopson, M.; Gilroy, C.; Whitney, H.G.; Lang, A.S. Distribution and Diversity of Dog Parvoviruses in Wild, Free-Roaming and Domestic Canids of Newfoundland and Labrador, Canada. Transbound. Emerg. Dis. 2022, 69, e2694–e2705. [Google Scholar] [CrossRef] [PubMed]
- Nur-Farahiyah, A.N.; Kumar, K.; Yasmin, A.R.; Omar, A.R.; Camalxaman, S.N. Isolation and Genetic Characterization of Canine Parvovirus in a Malayan Tiger. Front. Vet. Sci. 2021, 8, 660046. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Fry, K.; Cluff, H.D.; Mira, F.; Fenton, H.; Lang, A.S. Co-Circulation of Five Species of Dog Parvoviruses and Canine Adenovirus Type 1 among Gray Wolves (Canis lupus) in Northern Canada. Transbound. Emerg. Dis. 2022, 69, e1417–e1433. [Google Scholar] [CrossRef]
- Wang, S.-L.; Tu, Y.-C.; Lee, M.-S.; Wu, L.-H.; Chen, T.-Y.; Wu, C.-H.; Tsao, E.H.-S.; Chin, S.-C.; Li, W.-T. Fatal Canine Parvovirus-2 (CPV-2) Infection in a Rescued Free-Ranging Taiwanese Pangolin (Manis pentadactyla pentadactyla). Transbound. Emerg. Dis. 2020, 67, 1074–1081. [Google Scholar] [CrossRef]
- Kurucay, H.N.; Tamer, C.; Muftuoglu, B.; Elhag, A.E.; Gozel, S.; Cicek-Yildiz, Y.; Demirtas, S.; Ozan, E.; Albayrak, H.; Okur-Gumusova, S.; et al. First Isolation and Molecular Characterization of Canine Parvovirus-Type 2b (CPV-2b) from Red Foxes (Vulpes vulpes) Living in the Wild Habitat of Turkey. Virol. J. 2023, 20, 27. [Google Scholar] [CrossRef]
- Lina, Z.; Kai, W.; Fuyu, A.; Dongliang, Z.; Hailing, Z.; Xuelin, X.; Ce, G.; Hongmei, Y.; Yingjie, K.; Zhidong, Z.; et al. Fatal Canine Parvovirus Type 2a and 2c Infections in Wild Chinese Pangolins (Manis pentadactyla) in Southern China. Transbound. Emerg. Dis. 2022, 69, 4002–4008. [Google Scholar] [CrossRef]
- Allison, A.B.; Organtini, L.J.; Zhang, S.; Hafenstein, S.L.; Holmes, E.C.; Parrish, C.R. Single Mutations in the VP2 300 Loop Region of the Three-Fold Spike of the Carnivore Parvovirus Capsid Can Determine Host Range. J. Virol. 2016, 90, 753–767. [Google Scholar] [CrossRef]
- Parrish, C.R.; Aquadro, C.F.; Strassheim, M.L.; Evermann, J.F.; Sgro, J.Y.; Mohammed, H.O. Rapid Antigenic-Type Replacement and DNA Sequence Evolution of Canine Parvovirus. J. Virol. 1991, 65, 6544–6552. [Google Scholar] [CrossRef]
- Buonavoglia, C.; Martella, V.; Pratelli, A.; Tempesta, M.; Cavalli, A.; Buonavoglia, D.; Bozzo, G.; Elia, G.; Decaro, N.; Carmichael, L. Evidence for Evolution of Canine Parvovirus Type 2 in Italy. J. Gen. Virol. 2001, 82, 3021–3025. [Google Scholar] [CrossRef]
- Wilson, S.; Illambas, J.; Siedek, E.; Stirling, C.; Thomas, A.; Plevová, E.; Sture, G.; Salt, J. Vaccination of Dogs with Canine Parvovirus Type 2b (CPV-2b) Induces Neutralising Antibody Responses to CPV-2a and CPV-2c. Vaccine 2014, 32, 5420–5424. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Zeng, W.; Zhang, X.; Li, S. The Genetic Evolution of Canine Parvovirus—A New Perspective. PLoS ONE 2017, 12, e0175035. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; He, Y.; Wang, C.; Xiao, W.; Liu, R.; Xiao, X.; Zhou, P.; Li, S. The Increasing Prevalence of CPV-2c in Domestic Dogs in China. PeerJ 2020, 8, e9869. [Google Scholar] [CrossRef]
- Voorhees, I.E.H.; Lee, H.; Allison, A.B.; Lopez-Astacio, R.; Goodman, L.B.; Oyesola, O.O.; Omobowale, O.; Fagbohun, O.; Dubovi, E.J.; Hafenstein, S.L.; et al. Limited Intrahost Diversity and Background Evolution Accompany 40 Years of Canine Parvovirus Host Adaptation and Spread. J. Virol. 2019, 94, e01162-19. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Mira, F.; Canuti, M.; Vullo, S.; Purpari, G.; Chiaramonte, G.; Di Bella, S.; Cannella, V.; Randazzo, V.; Castronovo, C.; et al. Identification and Molecular Characterization of a Divergent Asian-like Canine Parvovirus Type 2b (CPV-2b) Strain in Southern Italy. Int. J. Mol. Sci. 2022, 23, 11240. [Google Scholar] [CrossRef] [PubMed]
- Mira, F.; Canuti, M.; Purpari, G.; Cannella, V.; Di Bella, S.; Occhiogrosso, L.; Schirò, G.; Chiaramonte, G.; Barreca, S.; Pisano, P.; et al. Molecular Characterization and Evolutionary Analyses of Carnivore protoparvovirus 1 NS1 Gene. Viruses 2019, 11, 308. [Google Scholar] [CrossRef]
- Mira, F.; Purpari, G.; Lorusso, E.; Di Bella, S.; Gucciardi, F.; Desario, C.; Macaluso, G.; Decaro, N.; Guercio, A. Introduction of Asian Canine Parvovirus in Europe through Dog Importation. Transbound. Emerg. Dis. 2018, 65, 16–21. [Google Scholar] [CrossRef]
- Mira, F.; Purpari, G.; Bella, S.D.; Colaianni, M.L.; Schirò, G.; Chiaramonte, G.; Gucciardi, F.; Pisano, P.; Lastra, A.; Decaro, N.; et al. Spreading of Canine Parvovirus Type 2c Mutants of Asian Origin in Southern Italy. Transbound. Emerg. Dis. 2019, 66, 2297. [Google Scholar] [CrossRef]
- Balboni, A.; Niculae, M.; Di Vito, S.; Urbani, L.; Terrusi, A.; Muresan, C.; Battilani, M. The Detection of Canine Parvovirus Type 2c of Asian Origin in Dogs in Romania Evidenced Its Progressive Worldwide Diffusion. BMC Vet. Res. 2021, 17, 206. [Google Scholar] [CrossRef]
- Mon, P.P.; Thurain, K.; Charoenkul, K.; Nasamran, C.; Wynn, M.; Tun, T.N.; Amonsin, A. Emergence of Canine Parvovirus Type 2c (CPV-2c) of Asian Origin in Domestic Dogs in Myanmar. Comp. Immunol. Microbiol. Infect. Dis. 2022, 90–91, 101901. [Google Scholar] [CrossRef]
- Ogbu, K.I.; Mira, F.; Purpari, G.; Nwosuh, C.; Loria, G.R.; Schirò, G.; Chiaramonte, G.; Tion, M.T.; Bella, S.D.; Ventriglia, G.; et al. Nearly Full-length Genome Characterization of Canine Parvovirus Strains Circulating in Nigeria. Transbound. Emerg. Dis. 2020, 67, 635. [Google Scholar] [CrossRef] [PubMed]
- Mira, F.; Schirò, G.; Franzo, G.; Canuti, M.; Purpari, G.; Giudice, E.; Decaro, N.; Vicari, D.; Antoci, F.; Guercio, A. Evaluation of Canine Parvovirus Type 2 (CPV-2) Molecular Epidemiology in Sicily, Southern Italy: A Geographical Island, an Epidemiological Continuum. Heliyon 2023. submitted. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple Alignment of Nucleotide Sequences Guided by Amino Acid Translations. Nucleic. Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [PubMed]
- IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies|Molecular Biology and Evolution | Oxford Academic. Available online: https://academic.oup.com/mbe/article/32/1/268/2925592 (accessed on 25 July 2023).
- RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies|Bioinformatics|Oxford Academic. Available online: https://academic.oup.com/bioinformatics/article/30/9/1312/238053 (accessed on 25 July 2023).
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Hill, V.; Baele, G. Bayesian Estimation of Past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis Testing Using Phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Frost, S.D.W. Not so Different after All: A Comparison of Methods for Detecting Amino Acid Sites under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian Approximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Franzo, G.; De Villiers, L.; De Villiers, M.; Ravandi, A.; Gyani, K.; Van Zyl, L.; Coetzee, L.M.; Khaiseb, S.; Molini, U. Molecular Epidemiology of Canine Parvovirus in Namibia: Introduction Pathways and Local Persistence. Prev. Vet. Med. 2022, 209, 105780. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Chiang, S.-Y.; Wu, H.-Y.; Lin, J.-H.; Chiou, M.-T.; Liu, H.-F.; Lin, C.-N. Phylodynamic and Genetic Diversity of Canine Parvovirus Type 2c in Taiwan. Int. J. Mol. Sci. 2017, 18, 2703. [Google Scholar] [CrossRef]
- Nguyen Manh, T.; Piewbang, C.; Rungsipipat, A.; Techangamsuwan, S. Molecular and Phylogenetic Analysis of Vietnamese Canine Parvovirus 2C Originated from Dogs Reveals a New Asia-IV Clade. Transbound. Emerg. Dis. 2021, 68, 1445–1453. [Google Scholar] [CrossRef]
- Wardhani, S.W.; Wongsakul, B.; Kasantikul, T.; Piewbang, C.; Techangamsuwan, S. Molecular and Pathological Investigations of Selected Viral Neuropathogens in Rabies-Negative Brains of Cats and Dogs Revealed Neurotropism of Carnivore Protoparvovirus-1. Front. Vet. Sci. 2021, 8, 710701. [Google Scholar] [CrossRef]
- Temuujin, U.; Tserendorj, A.; Fujiki, J.; Sakoda, Y.; Tseren-Ochir, E.-O.; Okamatsu, M.; Matsuno, K.; Sharav, T.; Horiuchi, M.; Umemura, T.; et al. The First Isolation and Identification of Canine Parvovirus (CPV) Type 2c Variants during 2016–2018 Genetic Surveillance of Dogs in Mongolia. Infect. Genet. Evol. 2019, 73, 269–275. [Google Scholar] [CrossRef]
- Carrino, M.; Tassoni, L.; Campalto, M.; Cavicchio, L.; Mion, M.; Corrò, M.; Natale, A.; Beato, M.S. Molecular Investigation of Recent Canine Parvovirus-2 (CPV-2) in Italy Revealed Distinct Clustering. Viruses 2022, 14, 917. [Google Scholar] [CrossRef] [PubMed]
- Ndiana, L.A.; Lanave, G.; Zarea, A.A.K.; Desario, C.; Odigie, E.A.; Ehab, F.A.; Capozza, P.; Greco, G.; Buonavoglia, C.; Decaro, N. Molecular Characterization of Carnivore protoparvovirus 1 Circulating in Domestic Carnivores in Egypt. Front. Vet. Sci. 2022, 9, 932247. [Google Scholar] [CrossRef] [PubMed]
- Tegegne, D.; Tsegaye, G.; Faustini, G.; Franzo, G. First Genetic Detection and Characterization of Canine Parvovirus Type 2 (Carnivore protoparvovirus 1) in Southwestern Ethiopia. Vet. Res. Commun. 2023, 47, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Van, D.; Le, T.D.H.; Maeda, K. Transition of Dominant Canine Parvovirus Genotype from 2b to 2c in Vietnamese Dogs. Vet. Ital. 2022, 58, 199–206. [Google Scholar] [CrossRef]
- Hao, X.; Li, Y.; Xiao, X.; Chen, B.; Zhou, P.; Li, S. The Changes in Canine Parvovirus Variants over the Years. Int. J. Mol. Sci. 2022, 23, 11540. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, J.; Bi, Z.; Tan, Y.; Lv, L.; Zhao, H.; Xia, X.; Zhu, Y.; Wang, Y.; Qian, J. Molecular Epidemiology and Genetic Evolution of Canine Parvovirus in East China, during 2018–2020. Infect. Genet. Evol. 2021, 90, 104780. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, X.; Zhu, J.; Liao, L.; Bao, E. Molecular Epidemiological Survey of Canine Parvovirus Circulating in China from 2014 to 2019. Pathogens 2021, 10, 588. [Google Scholar] [CrossRef]
- Decaro, N.; Campolo, M.; Elia, G.; Buonavoglia, D.; Colaianni, M.L.; Lorusso, A.; Mari, V.; Buonavoglia, C. Infectious Canine Hepatitis: An “Old” Disease Reemerging in Italy. Res. Vet. Sci. 2007, 83, 269–273. [Google Scholar] [CrossRef]
- Martella, V.; Cirone, F.; Elia, G.; Lorusso, E.; Decaro, N.; Campolo, M.; Desario, C.; Lucente, M.S.; Bellacicco, A.L.; Blixenkrone-Møller, M.; et al. Heterogeneity within the Hemagglutinin Genes of Canine Distemper Virus (CDV) Strains Detected in Italy. Vet. Microbiol. 2006, 116, 301–309. [Google Scholar] [CrossRef]
- Alfano, F.; Lanave, G.; Lucibelli, M.G.; Miletti, G.; D’Alessio, N.; Gallo, A.; Auriemma, C.; Amoroso, M.G.; Lucente, M.S.; De Carlo, E.; et al. Canine Distemper Virus in Autochtonous and Imported Dogs, Southern Italy (2014–2021). Animals 2022, 12, 2852. [Google Scholar] [CrossRef]
- Willi, B.; Spiri, A.M.; Meli, M.L.; Grimm, F.; Beatrice, L.; Riond, B.; Bley, T.; Jordi, R.; Dennler, M.; Hofmann-Lehmann, R. Clinical and Molecular Investigation of a Canine Distemper Outbreak and Vector-Borne Infections in a Group of Rescue Dogs Imported from Hungary to Switzerland. BMC Vet. Res. 2015, 11, 154. [Google Scholar] [CrossRef]
- Maya, L.; Calleros, L.; Francia, L.; Hernández, M.; Iraola, G.; Panzera, Y.; Sosa, K.; Pérez, R. Phylodynamics Analysis of Canine Parvovirus in Uruguay: Evidence of Two Successive Invasions by Different Variants. Arch. Virol. 2013, 158, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Grecco, S.; Iraola, G.; Decaro, N.; Alfieri, A.; Alfieri, A.; Gallo Calderón, M.; da Silva, A.P.; Name, D.; Aldaz, J.; Calleros, L.; et al. Inter- and Intracontinental Migrations and Local Differentiation Have Shaped the Contemporary Epidemiological Landscape of Canine Parvovirus in South America. Virus Evol. 2018, 4, vey011. [Google Scholar] [CrossRef]
- Tucciarone, C.M.; Franzo, G.; Mazzetto, E.; Legnardi, M.; Caldin, M.; Furlanello, T.; Cecchinato, M.; Drigo, M. Molecular Insight into Italian Canine Parvovirus Heterogeneity and Comparison with the Worldwide Scenario. Infect. Genet. Evol. 2018, 66, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Tang, N.; Zhu, J.; Wang, M.; Liu, Y.; Lyu, Y. Molecular Characteristics and Genetic Evolutionary Analyses of Circulating Parvoviruses Derived from Cats in Beijing. BMC Vet. Res. 2022, 18, 195. [Google Scholar] [CrossRef] [PubMed]
- Charoenkul, K.; Tangwangvivat, R.; Janetanakit, T.; Boonyapisitsopa, S.; Bunpapong, N.; Chaiyawong, S.; Amonsin, A. Emergence of Canine Parvovirus Type 2c in Domestic Dogs and Cats from Thailand. Transbound. Emerg. Dis. 2019, 66, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Tucciarone, C.M.; Franzo, G.; Legnardi, M.; Lazzaro, E.; Zoia, A.; Petini, M.; Furlanello, T.; Caldin, M.; Cecchinato, M.; Drigo, M. Genetic Insights into Feline Parvovirus: Evaluation of Viral Evolutionary Patterns and Association between Phylogeny and Clinical Variables. Viruses 2021, 13, 1033. [Google Scholar] [CrossRef]
- Allison, A.B.; Harbison, C.E.; Pagan, I.; Stucker, K.M.; Kaelber, J.T.; Brown, J.D.; Ruder, M.G.; Keel, M.K.; Dubovi, E.J.; Holmes, E.C.; et al. Role of Multiple Hosts in the Cross-Species Transmission and Emergence of a Pandemic Parvovirus. J. Virol. 2012, 86, 865–872. [Google Scholar] [CrossRef]
- Boros, Á.; Albert, M.; Urbán, P.; Herczeg, R.; Gáspár, G.; Balázs, B.; Cságola, A.; Pankovics, P.; Gyenesei, A.; Reuter, G. Unusual “Asian-Origin” 2c to 2b Point Mutant Canine Parvovirus (Parvoviridae) and Canine Astrovirus (Astroviridae) Co-Infection Detected in Vaccinated Dogs with an Outbreak of Severe Haemorrhagic Gastroenteritis with High Mortality Rate in Hungary. Vet. Res. Commun. 2022, 46, 1355–1361. [Google Scholar] [CrossRef]
- Franzo, G.; Tucciarone, C.M.; Casagrande, S.; Caldin, M.; Cortey, M.; Furlanello, T.; Legnardi, M.; Cecchinato, M.; Drigo, M. Canine Parvovirus (CPV) Phylogeny Is Associated with Disease Severity. Sci. Rep. 2019, 9, 11266. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzo, G.; Mira, F.; Schirò, G.; Canuti, M. Not Asian Anymore: Reconstruction of the History, Evolution, and Dispersal of the “Asian” Lineage of CPV-2c. Viruses 2023, 15, 1962. https://doi.org/10.3390/v15091962
Franzo G, Mira F, Schirò G, Canuti M. Not Asian Anymore: Reconstruction of the History, Evolution, and Dispersal of the “Asian” Lineage of CPV-2c. Viruses. 2023; 15(9):1962. https://doi.org/10.3390/v15091962
Chicago/Turabian StyleFranzo, Giovanni, Francesco Mira, Giorgia Schirò, and Marta Canuti. 2023. "Not Asian Anymore: Reconstruction of the History, Evolution, and Dispersal of the “Asian” Lineage of CPV-2c" Viruses 15, no. 9: 1962. https://doi.org/10.3390/v15091962