
Emerging Genomic Trends on Rabies Virus in Davao Region, Philippines, 2018–2021
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. RABV Genome Enrichment
2.3. Library Preparation
2.4. Nanopore Sequencing
2.5. Bioinformatics Data Processing and Analysis
3. Results
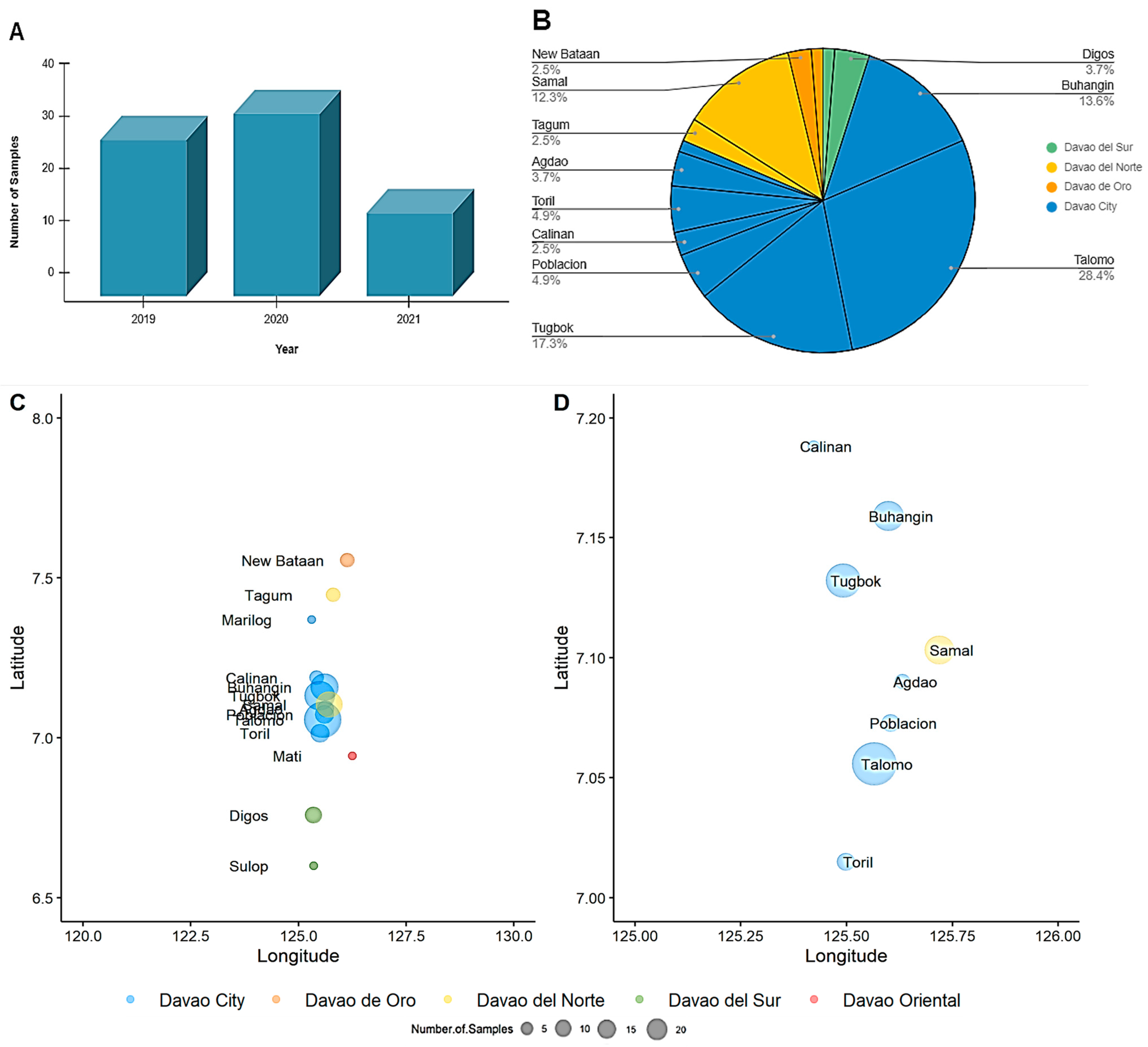
3.1. Sample Characteristics
3.2. RABV Whole Genome Sequences
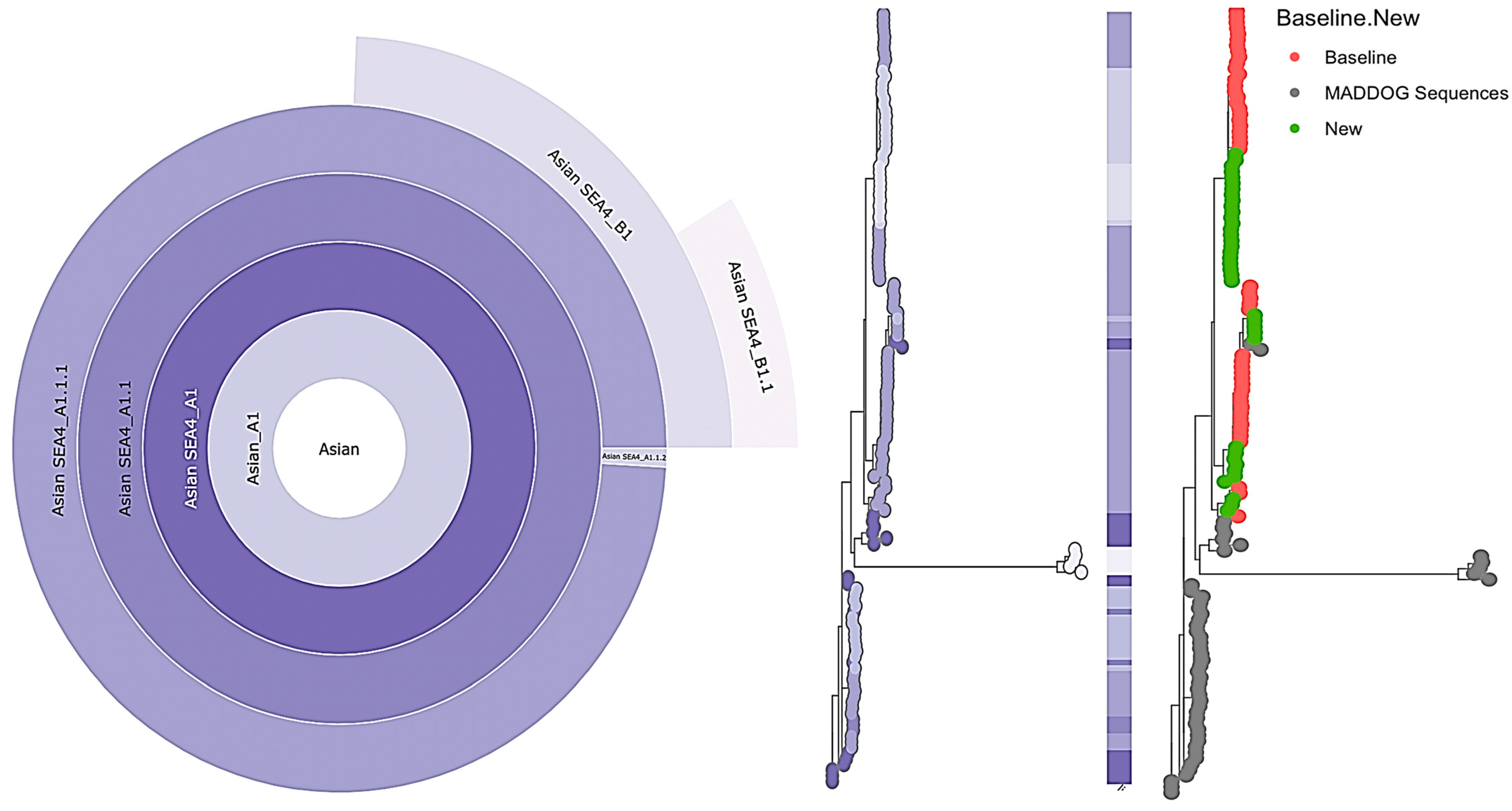
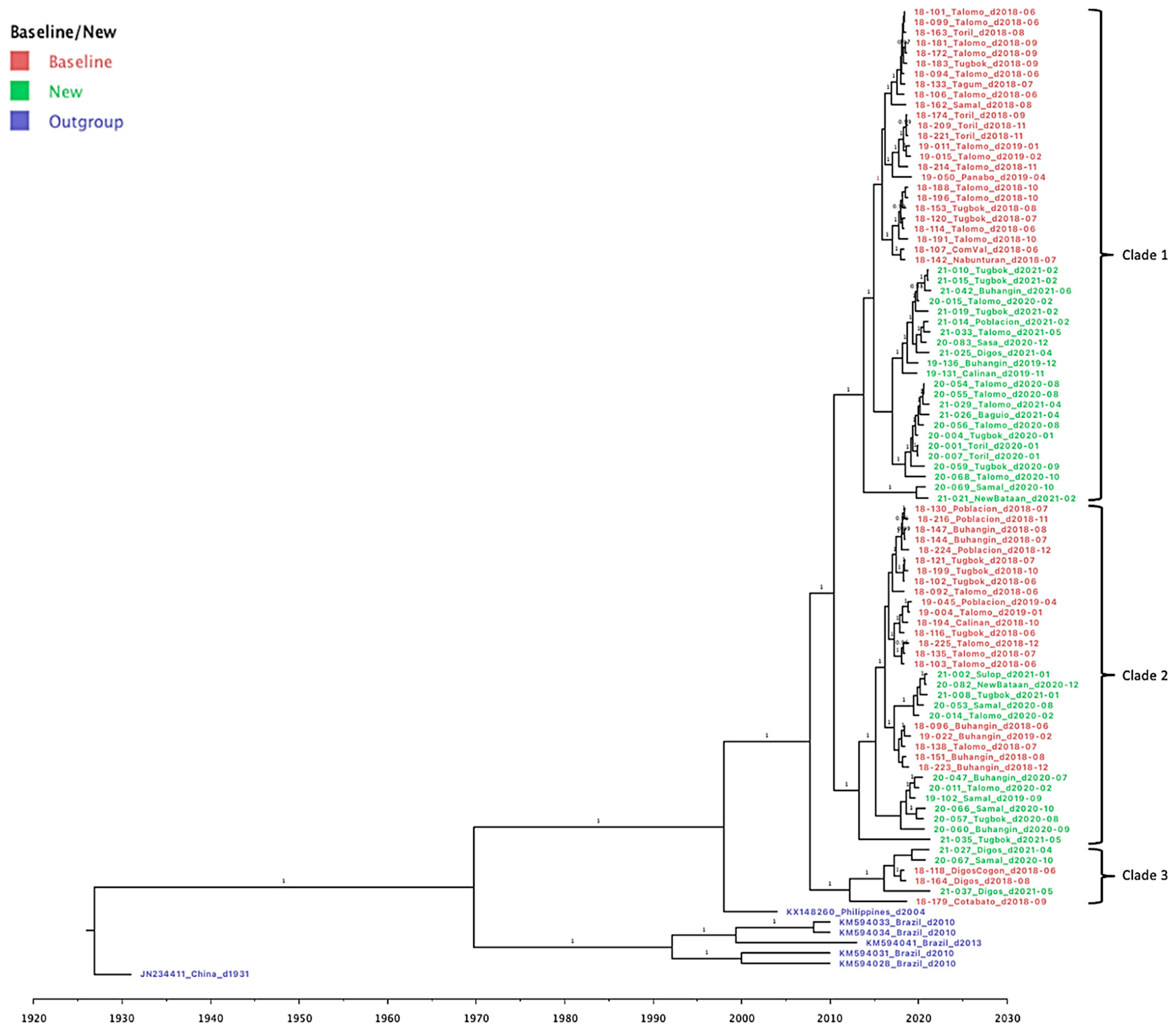
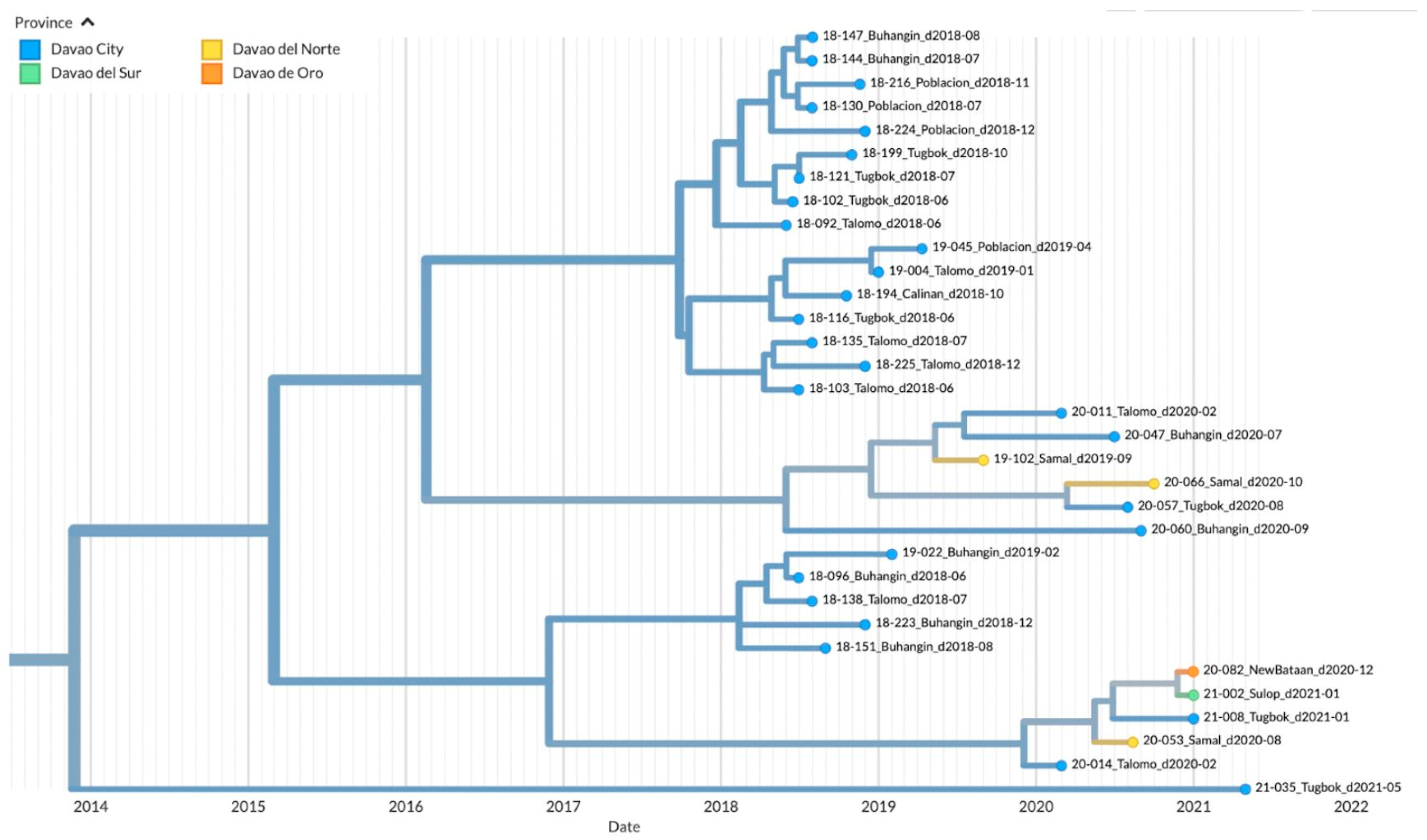
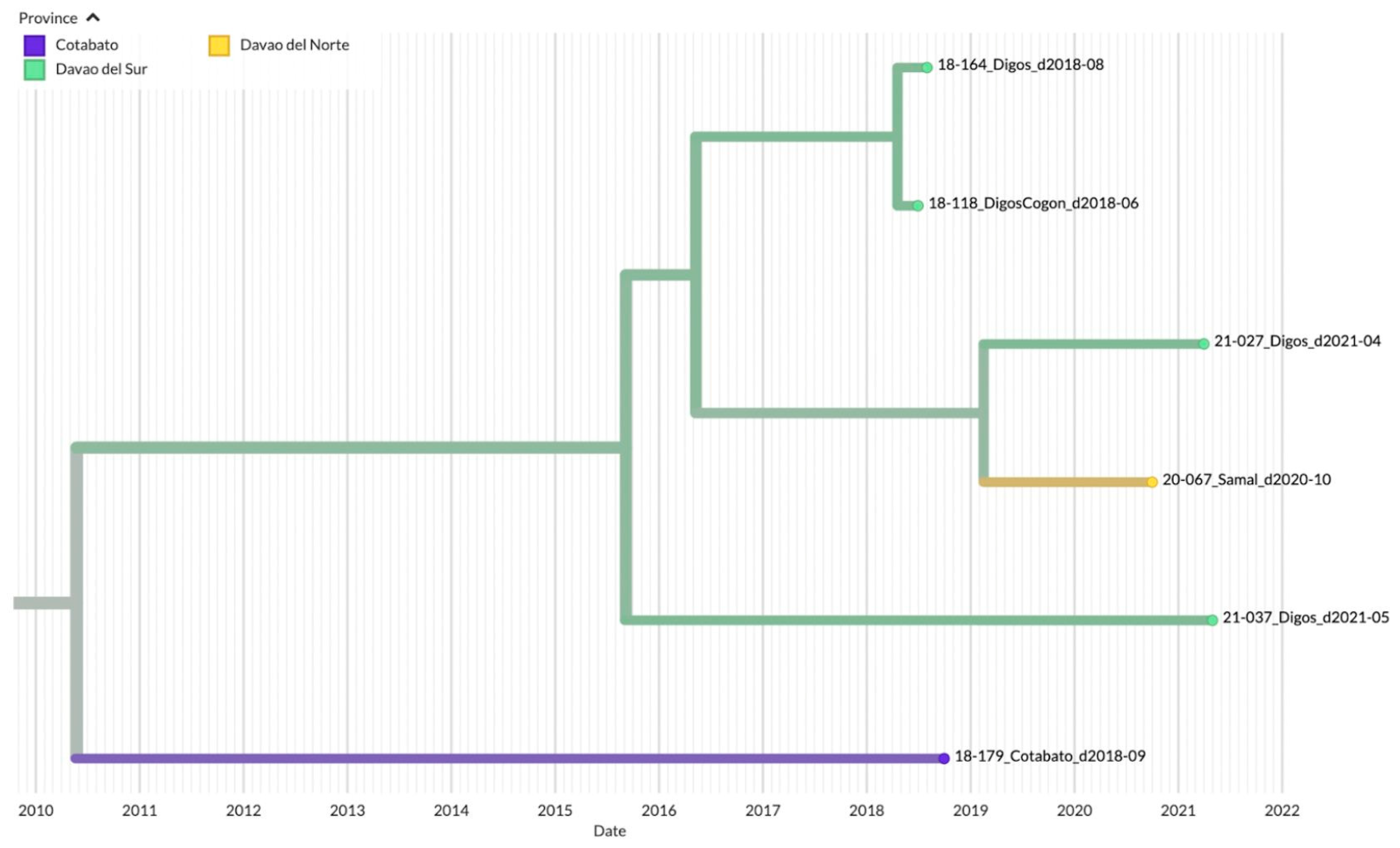
3.3. Genetic Diversity and Phylogeographic Distribution of RABV
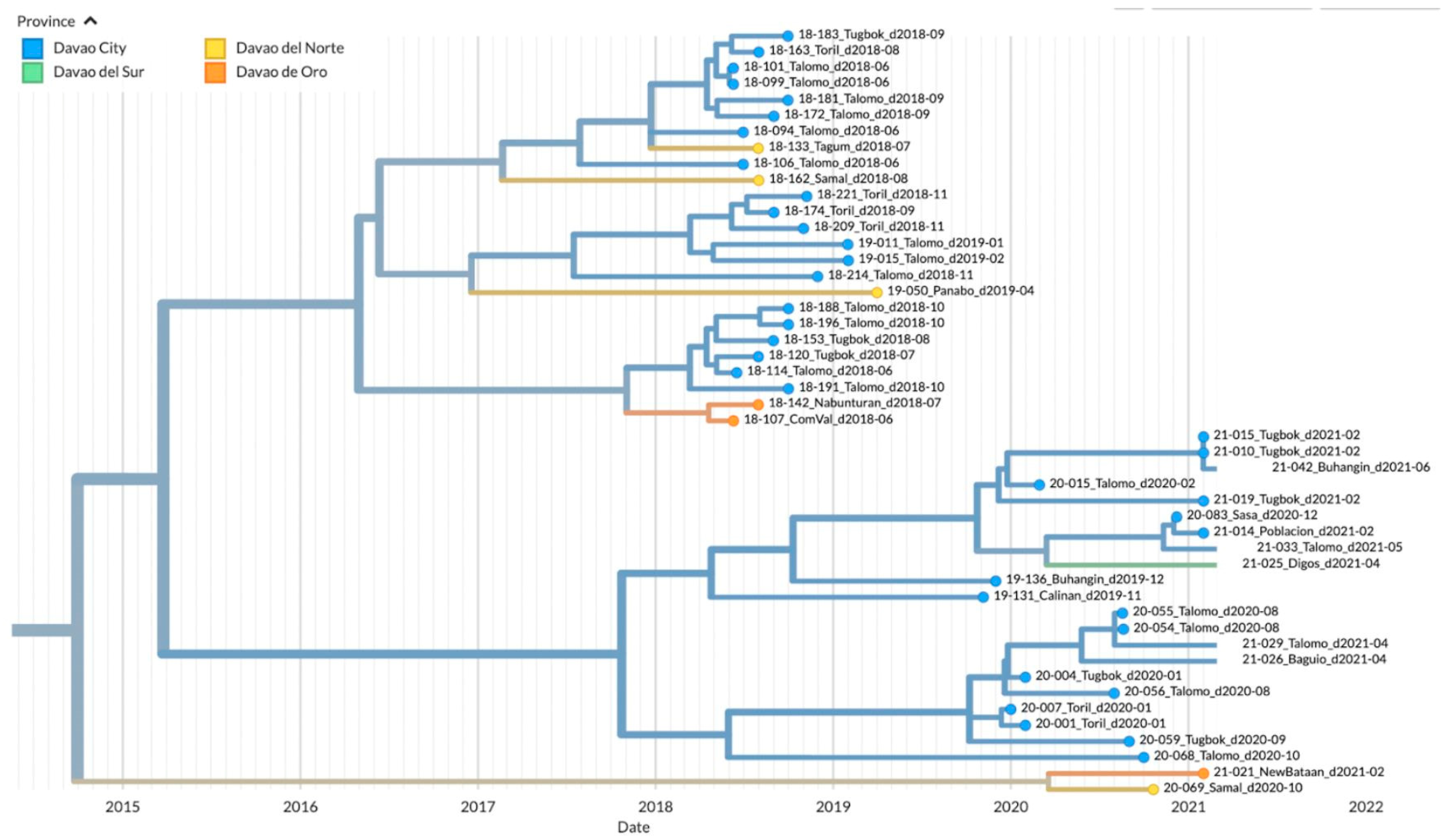
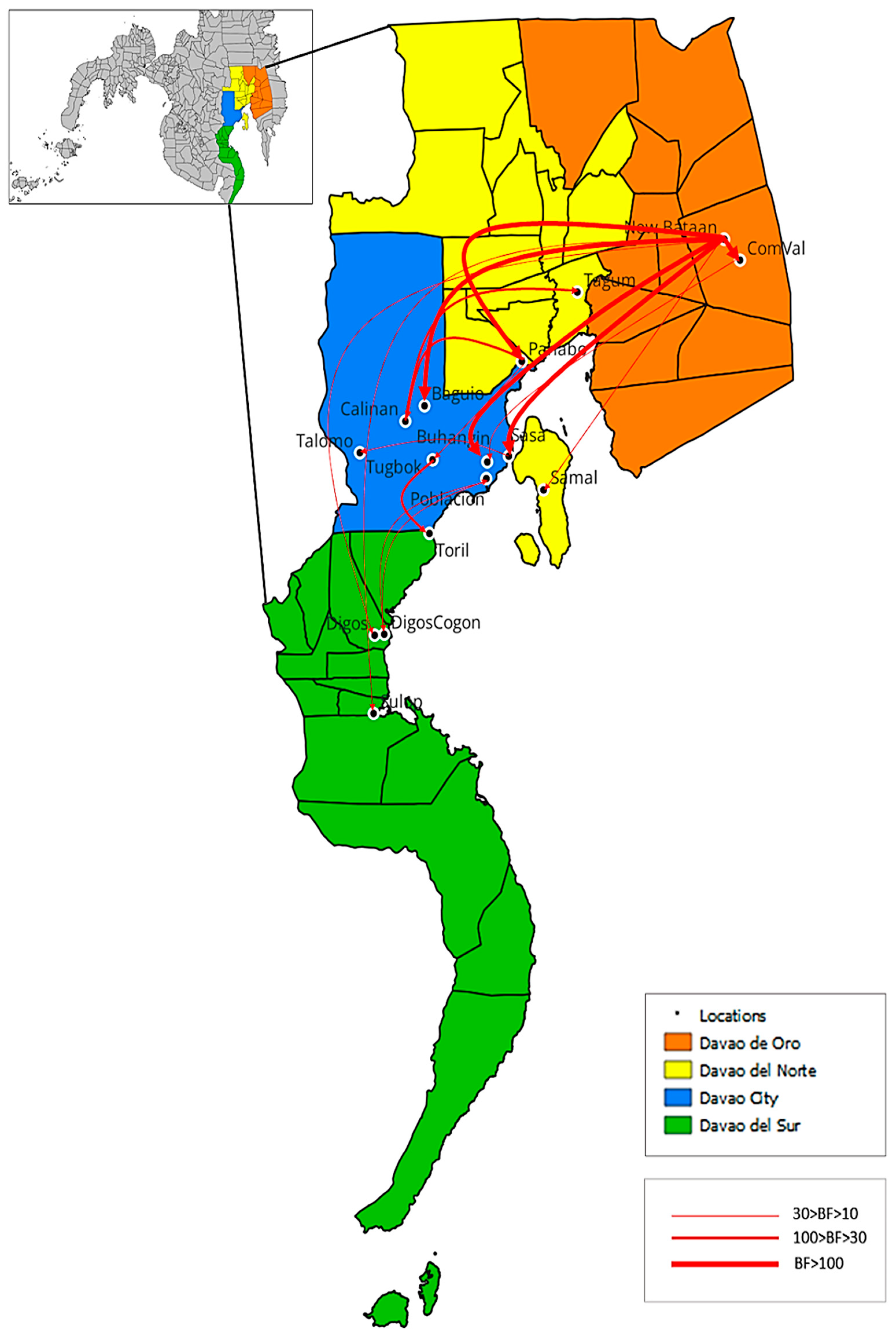
3.4. Phylodynamics of RABV in Davao Region
4. Discussion
4.1. Emerging Trends on RABV Diversity and Transmission Patterns
4.2. Genomic Surveillance in a Low Resource Setting
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Batch No. | Sample ID | Identified as RABV? | Closest Full Genome Ref Seq | Whole Genome Coverage | Breadth of Coverage | Coding Region Coverage | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | P | M | G | L | ||||||
| 1 | 27-21 | Yes | KX149259 | 94% | 98% | 100% | 100% | 100% | 100% | 91.2% |
| 29-21 | Yes | KX149259 | 96% | 99% | 100% | 100% | 100% | 100% | 94.4% | |
| 33-21 | Yes | KX149259 | 97% | 99% | 100% | 100% | 100% | 100% | 97.5% | |
| 35-21 | Yes | KX149259 | 92% | 99% | 100% | 100% | 100% | 100% | 90.9% | |
| 37-21 | Yes | KX149259 | 97% | 99% | 100% | 100% | 100% | 100% | 97.5% | |
| 42-21 | Yes | KX149259 | 97% | 99% | 100% | 100% | 100% | 100% | 97.5% | |
| 2 | 47-20 | Yes | KX149259 | 84% | 92% | 100% | 100% | 100% | 100% | 80.2% |
| 53-20 | Yes | KX149259 | 79% | 94% | 100% | 100% | 100% | 100% | 80.2% | |
| 54-20 | Yes | KX149259 | 91% | 97% | 100% | 100% | 100% | 100% | 90.9% | |
| 55-20 | Yes | KX149259 | 92% | 96% | 100% | 100% | 80.1% | 100% | 90.9% | |
| 56-20 | Yes | KX149259 | 94% | 96% | 100% | 100% | 100% | 100% | 94.5% | |
| 57-20 | Yes | KX149259 | 86% | 91% | 100% | 100% | 100% | 100% | 83.8% | |
| 59-20 | Yes | KX149259 | 93% | 96% | 100% | 100% | 100% | 100% | 94.5% | |
| 60-20 | Yes | KX149259 | 87% | 94% | 100% | 100% | 100% | 100% | 80.3% | |
| 66-20 | Yes | KX149259 | 90% | 97% | 100% | 100% | 100% | 100% | 83.8% | |
| 67-20 | Yes | KX149259 | 97% | 98% | 100% | 100% | 100% | 100% | 97.5% | |
| 68-20 | Yes | KX149259 | 96% | 98% | 100% | 100% | 80.1% | 87.1% | 94.3% | |
| 69-20 | Yes | KX149259 | 92% | 98% | 100% | 100% | 100% | 100% | 94.5% | |
| 82-20 | Yes | KX149259 | 88% | 99% | 100% | 100% | 100% | 100% | 83.4% | |
| 83-20 | Yes | KX149259 | 94% | 94% | 100% | 100% | 100% | 100% | 87.4% | |
| 02-21 | Yes | KX149259 | 87% | 96% | 100% | 100% | 100% | 100% | 83.4% | |
| 08-21 | Yes | KX149259 | 88% | 98% | 100% | 100% | 100% | 100% | 83.4% | |
| 10-21 | Yes | KX149259 | 95% | 99% | 100% | 100% | 100% | 100% | 94.5% | |
| 3 | 14-21 | Yes | KX149259 | 97% | 99% | 100% | 99.9% | 100% | 99.9% | 97.5% |
| 15-21 | Yes | KX149259 | 94% | 98% | 100% | 99.9% | 100% | 99.9% | 94.5% | |
| 19-21 | Yes | KX149259 | 96% | 99% | 100% | 99.9% | 100% | 99.9% | 94.5% | |
| 21-21 | Yes | KX149259 | 96% | 99% | 99.9% | 99.9% | 100% | 99.9% | 94.5% | |
| 25-21 | Yes | KX149259 | 94% | 98% | 100% | 99.9% | 100% | 99.9% | 94.5% | |
| 26-21 | Yes | KX149259 | 94% | 97% | 100% | 99.9% | 100% | 99.9% | 90.9% | |
| 4 | 98-19 | Yes b | KX149259 | 38% | 50% | 15.6% | 75.7% | 63.1% | 41.3% | 35.5% |
| 101-19 | Yes b | KX149259 | 25% | 33% | 15.6% | 75.7% | 31.2% | 17.0% | 21.6% | |
| 105-19 | Yes b | KX149259 | 25% | 32% | 15.6% | 75.7% | 31.2% | 17.0% | 21.6% | |
| 118-19 | Yes b | KX149259 | 32% | 51% | 0.0% | 71.3% | 31.2% | 41.3% | 35.5% | |
| 122-19 | Yes b | KX149259 | 47% | 56% | 15.6% | 75.7% | 63.1% | 60.8% | 49.9% | |
| 131-19 | Yes | KX149259 | 82% | 94% | 78.3% | 100% | 80.1% | 100% | 80.6% | |
| 102-19 | Yes | KX149259 | 85% | 93% | 83.8% | 100% | 100% | 100% | 83.4% | |
| 107-19 | Yes b | KX149259 | 69% | 89% | 63.3% | 100% | 80.1% | 69.8% | 63.7% | |
| 135-19 | Yes b | KX149259 | 80% | 89% | 83.9% | 100% | 100% | 100% | 72.3% | |
| 136-19 | Yes | KX149259 | 90% | 96% | 83.2% | 100% | 100% | 100% | 90.9% | |
| 01-20 | Yes | KX149259 | 88% | 94% | 83.9% | 100% | 100% | 100% | 84.9% | |
| 04-20 | Yes | KX149259 | 92% | 96% | 84.3% | 100% | 100% | 100% | 91.0% | |
| 07-20 | Yes | KX149259 | 95% | 98% | 98.9% | 100% | 100% | 100% | 94.5% | |
| 08-20 a | No a | NA a | NA a | 2% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | |
| 11-20 | Yes | KX149259 | 80% | 90% | 83.9% | 100% | 100% | 100% | 77.7% | |
| 14-20 | Yes | KX149259 | 88% | 93% | 100% | 100% | 100% | 100% | 83.4% | |
| 15-20 | Yes | KX149259 | 95% | 96% | 98.9% | 100% | 100% | 100% | 94.5% | |
| Sample ID | Country | Year | Lineage |
|---|---|---|---|
| AB573762 | Japan | 2006 | Asian SEA4 |
| AB573763 | Japan | 2006 | Asian SEA4 |
| EU086201 | Philippines | 1994 | Asian SEA4 |
| EU086202 | Philippines | 1994 | Asian SEA4 |
| EU086203 | Philippines | 2000 | Asian SEA4 |
| EU086204 | Philippines | 2001 | Asian SEA4 |
| KF620487 | Taiwan | 2012 | Asian SEA5 |
| KF620488 | Taiwan | 2012 | Asian SEA5 |
| KF620489 | Taiwan | 2013 | Asian SEA5 |
| KP860149 | Taiwan | 2013 | Asian SEA5 |
| KP860153 | Taiwan | 2013 | Asian SEA5 |
| KX148259 | Philippines | 1994 | Asian SEA4 |
| KX148260 | Philippines | 2004 | Asian SEA4 |
| KX148261 | Philippines | 1994 | Asian SEA4 |
| KX148262 | Philippines | 1994 | Asian SEA4 |
| KX148263 | Philippines | 1994 | Asian SEA4 |
| LC571945 | Japan | 2006 | Asian SEA4 |
| LC571946 | Japan | 2020 | Asian SEA4 |
| MN726836 | Philippines | 2013 | Asian SEA4 |
| MN726837 | Philippines | 2013 | Asian SEA4 |
| MN726838 | Philippines | 2013 | Asian SEA4 |
| MN726839 | Philippines | 2015 | Asian SEA4 |
| MN726840 | Philippines | 2015 | Asian SEA4 |
| MN726841 | Philippines | 2014 | Asian SEA4 |
| MN726843 | Philippines | 2012 | Asian SEA4 |
| MN726846 | Philippines | 2015 | Asian SEA4 |
| MN726847 | Philippines | 2017 | Asian SEA4 |
| MN726848 | Philippines | 2016 | Asian SEA4 |
| MN726851 | Philippines | 2014 | Asian SEA4 |
| MN726853 | Philippines | 2012 | Asian SEA4 |
| MN726854 | Philippines | 2015 | Asian SEA4 |
| MN726855 | Philippines | 2016 | Asian SEA4 |
| MN726856 | Philippines | 2012 | Asian SEA4 |
| MN726858 | Philippines | 2016 | Asian SEA4 |
| MN726861 | Philippines | 2015 | Asian SEA4 |
| MN726862 | Philippines | 2013 | Asian SEA4 |
| MN726865 | Philippines | 2014 | Asian SEA4 |
| MN726866 | Philippines | 2014 | Asian SEA4 |
| MN726867 | Philippines | 2012 | Asian SEA4 |
| MN726868 | Philippines | 2016 | Asian SEA4 |
| MN726869 | Philippines | 2013 | Asian SEA4 |
| MN726870 | Philippines | 2014 | Asian SEA4 |
| MN726871 | Philippines | 2013 | Asian SEA4 |
| MN726872 | Philippines | 2017 | Asian SEA4 |
| MN726873 | Philippines | 2014 | Asian SEA4 |
| MN726874 | Philippines | 2012 | Asian SEA4 |
| MN726879 | Philippines | 2012 | Asian SEA4 |
| MN726881 | Philippines | 2015 | Asian SEA4 |
| MN726882 | Philippines | 2019 | Asian SEA4 |
| MN857169 | Philippines | 2019 | Asian SEA4 |
| Sample ID | New/Baseline | Length | Year | Lineage |
|---|---|---|---|---|
| 21-002_Sulop_d2021-01 | New | 10,370 | 2021 | Asian SEA4_A1.1.2 |
| 21-008_Tugbok_d2021-01 | New | 10,551 | 2021 | Asian SEA4_A1.1.1 |
| 21-010_Tugbok_d2021-02 | New | 11,331 | 2021 | Asian SEA4_B1.1 |
| 20-047_Buhangin_d2020-07 | New | 9965 | 2020 | Asian SEA4_A1.1.1 |
| 20-053_Samal_d2020-08 | New | 9481 | 2020 | Asian SEA4_A1.1.1 |
| 20-054_Talomo_d2020-08 | New | 10,851 | 2020 | Asian SEA4_A1.1.1 |
| 20-055_Talomo_d2020-08 | New | 11,035 | 2020 | Asian SEA4_A1.1.1 |
| 20-056_Talomo_d2020-08 | New | 11,195 | 2020 | Asian SEA4_A1.1.1 |
| 20-057_Tugbok_d2020-08 | New | 10,219 | 2020 | Asian SEA4_A1.1.1 |
| 20-059_Tugbok_d2020-09 | New | 11,039 | 2020 | Asian SEA4_A1.1.1 |
| 20-060_Buhangin_d2020-09 | New | 10,347 | 2020 | Asian SEA4_A1.1.1 |
| 20-066_Samal_d2020-10 | New | 10,737 | 2020 | Asian SEA4_A1.1.1 |
| 20-067_Samal_d2020-10 | New | 11,626 | 2020 | Asian SEA4_A1.1.1 |
| 20-068_Talomo_d2020-10 | New | 11,397 | 2020 | Asian SEA4_A1.1.1 |
| 20-069_Samal_d2020-10 | New | 11,013 | 2020 | Asian SEA4_B1 |
| 20-082_NewBataan_d2020-12 | New | 10,550 | 2020 | Asian SEA4_A1.1.1 |
| 20-083_Sasa_d2020-12 | New | 10,473 | 2020 | Asian SEA4_B1.1 |
| 21-027_Digos_d2021-04 | New | 11,221 | 2021 | Asian SEA4_A1.1.1 |
| 21-029_Talomo_d2021-04 | New | 11,422 | 2021 | Asian SEA4_A1.1.1 |
| 21-033_Talomo_d2021-05 | New | 11,619 | 2021 | Asian SEA4_B1.1 |
| 21-035_Tugbok_d2021-05 | New | 10,958 | 2021 | Asian SEA4_A1.1.1 |
| 21-037_Digos_d2021-05 | New | 11,570 | 2021 | Asian SEA4_A1.1.1 |
| 21-042_Buhangin_d2021-06 | New | 11,587 | 2021 | Asian SEA4_B1.1 |
| 18-092_Talomo_d2018-06 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-094_Talomo_d2018-06 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-096_Buhangin_d2018-06 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-099_Talomo_d2018-06 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-101_Talomo_d2018-06 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-102_Tugbok_d2018-06 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-103_Talomo_d2018-06 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-106_Talomo_d2018-06 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-107_ComVal_d2018-06 | New | 11,800 | 2018 | Asian SEA4_B1 |
| 18-114_Talomo_d2018-06 | New | 11,800 | 2018 | Asian SEA4_B1 |
| 18-116_Tugbok_d2018-06 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-118_DigosCogon_d2018-06 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-120_Tugbok_d2018-07 | New | 11,800 | 2018 | Asian SEA4_B1 |
| 18-121_Tugbok_d2018-07 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-130_Poblacion_d2018-07 | New | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-133_Tagum_d2018-07 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-135_Talomo_d2018-07 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-138_Talomo_d2018-07 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-142_Nabunturan_d2018-07 | Baseline | 11,800 | 2018 | Asian SEA4_B1 |
| 18-144_Buhangin_d2018-07 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-147_Buhangin_d2018-08 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-151_Buhangin_d2018-08 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-153_Tugbok_d2018-08 | Baseline | 11,800 | 2018 | Asian SEA4_B1 |
| 18-162_Samal_d2018-08 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-163_Toril_d2018-08 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-164_Digos_d2018-08 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-172_Talomo_d2018-09 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-174_Toril_d2018-09 | Baseline | 11,800 | 2018 | Asian SEA4_B1 |
| 18-179_Cotabato_d2018-09 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-181_Talomo_d2018-09 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-183_Tugbok_d2018-09 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-188_Talomo_d2018-10 | Baseline | 11,800 | 2018 | Asian SEA4_B1 |
| 18-191_Talomo_d2018-10 | Baseline | 11,800 | 2018 | Asian SEA4_B1 |
| 18-194_Calinan_d2018-10 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-196_Talomo_d2018-10 | Baseline | 11,800 | 2018 | Asian SEA4_B1 |
| 18-199_Tugbok_d2018-10 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-209_Toril_d2018-11 | Baseline | 11,800 | 2018 | Asian SEA4_B1 |
| 18-214_Talomo_d2018-11 | Baseline | 11,800 | 2018 | Asian SEA4_B1 |
| 18-216_Poblacion_d2018-11 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-221_Toril_d2018-11 | Baseline | 11,800 | 2018 | Asian SEA4_B1 |
| 18-223_Buhangin_d2018-12 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-224_Poblacion_d2018-12 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 18-225_Talomo_d2018-12 | Baseline | 11,800 | 2018 | Asian SEA4_A1.1.1 |
| 19-004_Talomo_d2019-01 | Baseline | 11,800 | 2019 | Asian SEA4_A1.1.1 |
| 19-011_Talomo_d2019-01 | Baseline | 11,800 | 2019 | Asian SEA4_B1 |
| 19-015_Talomo_d2019-02 | Baseline | 11,800 | 2019 | Asian SEA4_B1 |
| 19-022_Buhangin_d2019-02 | Baseline | 11,800 | 2019 | Asian SEA4_A1.1.1 |
| 19-045_Poblacion_d2019-04 | Baseline | 11,800 | 2019 | Asian SEA4_A1.1.1 |
| 19-050_Panabo_d2019-04 | Baseline | 11,800 | 2019 | Asian SEA4_B1 |
| 19-102_Samal_d2019-09 | Baseline | 10,122 | 2019 | Asian SEA4_A1.1.1 |
| 19-131_Calinan_d2019-11 | Baseline | 9804 | 2019 | Asian SEA4_B1 |
| 19-136_Buhangin_d2019-12 | Baseline | 10,697 | 2019 | Asian SEA4_B1.1 |
| 20-001_Toril_d2020-01 | Baseline | 10,531 | 2020 | Asian SEA4_A1.1.1 |
| 20-004_Tugbok_d2020-01 | Baseline | 10,958 | 2020 | Asian SEA4_A1.1.1 |
| 20-007_Toril_d2020-01 | Baseline | 11,284 | 2020 | Asian SEA4_A1.1.1 |
| 20-011_Talomo_d2020-02 | Baseline | 9553 | 2020 | Asian SEA4_A1.1.1 |
| 20-014_Talomo_d2020-02 | Baseline | 10,535 | 2020 | Asian SEA4_A1.1.1 |
| 20-015_Talomo_d2020-02 | Baseline | 11,378 | 2020 | Asian SEA4_B1.1 |
| 21-014_Poblacion_d2021-02 | Baseline | 11,547 | 2021 | Asian SEA4_B1.1 |
| 21-015_Tugbok_d2021-02 | Baseline | 11,182 | 2021 | Asian SEA4_B1.1 |
| 21-019_Tugbok_d2021-02 | Baseline | 11,404 | 2021 | Asian SEA4_B1.1 |
| 21-021_NewBataan_d2021-02 | Baseline | 11,411 | 2021 | Asian SEA4_B1 |
| 21-025_Digos_d2021-04 | Baseline | 11,243 | 2021 | Asian SEA4_B1.1 |
| 21-026_Baguio_d2021-04 | Baseline | 11,163 | 2021 | Asian SEA4_A1.1.1 |
References
- World Health Organization; Food and Agriculture Organization of the United Nations; World Organisation for Animal Health. Zero by 30: The Global Strategic Plan to End Human Deaths from Dog-Mediated Rabies by 2030. 2018. Available online: https://apps.who.int/iris/handle/10665/272756 (accessed on 20 January 2022).
- Tarantola, A. Four thousand years of concepts relating to rabies in animals and humans, its prevention and its cure. Trop. Med. Infect. Dis. 2017, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Melo, G.D.; Sonthonnax, F.; Lepousez, G.; Jouvion, G.; Minola, A.; Zatta, F.; Larrous, F.; Kergoat, L.; Mazo, C.; Moigneu, C.; et al. A combination of two human monoclonal antibodies cures symptomatic rabies. EMBO Mol. Med. 2020, 12, e12628. [Google Scholar] [CrossRef] [PubMed]
- Cantaert, T.; Borand, L.; Kergoat, L.; Leng, C.; Ung, S.; In, S.; Peng, Y.; Phoeun, C.; Hing, C.; Taing, C.N.; et al. A 1-week intradermal dose-sparing regimen for rabies post-exposure prophylaxis (RESIST-2): An observational cohort study. Lancet Infect. Dis. 2019, 19, 1355–1362. [Google Scholar] [CrossRef]
- Department of Health. Anti-Rabies Act (RA 9482) and Other Issuances on Rabies Control and Prevention Programs. 2007. Available online: https://rabies.doh.gov.ph/images/PDF/IRR-INSIDE-REV2_new.pdf (accessed on 16 December 2022).
- Department of Health. National Rabies Prevention and Control Program. Strategic Plan 2020–2025. 2020. Available online: https://rr-asia.woah.org/wp-content/uploads/2020/03/final-mtp-rabies_philippines.pdf (accessed on 16 December 2022).
- Lachica, Z.P.T.; Evangelio, S.A.; Diamante, E.O.; Clemente, A.J.; Peralta, J.M.; Murao, L.A.E.; Mata, M.A.E.; Alviola, P.A., IV. Trends of canine rabies lyssavirus and impact of the intensified rabies control program in Davao city, Philippines: 2006–2017. Philipp. J. Sci. 2019, 148, 751–763. [Google Scholar]
- Klepac, P.; Metcalf, C.J.E.; McLean, A.R.; Hampson, K. Towards the endgame and beyond: Complexities and challenges for the elimination of infectious diseases. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotton, J.A.; Berriman, M.; Dalén, L.; Barnes, I. Eradication genomics—Lessons for parasite control. Science 2018, 361, 130–131. [Google Scholar]
- Balloux, F.; Brynildsrud, O.B.; van Dorp, L.; Shaw, L.P.; Chen, H.; Harris, K.A.; Wang, H.; Eldholm, V. From theory to practice: Translating whole-genome sequencing (WGS) into the clinic. Trends Microbiol. 2018, 26, 1035–1048. [Google Scholar] [CrossRef] [Green Version]
- Brunker, K.; Jaswant, G.; Thumbi, S.M.; Lushasi, K.; Lugelo, A.; Czupryna, A.M.; Ade, F.; Wambura, G.; Chuchu, V.; Steenson, R.; et al. Rapid in-country sequencing of whole virus genomes to inform rabies elimination programmes. Wellcome Open Res. 2020, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y. Advent of a new sequencing era: Long-read and on-site sequencing. J. Hum. Genet. 2020, 65, 1. [Google Scholar] [CrossRef]
- Mikheyev, A.S.; Tin, M.M.Y. A first look at the Oxford Nanopore MinION sequencer. Mol. Ecol. Resour. 2014, 14, 1097–1102. [Google Scholar] [CrossRef]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION sequencing device for microbial whole genome sequencing applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef] [Green Version]
- Bacus, M.G.; Buenaventura, S.G.C.; Mamites, A.M.C.; Elizagaque, H.G.; Labrador, C.C.; Delfin, F.C.; Eng, M.N.J.; Lagare, A.P.; Marquez, G.N.; Murao, L.A.E. Genome-based local dynamics of canine rabies virus epidemiology, transmission, and evolution in Davao City, Philippines, 2018–2019. Infect. Genet. Evol. 2021, 92, 104868. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 2010, 26, 1899–1900. [Google Scholar] [CrossRef] [Green Version]
- Penir, S. Calculating Mapping Statistics from a SAM/BAM File Using SAMtools and Awk. 2017. Available online: https://sarahpenir.github.io/bioinformatics/awk/calculating-mapping-stats-from-a-bam-file-using-samtools-and-awk/ (accessed on 16 December 2022).
- Morobe, J.M.; Pool, B.; Marie, L.; Didon, D.; Lambisia, A.W.; Makori, T.; Mohammed, K.S.; de Laurent, Z.R.; Ndwiga, L.; Mburu, M.W.; et al. Genomic Epidemiology of SARS-CoV-2 in Seychelles, 2020–2021. Viruses 2022, 14, 1318. [Google Scholar] [CrossRef] [PubMed]
- Ruis, C. Bammix v1.0.0. 2022. Available online: https://github.com/chrisruis/bammix (accessed on 12 January 2023).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.; Gifford, R.J.; Singer, J.; Hill, V.; O’toole, A.; Rambaut, A.; Hampson, K.; Brunker, K. Making genomic surveillance deliver: A lineage classification and nomenclature system to inform rabies elimination. PLoS Pathog. 2022, 18, e1010023. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Plessis, L.D.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Philippine Statistics Authority Gross Regional Domestic Product by Year Published. 2020. Available online: https://psa.gov.ph/grdp/tables (accessed on 22 June 2022).
- Denduangboripant, J.; Wacharapluesadee, S.; Lumlertdacha, B.; Ruankaew, N.; Hoonsuwan, W.; Puanghat, A.; Hemachudha, T. Transmission dynamics of rabies virus in Thailand: Implications for disease control. BMC Infect. Dis. 2005, 5, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohma, K.; Saito, M.; Kamigaki, T.; Tuason, L.T.; Demetria, C.S.; Orbina, J.R.C.; Manalo, D.L.; Miranda, M.E.; Noguchi, A.; Inoue, S.; et al. Phylogeographic analysis of rabies viruses in the Philippines. Infect. Genet. Evol. 2014, 23, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbi, C.; Lemey, P.; Suchard, M.A.; Abdelatif, E.; Elharrak, M.; Jalal, N.; Faouzi, A.; Echevarría, J.E.; Morón, S.V.; Rambaut, A.; et al. Phylodynamics and human-mediated dispersal of a zoonotic virus. PLoS Pathog. 2010, 6, e1001166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layan, M.; Dellicour, S.; Baele, G.; Cauchemez, S.; Bourhy, H. Mathematical modelling and phylodynamics for the study of dog rabies dynamics and control: A scoping review. PLoS Neglected Trop. Dis. 2021, 15, e0009449. [Google Scholar] [CrossRef] [PubMed]
- Zinsstag, J.; Lechenne, M.; Laager, M.; Mindekem, R.; Naïssengar, S.; Oussiguéré, A.; Bidjeh, K.; Rives, G.; Tessier, J.; Madjaninan, S.; et al. Vaccination of dogs in an African city interrupts rabies transmission and reduces human exposure. Sci. Transl. Med. 2017, 9, eaaf6984. [Google Scholar] [CrossRef] [Green Version]
- Bourhy, H.; Nakouné, E.; Hall, M.; Nouvellet, P.; Lepelletier, A.; Talbi, C.; Watier, L.; Holmes, E.C.; Cauchemez, S.; Lemey, P.; et al. Revealing the micro-scale signature of endemic zoonotic disease transmission in an African urban setting. PLoS Pathog. 2016, 12, e1005525. [Google Scholar] [CrossRef] [Green Version]
- Hayman, D.T.S.; Johnson, N.; Horton, D.L.; Hedge, J.; Wakeley, P.R.; Banyard, A.C.; Zhang, S.; Alhassan, A.; Fooks, A.R. Evolutionary history of rabies in Ghana. PLoS Neglected Trop. Dis. 2011, 5, e1001. [Google Scholar] [CrossRef] [Green Version]
- De Benedictis, P.; Sow, A.; Fusaro, A.; Veggiato, C.; Talbi, C.; Kaboré, A.; Dundon, W.G.; Bourhy, H.; Capua, I. Phylogenetic analysis of rabies viruses from Burkina Faso, 2007. Zoonoses Public Health 2010, 57, e42–e46. [Google Scholar] [CrossRef]
- Salomão, C.; Nacima, A.; Cuamba, L.; Gujral, L.; Amiel, O.; Baltazar, C.; Cliff, J.; Gudo, E.S. Epidemiology, clinical features and risk factors for human rabies and animal bites during an outbreak of rabies in Maputo and Matola cities, Mozambique, 2014: Implications for public health interventions for rabies control. PLoS Neglected Trop. Dis. 2017, 11, e0005787. [Google Scholar] [CrossRef] [Green Version]
- Beran, G.W. Ecology of dogs in the Central Philippines in relation to rabies control efforts. Comp. Immunol. Microbiol. Infect. Dis. 1982, 5, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Rysava, K.; Mancero, T.; Caldas, E.; de Carvalho, M.F.; Castro, A.P.B.; Gutiérrez, V.; Haydon, D.T.; Johnson, P.C.D.; Mancy, R.; Montebello, L.R.; et al. Towards the elimination of dog-mediated rabies: Development and application of an evidence-based management tool. BMC Infect. Dis. 2020, 20, 778. [Google Scholar] [CrossRef] [PubMed]
- Gigante, C.M.; Yale, G.; Condori, R.E.; Costa, N.C.; Van Long, N.; Minh, P.Q.; Chuong, V.D.; Tho, N.D.; Thanh, N.T.; Thin, N.X.; et al. Portable rabies virus sequencing in canine rabies endemic countries using the Oxford Nanopore MinION. Viruses 2020, 12, 1255. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Han, A.; Zlitni, S.; Brooks, E.F.; Vance, S.E.; Wolfe, M.; Singh, U.; Jagannathan, P.; Pinsky, B.A.; Boehm, A.; et al. Standardized preservation, extraction and quantification techniques for detection of fecal SARS-CoV-2 RNA. Nat. Commun. 2021, 12, 5753. [Google Scholar] [CrossRef]







| Procedure | Challenges Encountered | Original Protocol | Modification |
|---|---|---|---|
| RNA Extraction | Prescribed kit is unavailable | Zymo Research Quick-RNA miniprep kit, 2 mL ceramic tubes (1.4 mm), and Terralyzer | Biospin Viral DNA/RNA Extraction Kit, 2 mL ceramic tubes (2.8 mm), Bead Mill Homogenizer |
| Multiplex PCR | Low concentration of PCR products | Conventional multiplex PCR | Touchdown multiplex PCR |
| Library Preparation | Low concentration post barcode ligation | 5 ng DNA input per sample; 70% ethanol for cleanup wash step | 50 ng DNA input per sample; Freshly prepared 80% ethanol for cleanup wash step |
| Ultra II Ligation Module for Native Barcoding not available in purchased kit | Ultra II Ligation Master Mix and Ligation Enhancer | Blunt/TA Ligase Master Mix diluted with NFW | |
| Sequencing | Low read number Recycling of flow cells | Run can finish in six hours, basecalling mode on for fast basecalling n/a | Run extended to 16 h or more, basecalling mode off Yes, but excluded sequences that overlapped barcodes with samples from previous run |
| Sequence Batch No. | No. of Samples a | Concentration of Barcoded Amplicon Pool (ng/μL) | Concentration of Final Library (ng/μL) |
|---|---|---|---|
| 1 | 12 b | 3.78 | 0.956 |
| 2 | 17 | 1.39 | 1.28 |
| 3 | 17 | 1.18 | 1.21 |
| 4 | 17 | 0.84 | 0.14 |
| From | To | Support |
|---|---|---|
| Intra-city transmission | ||
| Sasa 1 | Talomo 1 | Strong |
| Tugbok 1 | Toril 1 | Very strong |
| Inter-city transmission | ||
| Poblacion 1 | DigosCogon 2 | Strong |
| ComVal 4 | Buhangin 1 | Strong |
| DigosCogon 2 | Poblacion 1 | Strong |
| Panabo 3 | Tugbok 1 | Strong |
| Calinan 1 | Tagum 3 | Very strong |
| Panabo 3 | Calinan 1 | Very strong |
| New Bataan 4 | Baguio 1 | Decisive |
| New Bataan 4 | Sasa 1 | Decisive |
| New Bataan 4 | Buhangin 1 | Decisive |
| Intra-province transmission | ||
| New Bataan 4 | ComVal 4 | Decisive |
| Inter-province transmission | ||
| New Bataan 4 | Digos 2 | Strong |
| New Bataan 4 | Sulop 2 | Strong |
| New Bataan 4 | Samal 3 | Strong |
| New Bataan 4 | Panabo 3 | Decisive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capin, J.B.G.; Sanque, A.J.C.; Eng, M.N.J.; Lagare, A.; Sepulveda, M.C.B.; Murao, L.A.E. Emerging Genomic Trends on Rabies Virus in Davao Region, Philippines, 2018–2021. Viruses 2023, 15, 1658. https://doi.org/10.3390/v15081658
Capin JBG, Sanque AJC, Eng MNJ, Lagare A, Sepulveda MCB, Murao LAE. Emerging Genomic Trends on Rabies Virus in Davao Region, Philippines, 2018–2021. Viruses. 2023; 15(8):1658. https://doi.org/10.3390/v15081658
Chicago/Turabian StyleCapin, Jessel Babe G., Angela Jahn C. Sanque, Maria Noreen J. Eng, Arlene Lagare, Maria Corazon B. Sepulveda, and Lyre Anni E. Murao. 2023. "Emerging Genomic Trends on Rabies Virus in Davao Region, Philippines, 2018–2021" Viruses 15, no. 8: 1658. https://doi.org/10.3390/v15081658