
Dynamic Evolution of SARS-CoV-2 in a Patient on Chemotherapy


Abstract
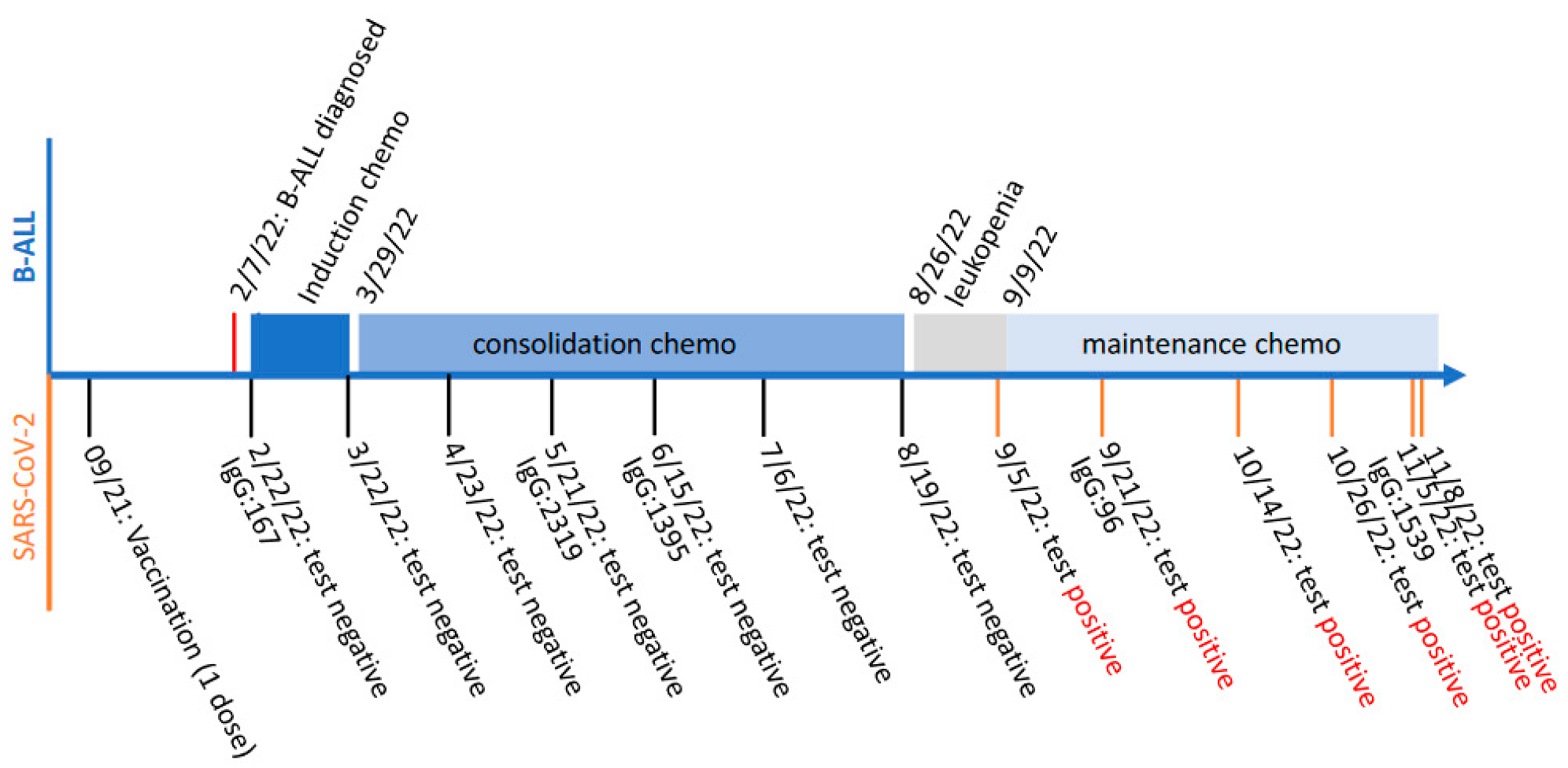
:1. Introduction
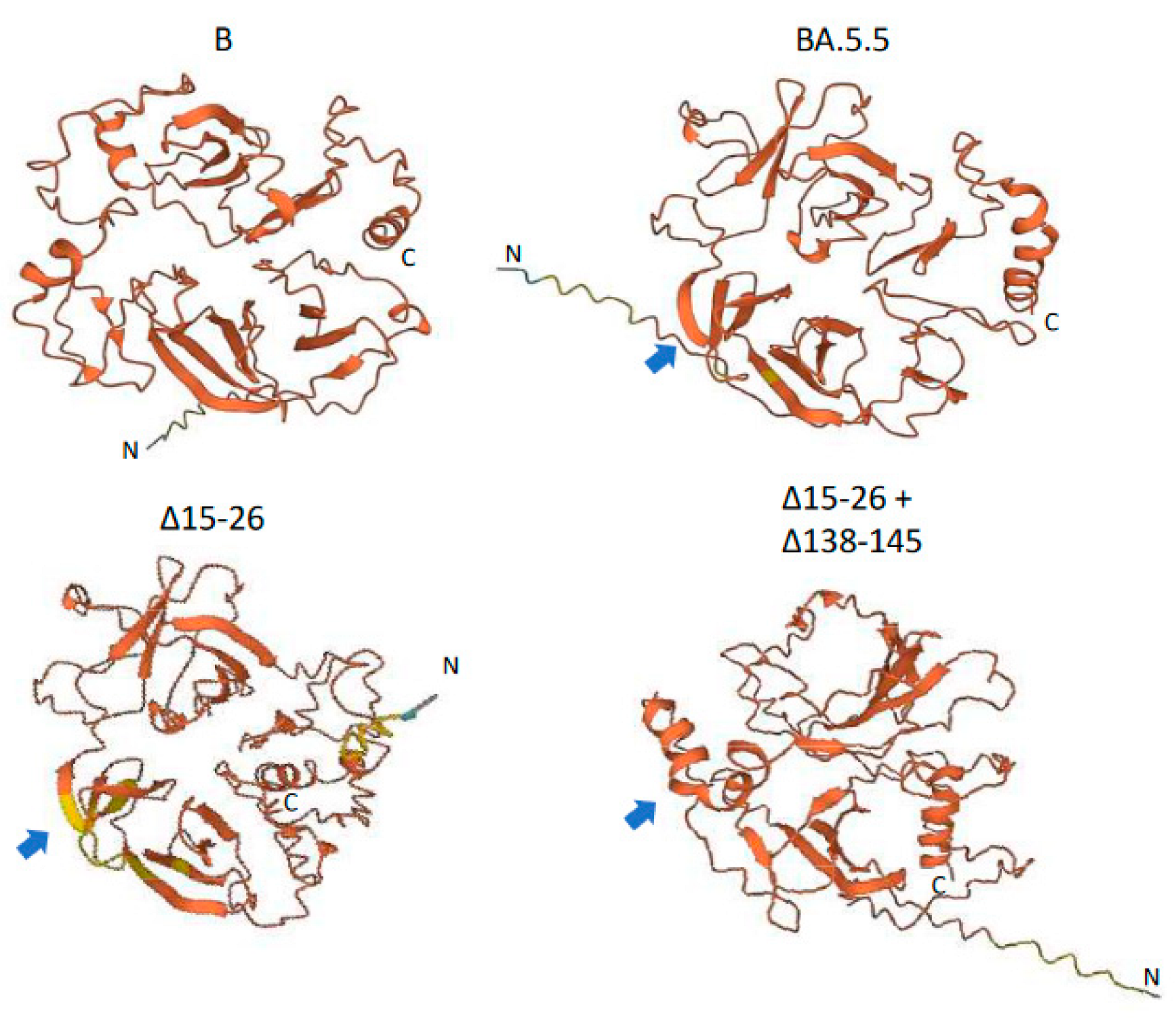
2. Results


3. Discussion
4. Materials and Methods
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradnık, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.E.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 2022, 185, 467–484 e15. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. How months-long COVID infections could seed dangerous new variants. Nature 2022, 606, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Magiorkinis, G. On the evolution of SARS-CoV-2 and the emergence of variants of concern. Trends Microbiol. 2022, 31, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.-H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Sonnleitner, S.T.; Prelog, M.; Sonnleitner, S.; Hinterbichler, E.; Halbfurter, H.; Kopecky, D.B.C.; Almanzar, G.; Koblmüller, S.; Sturmbauer, C.; Feist, L.; et al. Cumulative SARS-CoV-2 mutations and corresponding changes in immunity in an immunocompromised patient indicate viral evolution within the host. Nat. Commun. 2022, 13, 2560. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. Elife 2021, 10, e6909. [Google Scholar] [CrossRef] [PubMed]
- Itokawa, K.; Sekizuka, T.; Hashino, M.; Tanaka, R.; Kuroda, M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS ONE 2020, 15, e0239403. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic level protein structure with a language model. Science 2023, 379, 1123. [Google Scholar] [CrossRef] [PubMed]
- Pastorio, C.; Zech, F.; Jung, C.; Jacob, T.; Sanderson, T.; Sparrer, K.M.J.; Kirchhoff, F. Determinants of Spike infectivity, processing, and neutralization in SARS-CoV-2 Omicron subvariants BA.1 and BA.2. Cell Host Microbe. 2022, 30, 1255–1268.e5. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871 e13. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zost, S.J.; Greaney, A.J.; Starr, T.N.; Dingens, A.S.; Chen, E.C.; Chen, R.E.; Case, J.B.; Sutton, R.E.; Gilchuk, P.; et al. Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail. Nat. Microbiol. 2021, 6, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Z.; Ho, J.; Guo, Y.; Yeh, A.Y.; Mohri, H.; Liu, M.; Wang, M.; Yu, J.; Shah, J.G.; et al. Resistance of SARS-CoV-2 omicron subvariant BA.4.6 to antibody neutralisation. Lancet Infect. Dis. 2022, 22, 1666–1668. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Plessis, L.D.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Specimen | Collection Date | Deletion Site 1 | Deletion Site 2 | Deletion Site 3 | Site Mutation | Lineage | ||
|---|---|---|---|---|---|---|---|---|
| Δ24–26 | Δ15–26 | Δ69–70 | Δ139–145 | Δ138–145 | ||||
| Day 15 | 21 September2022 | 100% | - | 100% | - | - | - | BA.5.5 |
| Day 38 | 14 October 2022 | 43% | 57% | 100% | 10% | 4% | R346I | novel |
| Day 50 | 26 October 2022 | - | 100% | 100% | 9% | 6% | R346I | |
| Day 60 | 5 November 2022 | - | 100% | 100% | - | 100% | R346I | |
| Day 63 | 8 November 2022 | - | 100% | 100% | - | 24% | R346I | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, W.; Yin, C.; Briley, K.P.; Dalzell, W.A.B.; Fallon, J.T. Dynamic Evolution of SARS-CoV-2 in a Patient on Chemotherapy. Viruses 2023, 15, 1759. https://doi.org/10.3390/v15081759
Huang W, Yin C, Briley KP, Dalzell WAB, Fallon JT. Dynamic Evolution of SARS-CoV-2 in a Patient on Chemotherapy. Viruses. 2023; 15(8):1759. https://doi.org/10.3390/v15081759
Chicago/Turabian StyleHuang, Weihua, Changhong Yin, Kimberly P. Briley, William A. B. Dalzell, and John T. Fallon. 2023. "Dynamic Evolution of SARS-CoV-2 in a Patient on Chemotherapy" Viruses 15, no. 8: 1759. https://doi.org/10.3390/v15081759