
Foot-and-Mouth Disease Virus Serotype O Exhibits Phenomenal Genetic Lineage Diversity in India during 2018–2022

 , and
, and
Abstract
:1. Introduction
2. Materials and Method
2.1. Virus Isolation
2.2. VP1 Sequencing
2.3. Phylogenetic Analysis
2.4. Phylodynamic Analysis
2.5. Selection Pressure Analysis
2.6. Vaccine Matching Analysis
3. Results
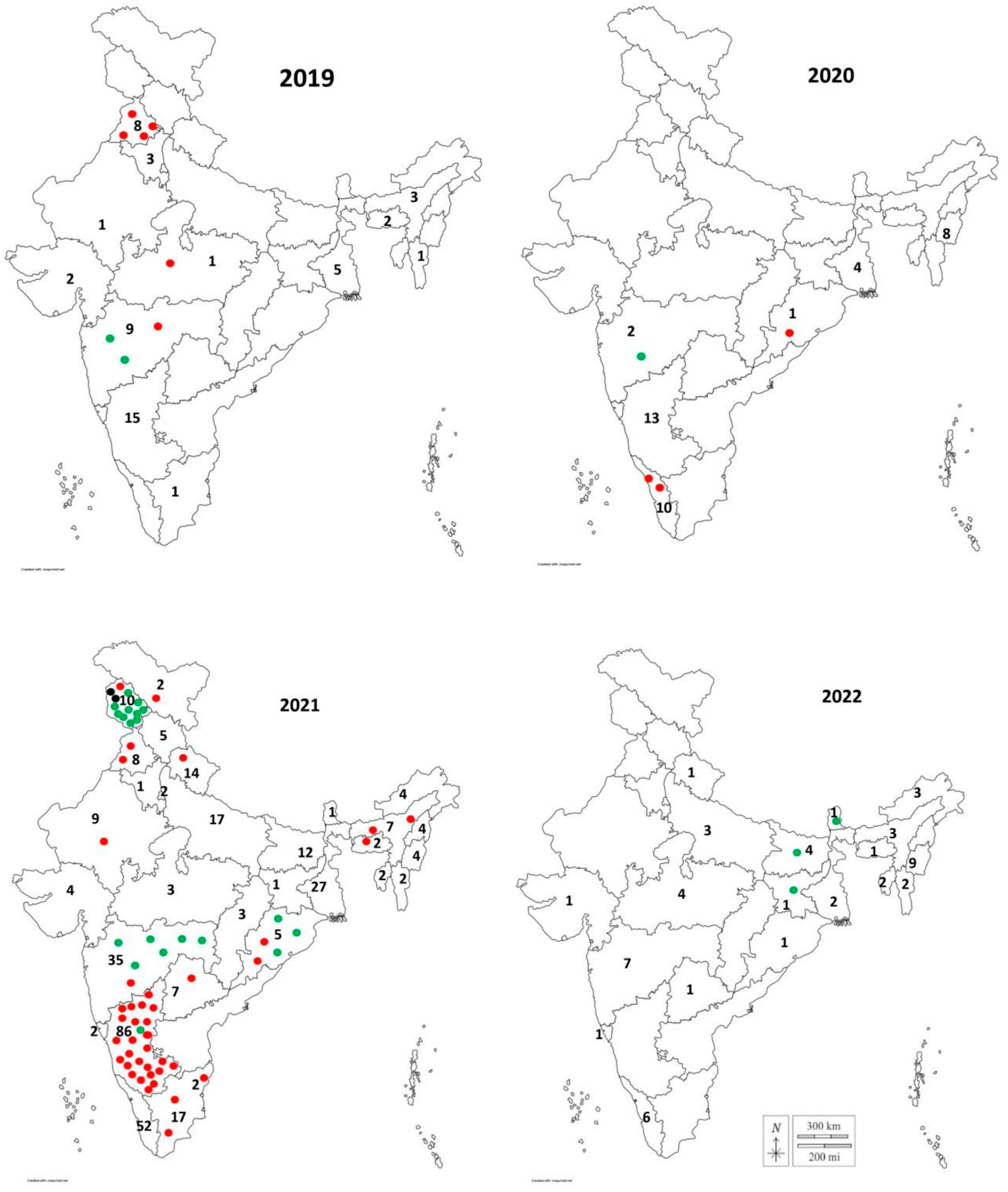
3.1. Serotype Detection and Virus Isolation
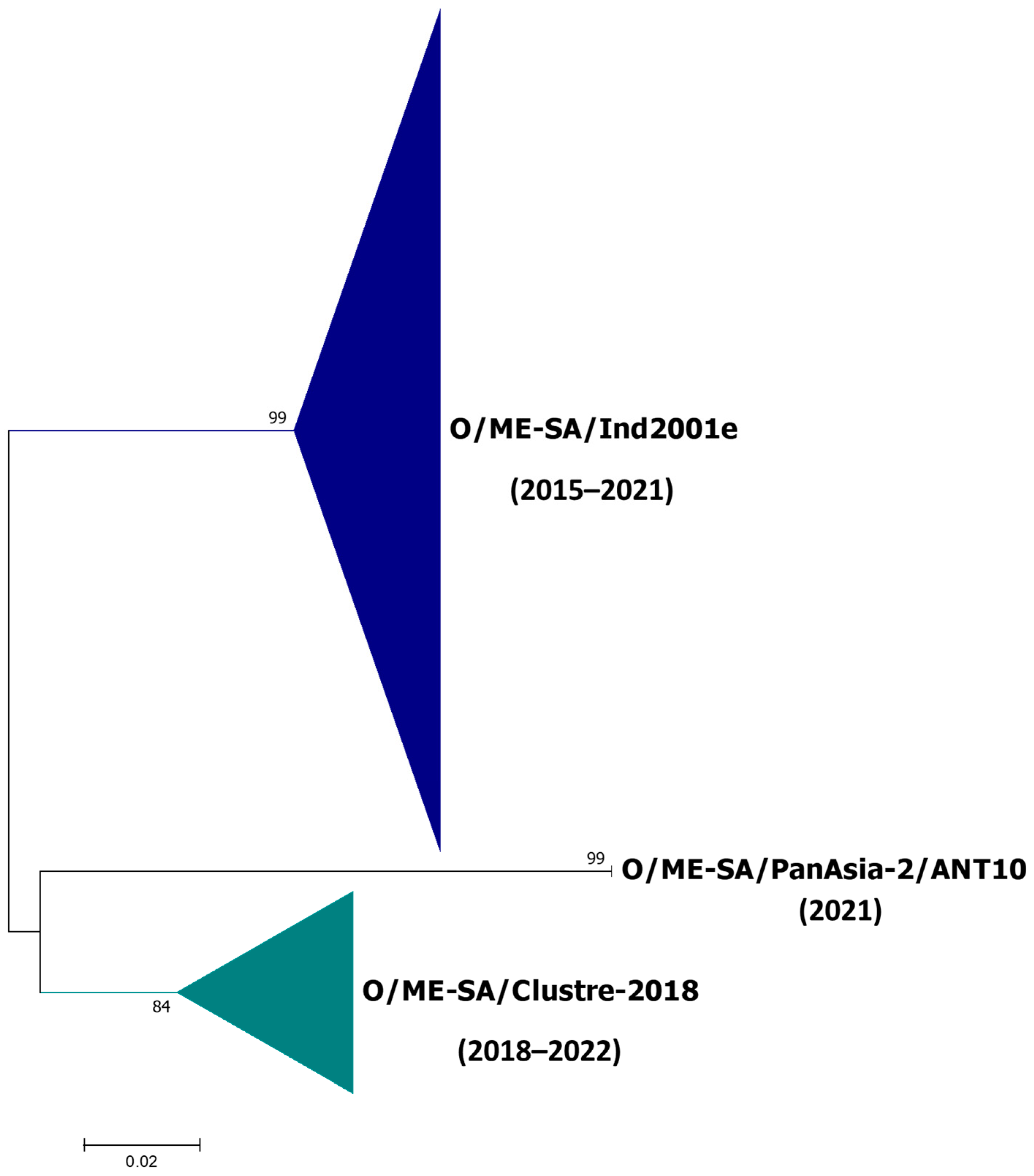
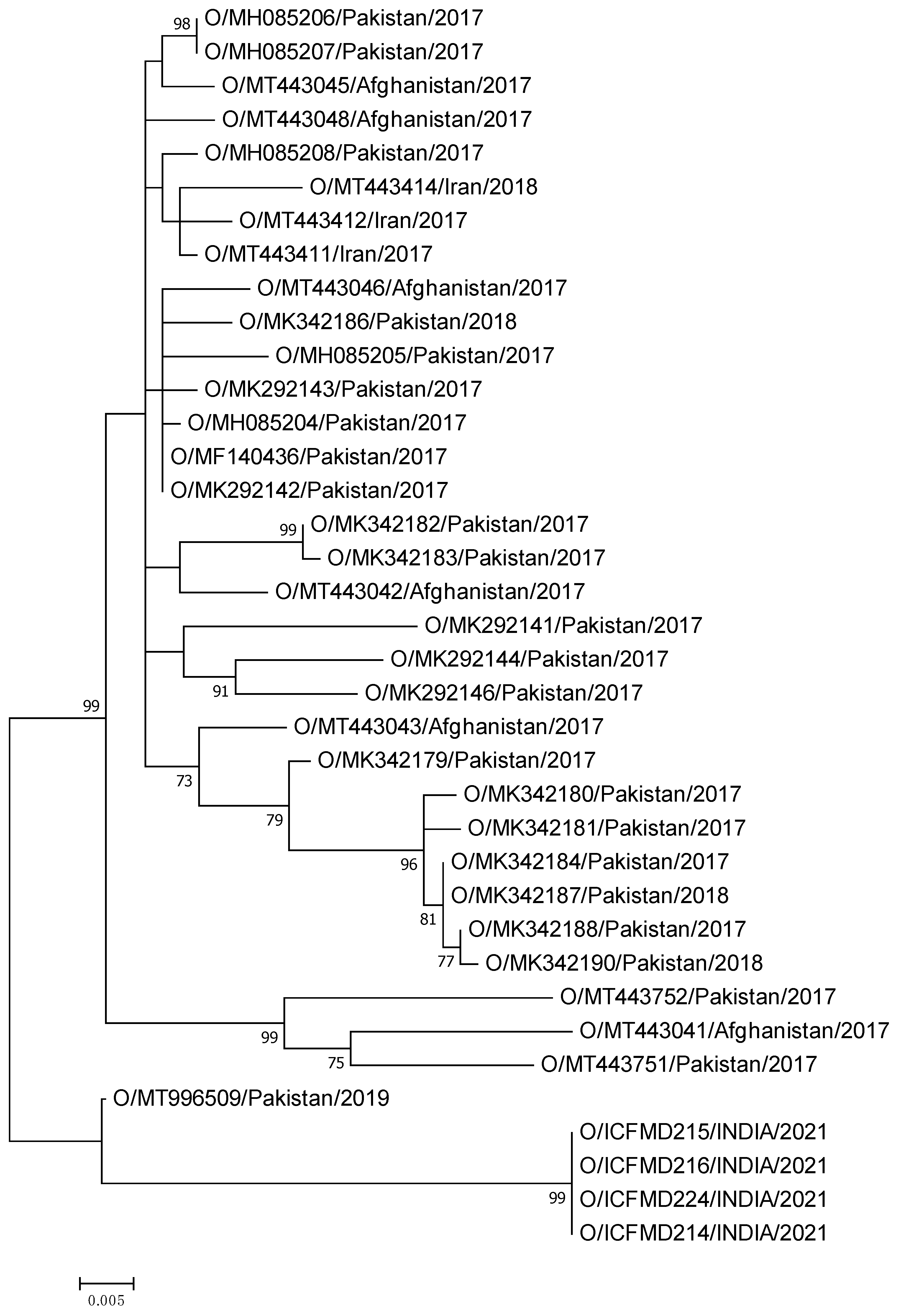
3.2. Phylogenetic Relationships
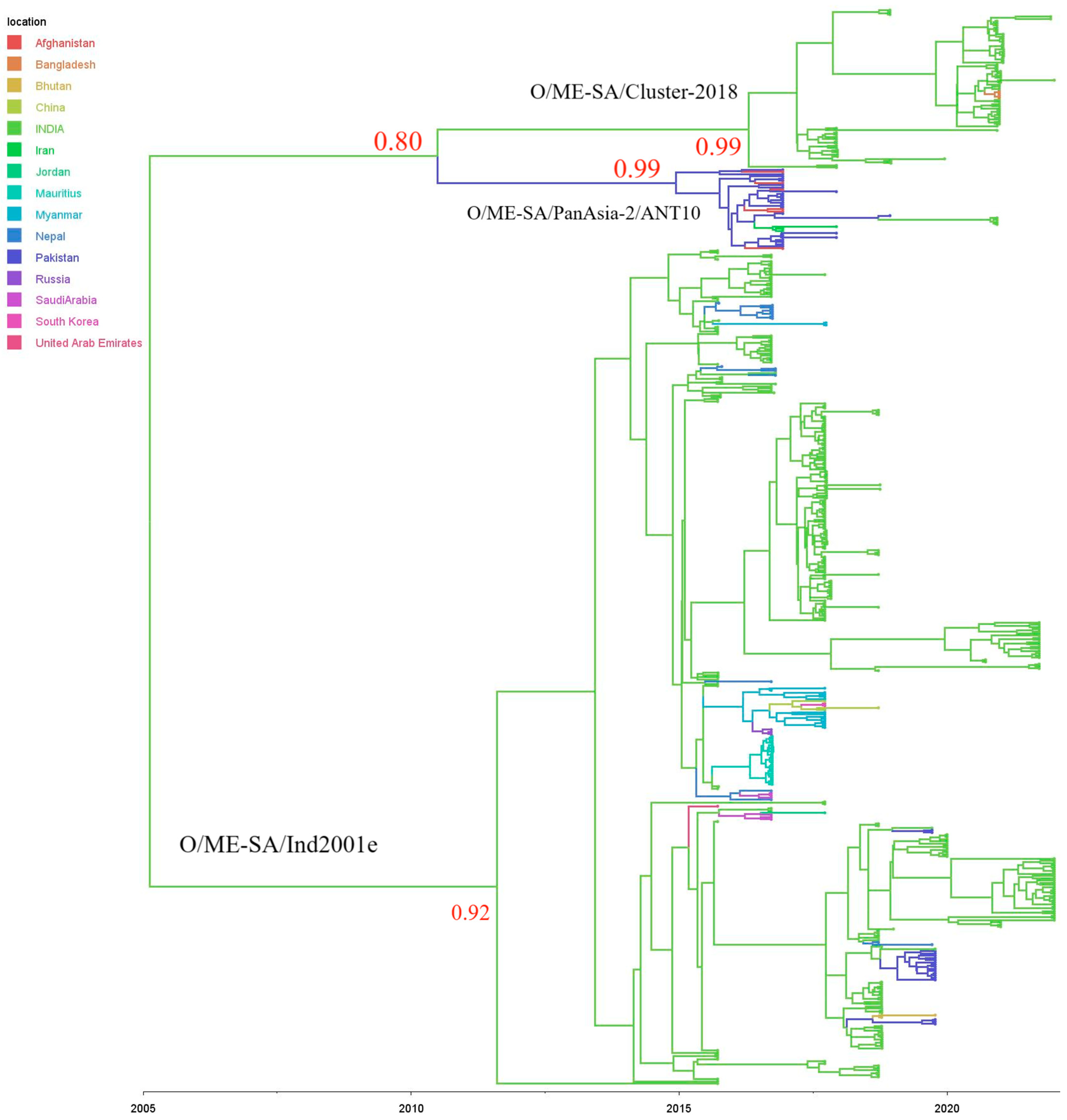
3.3. Phylodynamic Analyses
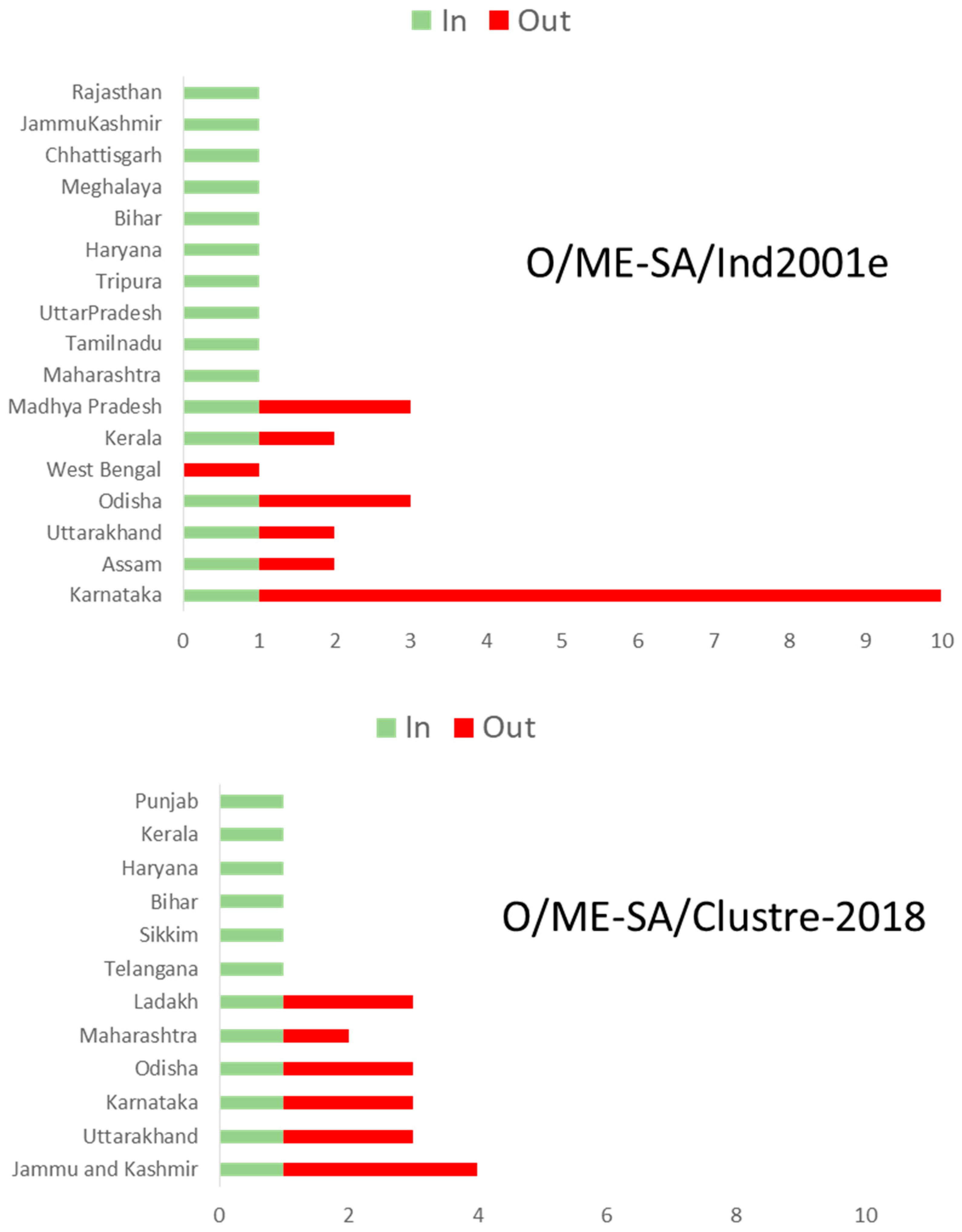
3.4. Lineage O/ME–SA/Ind2001e
3.5. Lineage O/ME–SA/Cluster-2018
3.6. Variations in Amino Acid
3.7. Vaccine Matching
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jamal, S.M.; Belsham, G.J. Foot-and-mouth disease: Past, present and future. Vet. Res. 2013, 44, 116. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, S.; Mohapatra, J.K.; Sahoo, N.R.; Sahoo, A.P.; Dahiya, S.S.; Rout, M.; Biswal, J.K.; Khulape, S.A.; Mallick, S.; Ranjan, R.; et al. Foot-and-mouth disease status in India during the second decade of the twenty-first century (2011–2020). Vet. Res. Commun. 2022, 46, 1011–1022. [Google Scholar] [CrossRef]
- Brito, B.P.; Mohapatra, J.K.; Subramaniam, S.; Pattnaik, B.; Rodriguez, L.L.; Moore, B.R.; Perez, A.M. Dynamics of widespread foot-and-mouth disease virus serotypes A, O and Asia-1 in southern Asia: A Bayesian phylogenetic perspective. Transbound. Emerg. Dis. 2017, 65, 696–710. [Google Scholar] [CrossRef]
- Subramaniam, S.; Mohapatra, J.K.; Sharma, G.K.; Biswal, J.K.; Ranjan, R.; Rout, M.; Das, B.; Dash, B.B.; Sanyal, A.; Pattnaik, B. Evolutionary dynamics of foot-and-mouth disease virus O/ME–SA/Ind2001 lineage. Vet. Microbiol. 2015, 178, 181–189. [Google Scholar] [CrossRef]
- Dahiya, S.S.; Subramaniam, S.; Biswal, J.K.; Das, B.; Prusty, B.R.; Ali, S.Z.; Khulape, S.A.; Mohapatra, J.K.; Singh, R.K. Genetic characterization of foot-and-mouth disease virus serotype O isolates collected during 2014–2018 revealed dominance of O/ME–SA/Ind2001e and the emergence of a novel lineage in India. Transbound. Emerg. Dis. 2021, 68, 3498–3508. [Google Scholar] [CrossRef]
- Bachanek-Bankowska, K.; Di Nardo, A.; Wadsworth, J.; Mioulet, V.; Pezzoni, G.; Grazioli, S.; Brocchi, E.; Kafle, S.C.; Hettiarachchi, R.; Kumarawadu, P.L.; et al. Reconstructing the evolutionary history of pandemic foot-and-mouth disease viruses: The impact of recombination within the emerging O/ME–SA/Ind-2001 lineage. Sci. Rep. 2018, 8, 14693. [Google Scholar] [CrossRef] [Green Version]
- Fish, I.; Stenfeldt, C.; Palinski, R.M.; Pauszek, S.J.; Arzt, J. Into the Deep (Sequence) of the Foot-and-Mouth Disease Virus Gene Pool: Bottlenecks and Adaptation during Infection in Naïve and Vaccinated Cattle. Pathogens 2020, 9, 208. [Google Scholar] [CrossRef] [Green Version]
- Mason, P.W.; Grubman, M.J.; Baxt, B. Molecular basis of pathogenesis of FMDV. Virus Res. 2003, 91, 9–32. [Google Scholar] [CrossRef]
- Knowles, N.J.; Samuel, A.R. Molecular epidemiology of foot-and-mouth disease virus. Virus Res. 2003, 91, 65–80. [Google Scholar] [CrossRef]
- Knowles, N.J.; Wadsworth, J.; Bachanek-Bankowska, K.; King, D.P. VP1 sequencing protocol for foot and mouth disease virus molecular epidemiology. Rev. Sci. Tech. 2016, 35, 741–755. [Google Scholar] [CrossRef] [Green Version]
- Hemadri, D.; Tosh, C.; Sanyal, A.; Venkataramanana, R. Emergences of a new strain of type O foot-and-mouth disease virus: Its phylogenetic and evolutionary relationship with PanAsia pandemic strain. Virus Genes 2002, 25, 23–34. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Rweyemamu, M.M.; Booth, J.C.; Head, M.; Pay, T.W. Micro-neutralization tests for serological typing and subtyping of foot-and-mouth disease virus strains. J. Hyg. 1978, 81, 107–123. [Google Scholar] [CrossRef] [Green Version]
- Ababneh, M.M.; Hananeh, W.; Bani Ismail, Z.; Hawawsheh, M.; Al-Zghoul, M.; Knowles, N.J.; Van Maanen, K. First detection of foot-and-mouth disease virus O/ME–SA/Ind2001e sublineage in Jordan. Transbound. Emerg. Dis. 2020, 67, 455–460. [Google Scholar] [CrossRef]
- Jamal, S.M.; Khan, S.; Knowles, N.J.; Wadsworth, J.; Hicks, H.M.; Mioulet, V.; Bin-Tarif, A.; Ludi, A.B.; Shah, S.A.A.; Abubakar, M.; et al. Foot-and-mouth disease viruses of the O/ME–SA/Ind-2001e sublineage in Pakistan. Transbound. Emerg. Dis. 2021, 68, 3126–3135. [Google Scholar] [CrossRef]
- Abubakar, M.; Syed, Z.; Manzoor, S.; Arshed, M.J. Deciphering Molecular Dynamics of Foot and Mouth Disease Virus (FMDV): A Looming Threat to Pakistan’s Dairy Industry. Dairy 2022, 3, 123–136. [Google Scholar] [CrossRef]
- Hagag, N.M.; Hassan, A.M.; Zaher, M.R.; Elnomrosy, S.M.; Shemies, O.A.; Hussein, H.A.; Ahmed, E.S.; Ali, M.H.; Ateay, M.; Abdel-Hakim, M.A.; et al. Molecular detection and phylogenetic analysis of newly emerging foot-and-mouth disease virus type A, Lineage EURO-SA in Egypt in 2022. Virus Res. 2022, 323, 198960. [Google Scholar] [CrossRef]
- Ranaweera, L.T.; Wijesundara, W.W.M.U.K.; Jayarathne, H.S.M.; Knowles, N.J.; Wadsworth, J.; Gray, A.; Adikari, A.M.J.B.; Weebadde, C.K.; Sooriyapathirana, S.D.S.S. Transboundary movements of foot and-mouth disease from India to Sri Lanka: A common pattern is shared by serotypes O and C. PLoS ONE 2019, 14, e0227126. [Google Scholar] [CrossRef]
- Arzt, J.; Fish, I.; Pauszek, S.J.; Johnson, S.L.; Chain, P.S.; Rai, D.K.; Rieder, E.; Goldberg, T.L.; Rodriguez, L.L.; Stenfeldt, C. The evolution of a super-swarm of foot-and-mouth disease virus in cattle. PLoS ONE 2019, 14, e0210847. [Google Scholar] [CrossRef] [Green Version]
- Paton, D.J.; Sumption, K.J.; Charleston, B. Options for control of foot-and-mouth disease: Knowledge, capability and policy. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2657–2667. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, M.; Parida, S. Foot and mouth disease vaccine strain selection: Current approaches and future perspectives. Expert Rev. Vaccines 2018, 17, 577–591. [Google Scholar] [CrossRef]
- Mahapatra, M.; Yuvaraj, S.; Madhanmohan, M.; Subramaniam, S.; Pattnaik, B.; Paton, D.J.; Srinivasan, V.A.; Parida, S. Antigenic and genetic comparison of foot-and-mouth disease virus serotype O Indian vaccine strain, O/IND/R2/75 against currently circulating viruses. Vaccine 2015, 33, 693–700. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Total Number of Clinical Samples Tested | Number of Samples Positive for Serotype O | Total Number of FMD Outbreaks | Number of Outbreaks by Serotype O |
|---|---|---|---|---|
| 2019 | 306 | 145 | 52 | 51 |
| 2020 | 215 | 94 | 46 | 38 |
| 2021 | 2824 | 1122 | 378 | 349 |
| 2022 | 411 | 141 | 55 | 51 |
| S No | Isolate ID | r-Value | Genetic Group |
|---|---|---|---|
| 1 | O/PD38/2018/Uttarakhand | 0.47 | O/ME–SA/Cluster-2018 |
| 2 | O/PD57/2018/Haryana | 0.33 | O/ME–SA/Cluster-2018 |
| 3 | O/PD224/2018/Kerala | 0.42 | O/ME–SA/Cluster-2018 |
| 4 | O/ICFMD202/2018/Tamilnadu | 0.71 | O/ME–SA/Cluster-2018 |
| 5 | O/ICFMD218/2018/Tamilnadu | 0.59 | O/ME–SA/Cluster-2018 |
| 6 | O/ICFMD/184/2021/Jammu and Kashmir | 0.93 | O/ME–SA/Cluster-2018 |
| 7 | O/ICFMD/189/2021/Jammu and Kashmir | 1 | O/ME–SA/Cluster-2018 |
| 8 | O/ICFMD/244/2021/Jammu and Kashmir | 1 | O/ME–SA/Cluster-2018 |
| 9 | O/ICFMD/345/2021/Jammu and Kashmir | 0.467 | O/ME–SA/Cluster-2018 |
| 10 | O/ICFMD/357/2021/Jammu and Kashmir | 1 | O/ME–SA/Cluster-2018 |
| 11 | O/ICFMD/441/2021/Jammu and Kashmir | 1 | O/ME–SA/Cluster-2018 |
| 12 | O/ICFMD/271/2021/Ladakh | 1 | O/ME–SA/Cluster-2018 |
| 13 | O/ICFMD/280/2021/Ladakh | 1 | O/ME–SA/Cluster-2018 |
| 14 | O/ICFMD/285/2021/Ladakh | 1 | O/ME–SA/Cluster-2018 |
| 15 | O/ICFMD/291/2021/Ladakh | 1 | O/ME–SA/Cluster-2018 |
| 16 | O/ICFMD/302/2021/Ladakh | 0.606 | O/ME–SA/Cluster-2018 |
| 17 | O/ICFMD/344/2021/Punjab | 1 | O/ME–SA/Cluster-2018 |
| 18 | O/ICFMD/366/2021/Punjab | 1 | O/ME–SA/Cluster-2018 |
| 19 | O/ICFMD/369/2021/Punjab | 1 | O/ME–SA/Cluster-2018 |
| 20 | O/ICFMD/350/2021/Maharashtra | 0.5 | O/ME–SA/Cluster-2018 |
| 21 | O/ICFMD/513/2021/Maharashtra | 1 | O/ME–SA/Cluster-2018 |
| 22 | O/ICFMD/522/2021/Maharashtra | 1 | O/ME–SA/Cluster-2018 |
| 23 | O/ICFMD/958/2021/Maharashtra | 0.834 | O/ME–SA/Cluster-2018 |
| 24 | O/ICFMD/850/2021/Rajasthan | 1 | O/ME–SA/Cluster-2018 |
| 25 | ICFMD/47/2022/Jharkhand | 1 | O/ME–SA/Cluster-2018 |
| 26 | ICFMD/160/2022/Sikkim | 0.901 | O/ME–SA/Cluster-2018 |
| 27 | ICFMD/182/2022/Bihar | 1 | O/ME–SA/Cluster-2018 |
| 28 | O/PD214/2018/HimachalPradesh | 0.31 | O/ME–SA/Ind2001e |
| 29 | O/PD400/2018/Uttarakhand | 0.53 | O/ME–SA/Ind2001e |
| 30 | O/PD403/2018/Uttarakhand | 0.51 | O/ME–SA/Ind2001e |
| 31 | O/ICFMD19/2019/Punjab | 0.71 | O/ME–SA/Ind2001e |
| 32 | O/ICFMD21/2019/Punjab | 0.43 | O/ME–SA/Ind2001e |
| 33 | O/ICFMD22/2019/Punjab | 0.52 | O/ME–SA/Ind2001e |
| 34 | O/ICFMD/241/2021/Odisha | 1 | O/ME–SA/Ind2001e |
| 35 | O/ICFMD/531/2021/Maharashtra | 0.655 | O/ME–SA/Ind2001e |
| 36 | O/ICFMD/472/2021/Tamilnadu | 1 | O/ME–SA/Ind2001e |
| 37 | O/ICFMD/534/2021/Tamilnadu | 0.841 | O/ME–SA/Ind2001e |
| 38 | O/ICFMD/548/2021/Karnataka | 1 | O/ME–SA/Ind2001e |
| 39 | O/ICFMD/556/2021/Karnataka | 0.501 | O/ME–SA/Ind2001e |
| 40 | O/ICFMD/572/2021/Karnataka | 1 | O/ME–SA/Ind2001e |
| 41 | O/ICFMD/591/2021/Karnataka | 0.88 | O/ME–SA/Ind2001e |
| 42 | O/ICFMD/594/2021/Karnataka | 1 | O/ME–SA/Ind2001e |
| 43 | O/ICFMD/606/2021/Karnataka | 0.726 | O/ME–SA/Ind2001e |
| 44 | O/ICFMD/622/2021/Karnataka | 0.72 | O/ME–SA/Ind2001e |
| 45 | O/ICFMD/643/2021/Karnataka | 0.785 | O/ME–SA/Ind2001e |
| 46 | O/ICFMD/658/2021/Karnataka | 0.9 | O/ME–SA/Ind2001e |
| 47 | O/ICFMD/669/2021/Karnataka | 0.649 | O/ME–SA/Ind2001e |
| 48 | O/ICFMD/688/2021/Karnataka | 0.88 | O/ME–SA/Ind2001e |
| 49 | O/ICFMD/689/2021/Karnataka | 1 | O/ME–SA/Ind2001e |
| 50 | O/ICFMD/755/2021/Karnataka | 0.841 | O/ME–SA/Ind2001e |
| 51 | O/ICFMD/770/2021/Karnataka | 0.72 | O/ME–SA/Ind2001e |
| 52 | O/ICFMD/803/2021/Karnataka | 1 | O/ME–SA/Ind2001e |
| 53 | O/ICFMD/820/2021/Karnataka | 0.9 | O/ME–SA/Ind2001e |
| 54 | O/ICFMD/828/2021/Karnataka | 1 | O/ME–SA/Ind2001e |
| 55 | O/ICFMD/829/2021/Karnataka | 0.856 | O/ME–SA/Ind2001e |
| 56 | O/ICFMD/830/2021/Karnataka | 0.85 | O/ME–SA/Ind2001e |
| 57 | OICFMD/890/2021/Assam | 0.494 | O/ME–SA/Ind2001e |
| 58 | O/ICFMD/214/2021/Jammu and Kashmir | 0.878 | O/ME–SA/PanAsia-2/ANT10 |
| 59 | O/ICFMD/224/2021/Jammu and Kashmir | 0.834 | O/ME–SA/PanAsia-2/ANT10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahiya, S.S.; Subramaniam, S.; Mohapatra, J.K.; Rout, M.; Biswal, J.K.; Giri, P.; Nayak, V.; Singh, R.P. Foot-and-Mouth Disease Virus Serotype O Exhibits Phenomenal Genetic Lineage Diversity in India during 2018–2022. Viruses 2023, 15, 1529. https://doi.org/10.3390/v15071529
Dahiya SS, Subramaniam S, Mohapatra JK, Rout M, Biswal JK, Giri P, Nayak V, Singh RP. Foot-and-Mouth Disease Virus Serotype O Exhibits Phenomenal Genetic Lineage Diversity in India during 2018–2022. Viruses. 2023; 15(7):1529. https://doi.org/10.3390/v15071529
Chicago/Turabian StyleDahiya, Shyam Singh, Saravanan Subramaniam, Jajati Keshari Mohapatra, Manoranjan Rout, Jitendra Kumar Biswal, Priyabrata Giri, Vinayak Nayak, and Rabindra Prasad Singh. 2023. "Foot-and-Mouth Disease Virus Serotype O Exhibits Phenomenal Genetic Lineage Diversity in India during 2018–2022" Viruses 15, no. 7: 1529. https://doi.org/10.3390/v15071529