
Genotype and Phenotype Characterization of Rhinolophus sp. Sarbecoviruses from Vietnam: Implications for Coronavirus Emergence
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical and Regulatory Statements
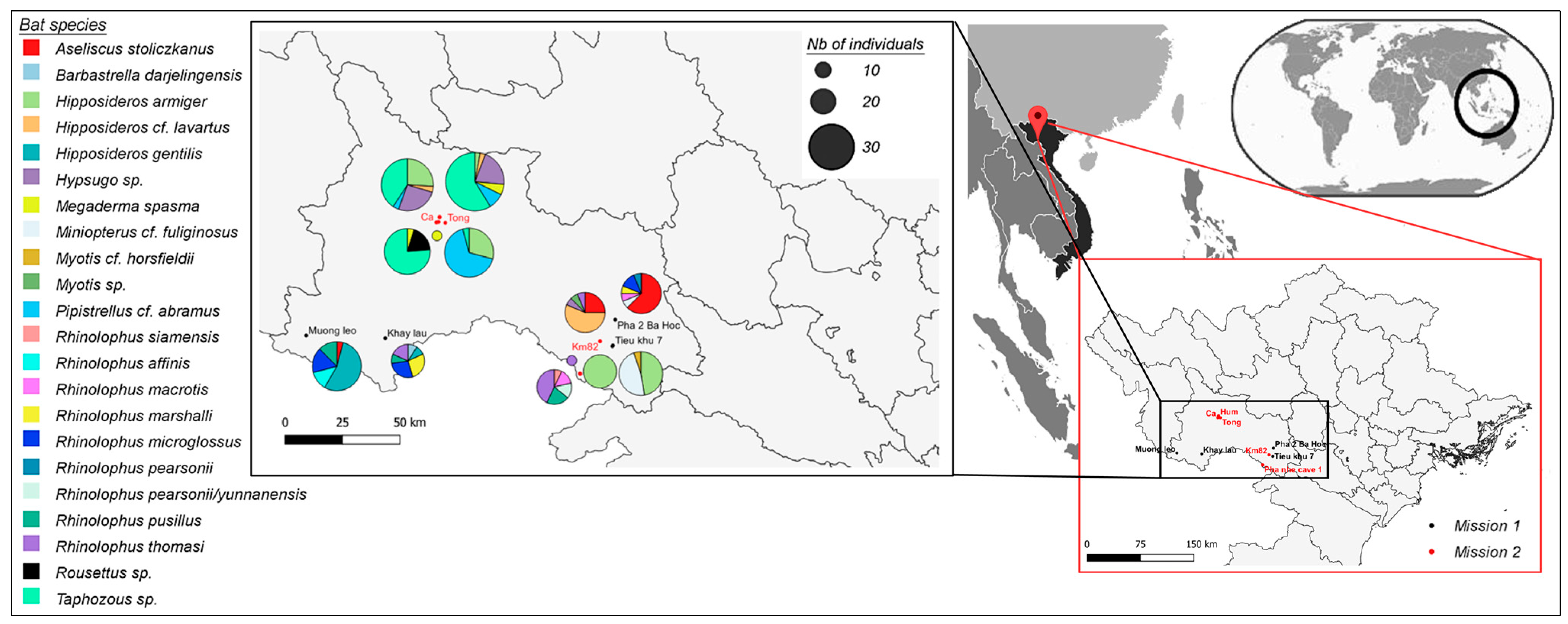
2.2. Bat Capture and Sample Collection
2.3. RNA Extraction and Pan-Coronavirus PCR Screening
2.4. Whole Genome Sequencing of Bat Sarbecoviruses
2.5. Molecular and Phylogenetic Analyses
2.6. In Silico Evaluation of RBD-ACE2 Binding Free Energy
- We used AlphaFold2 [66] to build structural models of ACE2 and RBDs. For each sequence, we generated 5 different models and selected the best model based on the AlphaFold2 pLDDT score.
- For each potential RBD-ACE2 complex, we created 200 models of the complex using HADDOCK [67]. ACE2 and RBDs were first docked into a complex with the help of inter-subunit distance restraints. These restraints were defined between each pair of atoms at the interface of the SARS-CoV-2 RBD and human ACE2 based on the X-ray structure of this complex (PDB code 6M0J [68]). The generated models were then refined via molecular dynamics simulations in explicit solvent during the last step of the HADDOCK modeling pipeline.
- The FoldX v. 5 scoring function [69] was used to estimate the RBD-ACE2 binding free energy from the ensemble of models generated with HADDOCK.
2.7. Generation of Lentiviral Pseudoviruses and ACE2-Dependent Entry Assays
3. Results
3.1. Sarbecovirus Detection Rate in Vietnamese Bats
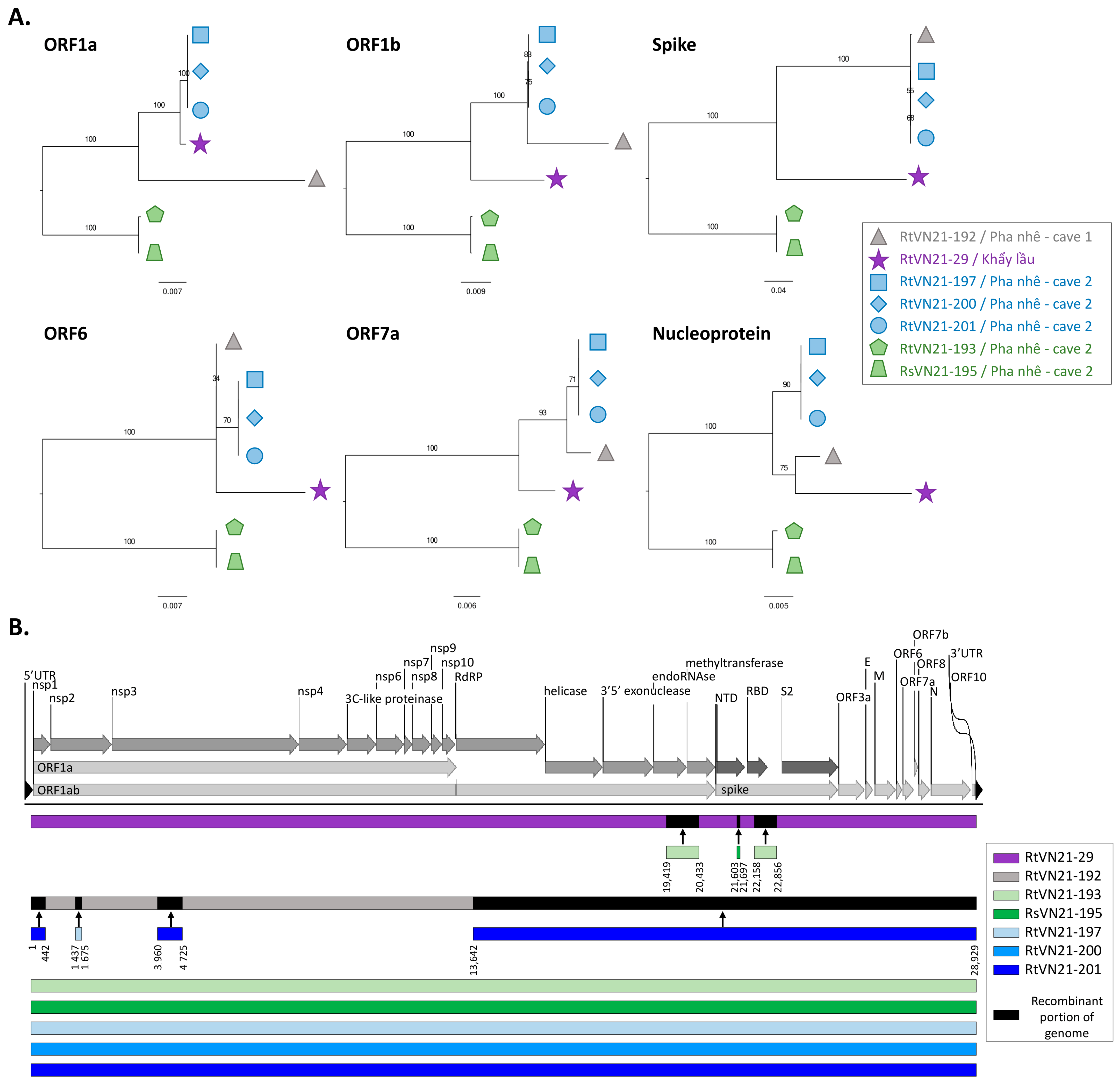
3.2. Genomic Characterization of Rhinolophus sp. Sarbecoviruses
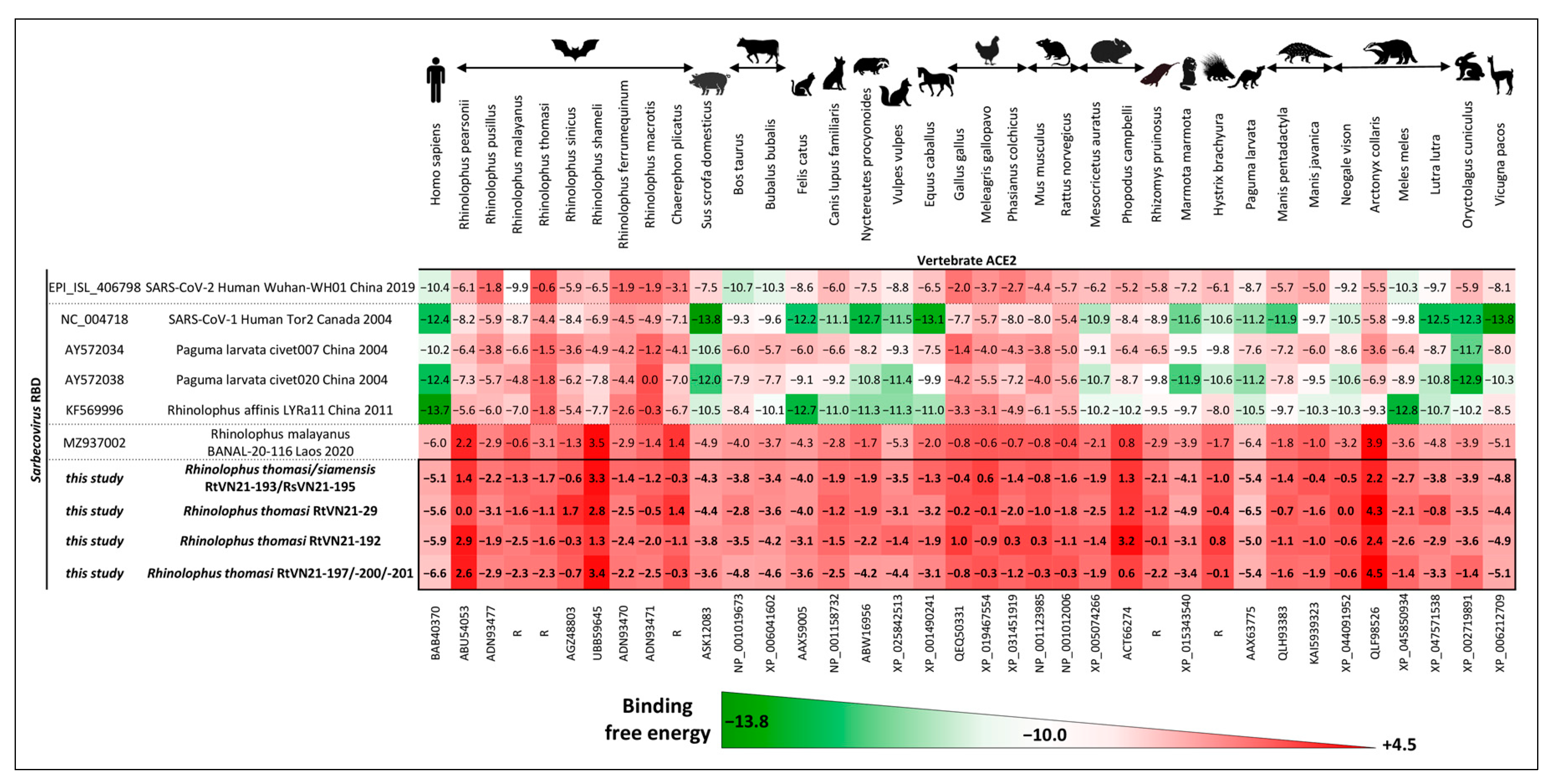
3.3. Prediction of Zoonotic Potential of Vietnamese Sarbecoviruses
3.3.1. In Silico Evaluation of RBD-ACE2 Binding Free Energy
3.3.2. ACE2-Dependent Entry Assay of Vietnamese Sarbecoviruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, H.J.; Wen, H.L.; Zhou, C.M.; Chen, F.F.; Luo, L.M.; Liu, J.W.; Yu, X.J. Bats as reservoirs of severe emerging infectious diseases. Virus Res. 2015, 205, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Anthony, S.J.; Gilardi, K.; Menachery, V.D.; Goldstein, T.; Ssebide, B.; Mbabazi, R.; Navarrete-Macias, I.; Liang, E.; Wells, H.; Hicks, A.; et al. Further Evidence for Bats as the Evolutionary Source of Middle East Respiratory Syndrome Coronavirus. mBio 2017, 8, e00373-17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Keusch, G.T.; Amuasi, J.H.; Anderson, D.E.; Daszak, P.; Eckerle, I.; Field, H.; Koopmans, M.; Lam, S.K.; Das Neves, C.G.; Peiris, M.; et al. Pandemic origins and a One Health approach to preparedness and prevention: Solutions based on SARS-CoV-2 and other RNA viruses. Proc. Natl. Acad. Sci. USA 2022, 119, e2202871119. [Google Scholar] [CrossRef]
- Ruiz-Aravena, M.; McKee, C.; Gamble, A.; Lunn, T.; Morris, A.; Snedden, C.E.; Yinda, C.K.; Port, J.R.; Buchholz, D.W.; Yeo, Y.Y.; et al. Ecology, evolution and spillover of coronaviruses from bats. Nat. Rev. Microbiol. 2022, 20, 299–314. [Google Scholar] [CrossRef]
- Rahman, S.; Ullah, S.; Shinwari, Z.K.; Ali, M. Bats-associated beta-coronavirus detection and characterization: First report from Pakistan. Infect. Genet Evol. 2023, 108, 105399. [Google Scholar] [CrossRef]
- Speranskaya, A.S.; Artiushin, I.V.; Samoilov, A.E.; Korneenko, E.V.; Khabudaev, K.V.; Ilina, E.N.; Yusefovich, A.P.; Safonova, M.V.; Dolgova, A.S.; Gladkikh, A.S.; et al. Identification and Genetic Characterization of MERS-Related Coronavirus Isolated from Nathusius’ Pipistrelle (Pipistrellus nathusii) near Zvenigorod (Moscow Region, Russia). Int. J. Environ. Res. Public Health 2023, 20, 3702. [Google Scholar] [CrossRef]
- Wang, J.; Pan, Y.F.; Yang, L.F.; Yang, W.H.; Lv, K.; Luo, C.M.; Wang, J.; Kuang, G.P.; Wu, W.C.; Gou, Q.Y.; et al. Individual bat virome analysis reveals co-infection and spillover among bats and virus zoonotic potential. Nat. Commun. 2023, 14, 4079. [Google Scholar] [CrossRef]
- Van Brussel, K.; Mahar, J.E.; Ortiz-Baez, A.S.; Carrai, M.; Spielman, D.; Boardman, W.S.J.; Baker, M.L.; Beatty, J.A.; Geoghegan, J.L.; Barrs, V.R.; et al. Faecal virome of the Australian grey-headed flying fox from urban/suburban environments contains novel coronaviruses, retroviruses and sapoviruses. Virology 2022, 576, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Muzeniek, T.; Perera, T.; Siriwardana, S.; Bas, D.; Kaplan, F.; Öruc, M.; Becker-Ziaja, B.; Perera, I.; Weerasena, J.; Handunnetti, S.; et al. Full Genome of batCoV/MinFul/2018/SriLanka, a Novel Alpha-Coronavirus Detected in Miniopterus fuliginosus, Sri Lanka. Viruses 2022, 14, 337. [Google Scholar] [PubMed]
- Wang, N.; Luo, C.M.; Yang, X.L.; Liu, H.Z.; Zhang, L.B.; Zhang, W.; Li, B.; Zhu, Y.; Peng, C.; Shi, Z.L.; et al. Genomic Characterization of Diverse Bat Coronavirus HKU10 in Hipposideros Bats. Viruses 2021, 13, 1962. [Google Scholar] [CrossRef]
- Hardmeier, I.; Aeberhard, N.; Qi, W.; Schoenbaechler, K.; Kraettli, H.; Hatt, J.M.; Fraefel, C.; Kubacki, J. Metagenomic analysis of fecal and tissue samples from 18 endemic bat species in Switzerland revealed a diverse virus composition including potentially zoonotic viruses. PLoS ONE 2021, 16, e0252534. [Google Scholar] [CrossRef] [PubMed]
- Daszak, P.; Keusch, G.T.; Phelan, A.L.; Johnson, C.K.; Osterholm, M.T. Infectious Disease Threats: A Rebound to Resilience. Health Aff. 2021, 40, 204–211. [Google Scholar] [CrossRef]
- Wang, N.; Li, S.Y.; Yang, X.L.; Huang, H.M.; Zhang, Y.J.; Guo, H.; Luo, C.M.; Miller, M.; Zhu, G.; Chmura, A.A.; et al. Serological Evidence of Bat SARS-Related Coronavirus Infection in Humans, China. Virol. Sin. 2018, 33, 104–107. [Google Scholar] [CrossRef]
- Li, H.; Mendelsohn, E.; Zong, C.; Zhang, W.; Hagan, E.; Wang, N.; Li, S.; Yan, H.; Huang, H.; Zhu, G.; et al. Human-animal interactions and bat coronavirus spillover potential among rural residents in Southern China. Biosaf. Health 2019, 1, 84–90. [Google Scholar] [CrossRef]
- Evans, T.S.; Tan, C.W.; Aung, O.; Phyu, S.; Lin, H.; Coffey, L.L.; Toe, A.T.; Aung, P.; Aung, T.H.; Aung, N.T.; et al. Exposure to diverse sarbecoviruses indicates frequent zoonotic spillover in human communities interacting with wildlife. Int. J. Infect. Dis. 2023, 131, 57–64. [Google Scholar] [CrossRef]
- Temmam, S.; Montagutelli, X.; Herate, C.; Donati, F.; Regnault, B.; Attia, M.; Baquero Salazar, E.; Chretien, D.; Conquet, L.; Jouvion, G.; et al. SARS-CoV-2-related bat virus behavior in human-relevant models sheds light on the origin of COVID-19. EMBO Rep. 2023, 24, e56055. [Google Scholar] [CrossRef]
- Tan, C.C.S.; Trew, J.; Peacock, T.P.; Mok, K.Y.; Hart, C.; Lau, K.; Ni, D.; Orme, C.D.L.; Ransome, E.; Pearse, W.D.; et al. Genomic screening of 16 UK native bat species through conservationist networks uncovers coronaviruses with zoonotic potential. Nat. Commun. 2023, 14, 3322. [Google Scholar] [CrossRef]
- Apaa, T.; Withers, A.J.; Staley, C.; Blanchard, A.; Bennett, M.; Bremner-Harrison, S.; Chadwick, E.A.; Hailer, F.; Harrison, S.W.R.; Loose, M.; et al. Sarbecoviruses of British horseshoe bats; sequence variation and epidemiology. J. Gen. Virol. 2023, 104, 001859. [Google Scholar] [CrossRef]
- Alkhovsky, S.; Lenshin, S.; Romashin, A.; Vishnevskaya, T.; Vyshemirsky, O.; Bulycheva, Y.; Lvov, D.; Gitelman, A. SARS-like Coronaviruses in Horseshoe Bats (Rhinolophus spp.) in Russia, 2020. Viruses 2022, 14, 113. [Google Scholar] [CrossRef]
- Wells, H.L.; Letko, M.; Lasso, G.; Ssebide, B.; Nziza, J.; Byarugaba, D.K.; Navarrete-Macias, I.; Liang, E.; Cranfield, M.; Han, B.A.; et al. The evolutionary history of ACE2 usage within the coronavirus subgenus Sarbecovirus. Virus Evol. 2021, 7, veab007. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Tong, S. Complete Genome Sequence of a Severe Acute Respiratory Syndrome-Related Coronavirus from Kenyan Bats. Microbiol. Resour. Announc. 2019, 8, e00548-19. [Google Scholar] [CrossRef]
- Latinne, A.; Hu, B.; Olival, K.J.; Zhu, G.; Zhang, L.; Li, H.; Chmura, A.A.; Field, H.E.; Zambrana-Torrelio, C.; Epstein, J.H.; et al. Origin and cross-species transmission of bat coronaviruses in China. Nat. Commun. 2020, 11, 4235. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef] [PubMed]
- Temmam, S.; Vongphayloth, K.; Baquero, E.; Munier, S.; Bonomi, M.; Regnault, B.; Douangboubpha, B.; Karami, Y.; Chrétien, D.; Sanamxay, D.; et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 2022, 604, 330–336. [Google Scholar] [CrossRef]
- Wacharapluesadee, S.; Tan, C.W.; Maneeorn, P.; Duengkae, P.; Zhu, F.; Joyjinda, Y.; Kaewpom, T.; Chia, W.N.; Ampoot, W.; Lim, B.L.; et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 2021, 12, 972. [Google Scholar] [CrossRef]
- Delaune, D.; Hul, V.; Karlsson, E.A.; Hassanin, A.; Ou, T.P.; Baidaliuk, A.; Gámbaro, F.; Prot, M.; Tu, V.T.; Chea, S.; et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat. Commun. 2021, 12, 6563. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Son, K.; Kim, Y.S.; Lee, S.Y.; Jheong, W.; Oem, J.K. Complete genome analysis of a SARS-like bat coronavirus identified in the Republic of Korea. Virus Genes 2019, 55, 545–549. [Google Scholar] [CrossRef]
- Murakami, S.; Kitamura, T.; Suzuki, J.; Sato, R.; Aoi, T.; Fujii, M.; Matsugo, H.; Kamiki, H.; Ishida, H.; Takenaka-Uema, A.; et al. Detection and Characterization of Bat Sarbecovirus Phylogenetically Related to SARS-CoV-2, Japan. Emerg. Infect. Dis. 2020, 26, 3025–3029. [Google Scholar] [CrossRef]
- Starr, T.N.; Zepeda, S.K.; Walls, A.C.; Greaney, A.J.; Alkhovsky, S.; Veesler, D.; Bloom, J.D. ACE2 binding is an ancestral and evolvable trait of sarbecoviruses. Nature 2022, 603, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, G.; Wang, Y.; Ren, W.; Zhao, X.; Ji, F.; Zhu, Y.; Feng, F.; Gong, M.; Ju, X.; et al. Functional and genetic analysis of viral receptor ACE2 orthologs reveals a broad potential host range of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2025373118. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef]
- Xiao, K.; Zhai, J.; Feng, Y.; Zhou, N.; Zhang, X.; Zou, J.J.; Li, N.; Guo, Y.; Li, X.; Shen, X.; et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 2020, 583, 286–289. [Google Scholar] [CrossRef]
- Ren, W.; Lan, J.; Ju, X.; Gong, M.; Long, Q.; Zhu, Z.; Yu, Y.; Wu, J.; Zhong, J.; Zhang, R.; et al. Mutation Y453F in the spike protein of SARS-CoV-2 enhances interaction with the mink ACE2 receptor for host adaption. PLoS Pathog. 2021, 17, e1010053. [Google Scholar] [CrossRef]
- Guo, H.; Hu, B.; Si, H.R.; Zhu, Y.; Zhang, W.; Li, B.; Li, A.; Geng, R.; Lin, H.F.; Yang, X.L.; et al. Identification of a novel lineage bat SARS-related coronaviruses that use bat ACE2 receptor. Emerg. Microbes Infect. 2021, 10, 1507–1514. [Google Scholar] [CrossRef]
- Mou, H.; Quinlan, B.D.; Peng, H.; Liu, G.; Guo, Y.; Peng, S.; Zhang, L.; Davis-Gardner, M.E.; Gardner, M.R.; Crynen, G.; et al. Mutations derived from horseshoe bat ACE2 orthologs enhance ACE2-Fc neutralization of SARS-CoV-2. PLoS Pathog. 2021, 17, e1009501. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, K.; Han, P.; Xu, Z.; Zheng, A.; Pan, X.; Jia, Y.; Su, C.; Tang, L.; Wu, L.; et al. Host range and structural analysis of bat-origin RshSTT182/200 coronavirus binding to human ACE2 and its animal orthologs. EMBO J. 2023, 42, e111737. [Google Scholar] [CrossRef]
- Guo, H.; Li, A.; Dong, T.Y.; Su, J.; Yao, Y.L.; Zhu, Y.; Shi, Z.L.; Letko, M. ACE2-Independent Bat Sarbecovirus Entry and Replication in Human and Bat Cells. mBio 2022, 13, e0256622. [Google Scholar] [CrossRef] [PubMed]
- Aicher, S.M.; Streicher, F.; Chazal, M.; Planas, D.; Luo, D.; Buchrieser, J.; Nemcova, M.; Seidlova, V.; Zukal, J.; Serra-Cobo, J.; et al. Species-Specific Molecular Barriers to SARS-CoV-2 Replication in Bat Cells. J. Virol. 2022, 96, e0060822. [Google Scholar] [CrossRef]
- Fujima, S.; Kosuke, Y.; Kimura, I.; Tukunaga, K.; Ito, J.; Sato, K. Determination of the factors responsible for host tropism of SARS-CoV-2-related bat coronaviruses. bioRxiv 2023, preprint. [Google Scholar]
- Yan, H.; Jiao, H.; Liu, Q.; Zhang, Z.; Xiong, Q.; Wang, B.J.; Wang, X.; Guo, M.; Wang, L.F.; Lan, K.; et al. ACE2 receptor usage reveals variation in susceptibility to SARS-CoV and SARS-CoV-2 infection among bat species. Nat. Ecol. Evol. 2021, 5, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Berto, A.; Anh, P.H.; Carrique-Mas, J.J.; Simmonds, P.; Van Cuong, N.; Tue, N.T.; Van Dung, N.; Woolhouse, M.E.; Smith, I.; Marsh, G.A.; et al. Detection of potentially novel paramyxovirus and coronavirus viral RNA in bats and rats in the Mekong Delta region of southern Viet Nam. Zoonoses Public Health 2018, 65, 30–42. [Google Scholar] [CrossRef]
- Huong, N.Q.; Nga, N.T.T.; Long, N.V.; Luu, B.D.; Latinne, A.; Pruvot, M.; Phuong, N.T.; Quang, L.T.V.; Hung, V.V.; Lan, N.T.; et al. Coronavirus testing indicates transmission risk increases along wildlife supply chains for human consumption in Viet Nam, 2013–2014. PLoS ONE 2020, 15, e0237129. [Google Scholar] [CrossRef] [PubMed]
- Phan, M.V.T.; Ngo Tri, T.; Hong Anh, P.; Baker, S.; Kellam, P.; Cotten, M. Identification and characterization of Coronaviridae genomes from Vietnamese bats and rats based on conserved protein domains. Virus Evol. 2018, 4, vey035. [Google Scholar] [CrossRef]
- Latinne, A.; Nga, N.T.T.; Long, N.V.; Ngoc, P.T.B.; Thuy, H.B.; Long, P.T.; Phuong, N.T.; Quang, L.T.V.; Tung, N.; Nam, V.S.; et al. One Health Surveillance Highlights Circulation of Viruses with Zoonotic Potential in Bats, Pigs, and Humans in Viet Nam. Viruses 2023, 15, 790. [Google Scholar] [CrossRef]
- Sikes, R.S.; Animal Care and Use Committee of the American Society of Mammalogists. Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. J. Mammal. 2016, 97, 663–688. [Google Scholar] [CrossRef]
- Wilson, D.E.; Mittermeier, R.A. (Eds.) Handbook of the Mammals of the World. In Bats; Lynx Edicions: Barcelona, Spain, 2019; Volume 9. [Google Scholar]
- Thong, V.D. Taxonomy and Echolocation of Vietnamese Bats; Publishing House for Science and Technology: Hanoi, Vietnam, 2021; 258p. (In Vietnamese) [Google Scholar]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The Barcode of Life Data System (www.barcodinglife.org). Mol. Ecol. 2007, 7, 355–364. [Google Scholar]
- Chu, D.K.; Leung, C.Y.; Gilbert, M.; Joyner, P.H.; Ng, E.M.; Tse, T.M.; Guan, Y.; Peiris, J.S.; Poon, L.L. Avian coronavirus in wild aquatic birds. J. Virol. 2011, 85, 12815–12820. [Google Scholar]
- Wu, Z.; Han, Y.; Wang, Y.; Liu, B.; Zhao, L.; Zhang, J.; Su, H.; Zhao, W.; Liu, L.; Bai, S.; et al. A comprehensive survey of bat sarbecoviruses across China in relation to the origins of SARS-CoV and SARS-CoV-2. Natl. Sci. Rev. 2022, 10, nwac213. [Google Scholar]
- C2A.A2C. Available online: https://gitlab.pasteur.fr/GIPhy/C2A.A2C (accessed on 27 June 2023).
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [PubMed]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 27 June 2023).
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2020, 7, veaa087. [Google Scholar] [PubMed]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef]
- Crawford, K.H.D.; Eguia, R.; Dingens, A.S.; Loes, A.N.; Malone, K.D.; Wolf, C.R.; Chu, H.Y.; Tortorici, M.A.; Veesler, D.; Murphy, M.; et al. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 2020, 12, 513. [Google Scholar]
- Sánchez, C.A.; Li, H.; Phelps, K.L.; Zambrana-Torrelio, C.; Wang, L.F.; Zhou, P.; Shi, Z.L.; Olival, K.J.; Daszak, P. A strategy to assess spillover risk of bat SARS-related coronaviruses in Southeast Asia. Nat. Commun. 2022, 13, 4380. [Google Scholar]
- Hassanin, A.; Tu, V.T.; Curaudeau, M.; Csorba, G. Inferring the ecological niche of bat viruses closely related to SARS-CoV-2 using phylogeographic analyses of Rhinolophus species. Sci. Rep. 2021, 11, 14276. [Google Scholar]
- Lytras, S.; Hughes, J.; Martin, D.; Swanepoel, P.; de Klerk, A.; Lourens, R.; Kosakovsky Pond, S.L.; Xia, W.; Jiang, X.; Robertson, D.L. Exploring the Natural Origins of SARS-CoV-2 in the Light of Recombination. Genome Biol. Evol. 2022, 14, evac018. [Google Scholar]
- Muylaert, R.L.; Kingston, T.; Luo, J.; Vancine, M.H.; Galli, N.; Carlson, C.J.; John, R.S.; Rulli, M.C.; Hayman, D.T.S. Present and future distribution of bat hosts of sarbecoviruses: Implications for conservation and public health. Proc. Biol. Sci. 2022, 289, 20220397. [Google Scholar]
- Wang, Z.; Huang, G.; Huang, M.; Dai, Q.; Hu, Y.; Zhou, J.; Wei, F. Global patterns of phylogenetic diversity and transmission of bat coronavirus. Sci. China Life Sci. 2023, 66, 861–874. [Google Scholar]
- Beyer, R.M.; Manica, A.; Mora, C. Shifts in global bat diversity suggest a possible role of climate change in the emergence of SARS-CoV-1 and SARS-CoV-2. Sci. Total Environ. 2021, 767, 145413. [Google Scholar]
- Li, L.; Zhang, L.; Zhou, J.; He, X.; Yu, Y.; Liu, P.; Huang, W.; Xiang, Z.; Chen, J. Epidemiology and Genomic Characterization of Two Novel SARS-Related Coronaviruses in Horseshoe Bats from Guangdong, China. mBio 2022, 13, e0046322. [Google Scholar] [PubMed]
- Chen, W.; Yan, M.; Yang, L.; Ding, B.; He, B.; Wang, Y.; Liu, X.; Liu, C.; Zhu, H.; You, B.; et al. SARS-associated coronavirus transmitted from human to pig. Emerg. Infect. Dis. 2005, 11, 446–448. [Google Scholar] [CrossRef]
- Weingartl, H.M.; Copps, J.; Drebot, M.A.; Marszal, P.; Smith, G.; Gren, J.; Andova, M.; Pasick, J.; Kitching, P.; Czub, M. Susceptibility of pigs and chickens to SARS coronavirus. Emerg. Infect. Dis. 2004, 10, 179–184. [Google Scholar] [PubMed]
- Partridge, L.J.; Urwin, L.; Nicklin, M.J.H.; James, D.C.; Green, L.R.; Monk, P.N. ACE2-Independent Interaction of SARS-CoV-2 Spike Protein with Human Epithelial Cells Is Inhibited by Unfractionated Heparin. Cells 2021, 10, 1419. [Google Scholar] [CrossRef] [PubMed]
- Chionh, Y.T.; Cui, J.; Koh, J.; Mendenhall, I.H.; Ng, J.H.J.; Low, D.; Itahana, K.; Irving, A.T.; Wang, L.F. High basal heat-shock protein expression in bats confers resistance to cellular heat/oxidative stress. Cell Stress Chaperones 2019, 24, 835–849. [Google Scholar]
- O’Shea, T.J.; Cryan, P.M.; Cunningham, A.A.; Fooks, A.R.; Hayman, D.T.; Luis, A.D.; Peel, A.J.; Plowright, R.K.; Wood, J.L. Bat flight and zoonotic viruses. Emerg. Infect. Dis. 2014, 20, 741–745. [Google Scholar]
- Tian, J.; Sun, J.; Li, D.; Wang, N.; Wang, L.; Zhang, C.; Meng, X.; Ji, X.; Suchard, M.A.; Zhang, X.; et al. Emerging viruses: Cross-species transmission of coronaviruses, filoviruses, henipaviruses, and rotaviruses from bats. Cell Rep. 2022, 39, 110969. [Google Scholar]
- Guo, H.; Hu, B.J.; Yang, X.L.; Zeng, L.P.; Li, B.; Ouyang, S.; Shi, Z.L. Evolutionary Arms Race between Virus and Host Drives Genetic Diversity in Bat Severe Acute Respiratory Syndrome-Related Coronavirus Spike Genes. J. Virol. 2020, 94, e00902-20. [Google Scholar]
- Zhou, H.; Ji, J.; Chen, X.; Bi, Y.; Li, J.; Wang, Q.; Hu, T.; Song, H.; Zhao, R.; Chen, Y.; et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 2021, 184, 4380–4391.e14. [Google Scholar] [CrossRef]
- Wang, L.; Fu, S.; Cao, Y.; Zhang, H.; Feng, Y.; Yang, W.; Nie, K.; Ma, X.; Liang, G. Discovery and genetic analysis of novel coronaviruses in least horseshoe bats in southwestern China. Emerg. Microbes Infect. 2017, 6, e14. [Google Scholar] [CrossRef]
- Damas, J.; Hughes, G.M.; Keough, K.C.; Painter, C.A.; Persky, N.S.; Corbo, M.; Hiller, M.; Koepfli, K.P.; Pfenning, A.R.; Zhao, H.; et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc. Natl. Acad. Sci. USA 2020, 117, 22311–22322. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Zhang, M.; Chang, T.L. ACE2-Independent Alternative Receptors for SARS-CoV-2. Viruses 2022, 14, 2535. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Hao, W.; Ma, B.; Li, Z.; Wang, X.; Gao, X.; Li, Y.; Qin, B.; Shang, S.; Cui, S.; Tan, Z. Binding of the SARS-CoV-2 spike protein to glycans. Sci. Bull. 2021, 66, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, Q.; Ma, L.; Wu, D.; Gao, J.; Chen, G.; Li, H. Systematic profiling of ACE2 expression in diverse physiological and pathological conditions for COVID-19/SARS-CoV-2. J. Cell Mol. Med. 2020, 24, 9478–9482. [Google Scholar] [CrossRef]
- Schroeder, S.; Mache, C.; Kleine-Weber, H.; Corman, V.M.; Muth, D.; Richter, A.; Fatykhova, D.; Memish, Z.A.; Stanifer, M.L.; Boulant, S.; et al. Functional comparison of MERS-coronavirus lineages reveals increased replicative fitness of the recombinant lineage 5. Nat. Commun. 2021, 12, 5324. [Google Scholar] [CrossRef]
- Terada, Y.; Matsui, N.; Noguchi, K.; Kuwata, R.; Shimoda, H.; Soma, T.; Mochizuki, M.; Maeda, K. Emergence of pathogenic coronaviruses in cats by homologous recombination between feline and canine coronaviruses. PLoS ONE 2014, 9, e106534. [Google Scholar] [CrossRef]
- Laude, H.; Godet, M.; Bernard, S.; Gelfi, J.; Duarte, M.; Delmas, B. Functional domains in the spike protein of transmissible gastroenteritis virus. Adv. Exp. Med. Biol. 1995, 380, 299–304. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Strain | Province | District | Commune | Hamlet | Latitude (DD.ddddd’) | Longitude (DD.ddddd’) | Elevation (meters) | Bat Species | Bat COI Accession No. | Bat Weight (g) | Genome Coverage | CoV Accession No. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RtVN21-29 | Sơn La | Sốp cộp | Mường và | Khẩy lầu | 20.882820 | 103.695280 | 936 | Rhinolophus thomasi | OR427971 | 10.7 | 100% | OR261262 |
| RtVN21-192 | Sơn La | Mộc Châu | Lóng Sập | Pha nhê/cave 1 | 20.736283 | 104.504630 | 1059 | Rhinolophus thomasi | OR435217 | 8.0 | 100% | OR261263 |
| RtVN21-193 | Sơn La | Mộc Châu | Lóng Sập | Pha nhê/cave 2 | 20.734502 | 104.505343 | 1073 | Rhinolophus thomasi | OR427972 | 9.3 | 100% | OR261264 |
| RtVN21-194 | Sơn La | Mộc Châu | Lóng Sập | Pha nhê/cave 2 | 20.734502 | 104.505343 | 1073 | Rhinolophus thomasi | OR427973 | 8.2 | 10.35% | nd |
| RsVN21-195 | Sơn La | Mộc Châu | Lóng Sập | Pha nhê/cave 2 | 20.734502 | 104.505343 | 1073 | Rhinolophus siamensis | OR427974 | 7.1 | 100% | OR261265 |
| RtVN21-197 | Sơn La | Mộc Châu | Lóng Sập | Pha nhê/cave 2 | 20.734502 | 104.505343 | 1073 | Rhinolophus thomasi | OR427975 | 7.9 | 100% | OR261266 |
| RtVN21-200 | Sơn La | Mộc Châu | Lóng Sập | Pha nhê/cave 2 | 20.734502 | 104.505343 | 1073 | Rhinolophus thomasi | OR427976 | 8.0 | 100% | OR261267 |
| RtVN21-201 | Sơn La | Mộc Châu | Lóng Sập | Pha nhê/cave 2 | 20.734502 | 104.505343 | 1073 | Rhinolophus thomasi | OR427977 | 7.3 | 100% | OR261268 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Temmam, S.; Tu, T.C.; Regnault, B.; Bonomi, M.; Chrétien, D.; Vendramini, L.; Duong, T.N.; Phong, T.V.; Yen, N.T.; Anh, H.N.; et al. Genotype and Phenotype Characterization of Rhinolophus sp. Sarbecoviruses from Vietnam: Implications for Coronavirus Emergence. Viruses 2023, 15, 1897. https://doi.org/10.3390/v15091897
Temmam S, Tu TC, Regnault B, Bonomi M, Chrétien D, Vendramini L, Duong TN, Phong TV, Yen NT, Anh HN, et al. Genotype and Phenotype Characterization of Rhinolophus sp. Sarbecoviruses from Vietnam: Implications for Coronavirus Emergence. Viruses. 2023; 15(9):1897. https://doi.org/10.3390/v15091897
Chicago/Turabian StyleTemmam, Sarah, Tran Cong Tu, Béatrice Regnault, Massimiliano Bonomi, Delphine Chrétien, Léa Vendramini, Tran Nhu Duong, Tran Vu Phong, Nguyen Thi Yen, Hoang Ngoc Anh, and et al. 2023. "Genotype and Phenotype Characterization of Rhinolophus sp. Sarbecoviruses from Vietnam: Implications for Coronavirus Emergence" Viruses 15, no. 9: 1897. https://doi.org/10.3390/v15091897